
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mixing of exciton and charge-transfer states in light-harvesting complex Lhca4†
Vladimir I.
Novoderezhkin
*a,
Roberta
Croce
b,
Md.
Wahadoszamen
bc,
Iryna
Polukhina
b,
Elisabet
Romero
b and
Rienk
van Grondelle
b
aA. N. Belozersky Institute of Physico-Chemical Biology, Moscow State University, Leninskie Gory, 119992, Moscow, Russia. E-mail: novoder@belozersky.msu.ru
bDepartment of Biophysics, Faculty of Sciences, VU University Amsterdam, De Boelelaan 1081, 1081 HV Amsterdam, The Netherlands
cDepartment of Physics, University of Dhaka, Dhaka-1000, Bangladesh
First published on 23rd June 2016
Abstract
Lhca4 is one of the peripheral antennae of higher plant photosystem I and it is characterized by the presence of chlorophyll a with absorption and emission bands around 30 nm red-shifted compared to those of the other chlorophylls associated with plant complexes. In this work we have investigated the origin of this red shift by using the recent structure of Lhca4 (Qin et al., Science, 2015, 348, 989) to build an exciton model that includes a charge-transfer (CT) state mixed with the excited-state manifold. A simultaneous quantitative fit of absorption, linear dichroism, fluorescence, and Stark absorption spectra of the wild-type Lhca4 and NH mutant (where the sites involved in CT are affected) enables us to determine the origin of the CT state and explore its spectral signatures. A huge borrowing of dipole strength by the CT, accompanied by anomalous broadening and red-shifting of the fluorescence as well as dramatic changes in the Stark spectrum, can be accounted for by a model implying an exciton-type mixing between excited states and CT states.
Introduction
Photosystem I (PSI) is one of the two multi-pigment–protein complexes responsible for the first steps of oxygenic photosynthesis.1–3 In plants PSI is composed of a core complex, containing about 100 chlorophylls (Chls) including the reaction center, where charge separation occurs, and an outer antenna (LHCI), composed of 4 subunits (Lhca1–4) assembled on one side of the PSI-core4 in the form of two heterodimers, Lhca1–4 and Lhca2–3.5 Lhcas belong to the light-harvesting complex multigenic family6 and the recent crystal structures show that they share a very similar folding and coordinate 13–15 Chls (a and b) and 3–4 carotenoids each in highly conserved binding sites.7,8 However, their spectroscopic properties are largely different.2A typical feature of PSI is the presence of Chls that absorb/emit at an energy lower than the reaction center.9,10 These Chls extend the absorption of the complex to the far red, but they also slow down the transfer of excitation energy to the reaction center9,11–13 as energy migration needs to be thermally activated.14 In plants the most red forms (spectral components shifted to the red from the main absorption) are associated with the Lhca subunits15 and in particular with Lhca3 and Lhca4.16–18 In addition to a red-shifted spectrum, the red forms have peculiar properties. The Huang-Rhys factor for the red Chls of LHCI (S = 2.9) is at least three times larger than that for the bulk Chls.19 This observation has led to the suggestion that the red spectral forms originate from a mixing between the excited and charge transfer (CT) states within tightly packed Chl dimers.19–21
The red-most emission (with fluorescence (FL) peaking at ∼730 nm) originates from the Lhca4 subunit. Mutations affecting the ChlA4, A5 and B5 sites (or a602, a603, a609 in the notation of Qin et al.7 that will be used in this work) result in the disappearance of the broad 730 nm FL peak (together with the 710 nm shoulder present in absorption) and produce a narrow FL band peaking at 685 nm.22,23 Therefore, it was concluded that the red spectral forms are determined by interactions within the strongly coupled cluster a602–a603–a609, and predominantly between the a603 and a609 Chls.22,23 It was also shown that the absorption of the red forms can be tuned by changes in the protein environment close to these interacting Chls.22,24 The substitution of the natural ligand of Chl 603, Asn by His (in the following indicated as the NH mutant), showed that, although the new residue was still able to coordinate the Chl, the red absorption largely disappeared.22 More detailed studies of the red spectral forms (including FL line-narrowing) revealed a dramatic difference in homogeneous and inhomogeneous broadening and in reorganization energy between the wild-type (WT) Lhca4 and the NH mutant.20 This has supported the idea that the red form originates from the mixing of low energy excitonic transitions with a CT state.
The availability of the high resolution structure of Lhca47,8 opens the way to perform a quantitative modeling of the spectral properties of the WT Lhca4 and the NH mutant and to explore the mixing of exciton and CT states. Here we present an exciton model of Lhca4 obtained by extracting the site energies of the pigments (Chls a and b) from a simultaneous fit of the absorption (OD), linear dichroism (LD), Stark absorption (SA), and FL spectra at 77 K. Comparing the spectra of the WT and NH mutant we assign the origin of the CT state and define its parameters and the degree of mixing with the excited states. We conclude that the mixing of exciton and CT states has been used to control the spectral properties of plant LHCs by precisely modulating the positions and protein environments of the chlorophylls.
The model
The structural model of Lhca4 from the study of Qin et al.7 is shown in Fig. 1. According to structural data7 the Lhca4 complex contains 15 Chls when assembled as a hetero-dimer and associated with the PSI core. However, there is convincing evidence that the reconstituted complex used in our experiments, which is a monomer, does not contain some of the pigments, namely Chl 601, 617, and 618. Chl 601 is located at the periphery of the complex and it seems to be present in Lhc trimers25 and dimers,7,8 but not in monomers.26 Chl 618 is located at the interface between the two monomers in the Lhca1–Lhca4 heterodimer and it is thus very likely absent in the Lhca4 monomer. The same is true for Chl 617, which in the structure is coordinated to H99. Indeed, the mutation at this residue did not lead to a loss of Chls, indicating that this Chl is not present in the monomer24 and that its binding is probably stabilized by the dimerization.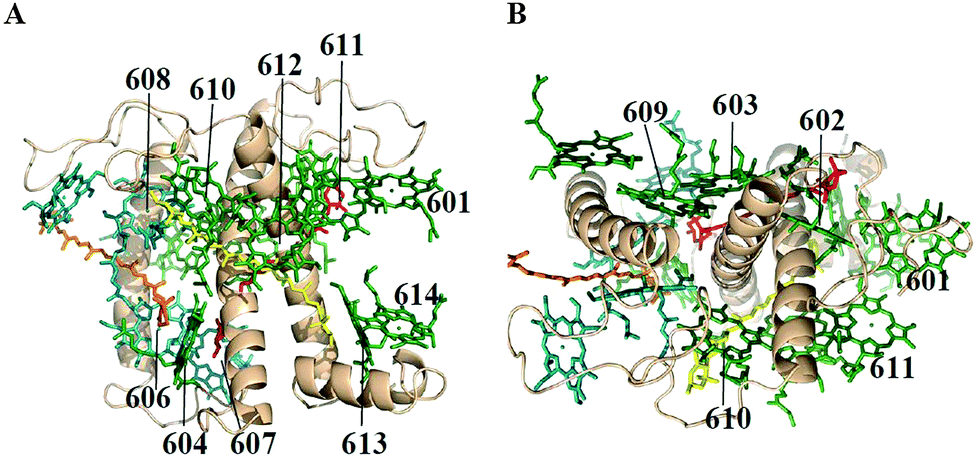 | ||
| Fig. 1 Structure of Lhca47 with a view perpendicular to the membrane normal (A) and along the membrane normal from the stromal side (B), including Chls a (green), Chls b (blue), lutein (yellow), violaxanthin (red), and β-carotene (orange). | ||
In our modeling we have used the full 15-state model of the complex, as well as the 12-state model of reconstituted Lhca4, containing 9 Chls a (602–604, 609–614) and 3 Chls b (606–608) based on the results of mutation analysis.23 We also include a charge-transfer (CT) state corresponding to a charge separation between two Chls a, i.e. a603 and a609. In order to check this assignment and to explore the properties of the CT state we perform a quantitative structure-based modeling of the steady-state spectra (including SA) of the WT Lhca4 and NH mutant in which the ligand for Chl a603 is substituted.22 In the mutant the Chl a603 is still present, but the two interacting Chls in the a603–609 dimer are expected to be in a different geometry, thus breaking the formation of the hypothesized CT state.
Exciton couplings have been calculated from the structural data in the point-dipole approximation supposing that the dipoles of the Chl Qy transitions are directed from NB to ND atoms and the distance between the dipoles is equal to the distance between the Mg atoms of the two Chls. The same approach was used in our modeling of the LHCII complex.27 Couplings calculated for LHCII using more sophisticated methods28–31 showed a good correlation with our earlier model,27 as was pointed out by Müh et al.29 We conclude that the point dipole approximation can be used as a first step in modeling of the Lhca4 complex whose structure is very similar to that of LHCII.32 The effective transition dipole moments are supposed to be 4 and 3.4 Debye (D) for Chls a and b, respectively (see ESI†). The static dipole moments of Chls a and b are supposed to be parallel to the corresponding transition dipoles. Their values are taken from the SA data for the photosystem II reaction center (PSII-RC)33 and LHCII34 and adjusted from the fit of the SA spectra of Lhca4, giving the values of 1 and 2.8 D for Chls a and b, respectively.
The formation of the CT state can occur within a tightly packed pair of Chls. The best candidate is the a603–609 dimer with shortest pigment–pigment spacing (in the Chls a region). In addition, the red spectral forms (reflecting a mixing with the CT state) are changed dramatically upon mutation affecting the environment of these two Chls. Therefore, we propose that the CT state corresponds to a charge separation within the a603–609 dimer. Coupling of the a603 and a609 sites to the CT state necessary to explain the observed spectral features is 150 cm−1, as estimated from the best fit of the spectra (see the detailed discussion in the ESI†). The diabatic CT state (i.e. the CT state in the absence of coupling to the excited states) is dipole forbidden. The static dipole moment for the CT state is directed from the a603 to the a609 pigment and equals to 20 D (this value is based on our estimate for the PSII-RC35 and adjusted from the fit of the Lhca4 SA). The transition dipole moment S1 → S2 corresponding to a double excitation of the pigment (that should be taken into account when calculating the SA signal35,36) is 0.7 of the S0 → S1 transition dipole. This value is based on modeling of nonlinear responses for LHCII27 and adjusted from the fit of the Lhca4 SA (see ESI†). No excited-state absorption from the CT state is included.
The unperturbed site energies of the 12 pigments and the energy of the CT state in monomeric Lhca4 have been extracted from a simultaneous quantitative fit of the OD, LD, FL, and SA spectra at 77 K both for the WT complex and the NH mutant. In the NH mutant the CT is absent, whereas the a603 and a609 sites are supposed to be blue shifted (due to the mutation) by 250 and 200 cm−1, respectively, as compared to the position of the a603 and a609 diabatic states in the WT.22 Other site energies in the NH mutant are taken to be the same as those in the WT complex. An evolutionary based search has led us to several plausible sets of site energies. The spectra are fitted using the modified Redfield theory in the pure exciton basis, supposing an exciton-type mixing between the CT and the excited states.35 Expressions needed to calculate the OD, LD, FL, and SA spectra are given in the ESI.†
For modeling of Lhca4 we have taken the same spectral density (Brownian oscillator) as for LHCII37 with some adjustment of the damping constant γ0 and coupling strength λ0 for the overdamped part and the total Huang-Rhys S factor for the 48 high-frequency vibrations (the S factor has been adjusted by uniform scaling of all the couplings to vibrations λj, where j = 1–48). For Chls a the thus adjusted values are: γ0 = 40 cm−1, λ0 = 200 cm−1, and S = 0.84. We suppose that the shape of the electron–phonon spectral density is the same for Chls a, Chls b, and CT states, but the couplings (λ0, λj) are different. From the fit of the spectra we have found that the λ0 and λj values determined for Chls a should be multiplied by 0.8, and 3.2 for Chls b, and CT states, respectively.
Coupling to slow conformations of the complex is accounted for using the model of the static disorder, implying uncorrelated random shifts of the site energies described by a Gaussian distribution with a width of σ (FWHM). The disorder values for the Chls a, Chls b, and CT states determined from the fit are σ = 96, 67, and 144 cm−1 for Chls a, Chls b, and CT states, respectively.
Results
The exciton couplings between the 12 pigments are listed in Table 1. They are very similar to the couplings calculated with the same approximations for the LHCII complex,38 which is expected based on the very similar pigment organization of the two complexes.7,8,32 At the stromal side there are two Chl a clusters, i.e. strongly coupled a602–603–609 and a610–611–612 trimers (the corresponding interaction energies are shown in bold in Table 1), and one Chl b, i.e. b608. Couplings between this Chl b and the two Chl a clusters are strong enough (italicized bold in Table 1), so that fast (sub-ps) b → a conversion is expected at the stromal side of the Lhca4 complex. The only essential difference with LHCII is the different identity of the 609 pigment which is Chl a in Lhca4. Thus, instead of the a602–603 dimer we now have the a602–603–609 trimer in the a-region. At the luminal side there is one Chl a dimer, i.e. a613–614 (with strong coupling shown in bold in Table 1) and one heterotrimer a604–b606–b607. Strong coupling within this trimer (as shown in Table 1) determines fast conversion from the b-region to the a604 site. Couplings between these two luminal-side clusters are weak as well as their couplings to the stromal side pigments (corresponding to slow ps migration).| a602 | a603 | a609 | a610 | a611 | a612 | b608 | a613 | a614 | a604 | b606 | b607 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a602 | 0 | 33.03 | −26.44 | −10.32 | 8.52 | 16.91 | −8.96 | −5.50 | 2.37 | 6.65 | 6.02 | 6.36 |
| a603 | 33.03 | 0 | 171.41 | 14.34 | −4.21 | −1.64 | 8.16 | 2.59 | −6.83 | −5.52 | −12.72 | 1.31 |
| a609 | −26.44 | 171.41 | 0 | 2.76 | 5.53 | −2.80 | 51.74 | −3.25 | 2.84 | −7.44 | −3.97 | −13.13 |
| a610 | −10.32 | 14.34 | 2.76 | 0 | −25.37 | 16.43 | 50.35 | 7.12 | −2.21 | −5.39 | −3.30 | 0.69 |
| a611 | 8.52 | −4.21 | 5.53 | −25.37 | 0 | 130.33 | 4.35 | −9.49 | 5.57 | −3.17 | −2.82 | −3.02 |
| a612 | 16.91 | −1.64 | −2.80 | 16.43 | 130.33 | 0 | −0.59 | −2.75 | 2.07 | 1.90 | 3.27 | 2.95 |
| b608 | −8.96 | 8.16 | 51.74 | 50.35 | 4.35 | −0.59 | 0 | −1.99 | 1.47 | −1.00 | −3.20 | −4.39 |
| a613 | −5.50 | 2.59 | −3.25 | 7.12 | −9.49 | −2.75 | −1.99 | 0 | −76.54 | 0.59 | 1.31 | 0.60 |
| a614 | 2.37 | −6.83 | 2.84 | −2.21 | 5.57 | 2.07 | 1.47 | −76.54 | 0 | −3.49 | −2.21 | −3.57 |
| a604 | 6.65 | −5.52 | −7.44 | −5.39 | −3.17 | 1.90 | −1.00 | 0.59 | −3.49 | 0 | 82.60 | 28.32 |
| b606 | 6.02 | −12.72 | −3.97 | −3.30 | −2.82 | 3.27 | −3.20 | 1.31 | −2.21 | 82.60 | 0 | 59.16 |
| b607 | 6.36 | 1.31 | −13.13 | 0.69 | −3.02 | 2.95 | −4.39 | 0.60 | −3.57 | 28.32 | 59.16 | 0 |
The character of the exciton mixing between the pigments is determined not only by the pigment–pigment couplings, but also strongly dependent on the site energies (including the spread of the unperturbed energies plus additional random shifts induced by the disorder). The search of the best fit of the data has led us to several sets of site energies listed in Table 2. In all these sets the a604 pigment is in the middle region between the b- and a-bands. In the a-region the red-most sites are a603 and a613, in contrast to LHCII, where the a610–611–612 sites are the red-most ones.
| a Configurations with an improved fit of the WT LD. | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Site no. | — | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | — | — | 13 |
| Pigment | 601 | 602 | 603 | 604 | 606 | 607 | 608 | 609 | 610 | 611 | 612 | 613 | 614 | 617 | 618 | CT |
| Chla/b | a | a | a | a | b | b | b | a | a | a | a | a | a | a | b | |
| E1 | — | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 172 172 |
15068 |
15494 |
15834 |
15788 |
15741 |
15259 |
15339 |
15225 |
15197 |
15090 |
15240 |
— | — | 14910 |
| E2 | — | 15205 |
15039 |
15453 |
15856 |
15793 |
15731 |
15232 |
15345 |
15223 |
15190 |
15068 |
15190 |
— | — | 14879 |
| E3 | — | 15132 |
14990 |
15511 |
15755 |
15899 |
15802 |
15274 |
15263 |
15223 |
15188 |
15058 |
15197 |
— | — | 14856 |
| E4 | — | 15204 |
15037 |
15440 |
15755 |
15879 |
15802 |
15287 |
15295 |
15186 |
15297 |
15084 |
15230 |
— | — | 14897 |
| E5a | — | 15163 |
15066 |
15315 |
15720 |
15820 |
15720 |
15232 |
15223 |
15177 |
15227 |
15043 |
15219 |
— | — | 14898 |
| E6a | — | 15093 |
15019 |
15438 |
15714 |
15832 |
15702 |
15186 |
15302 |
15160 |
15248 |
15026 |
15328 |
— | — | 14866 |
| E7a | — | 15142 |
15063 |
15447 |
15794 |
15748 |
15701 |
15238 |
15238 |
15191 |
15220 |
15000 |
15141 |
— | — | 14894 |
An example of a simultaneous fit of the 8 spectra is shown in Fig. 2. The measured spectra display at least five essential features present in the WT spectra and disappearing in the NH mutant, which can be considered as signatures of the CT state. The WT spectra display (i) a red shoulder in OD (at 690–720 nm), (ii) a shoulder in the LD in the same region, (iii) anomalously big Stokes shift of the FL (to 730 nm), (iv) anomalous broadening of the FL profile, and (v) three intense red peaks of the SA (in the 680–740 nm region). In the NH mutant the red shoulder in OD and LD disappears, the FL peak becomes narrow and moves to the ‘normal’ position near 680 nm, and the three red peaks of the SA disappear (or at least are significantly reduced and modified).
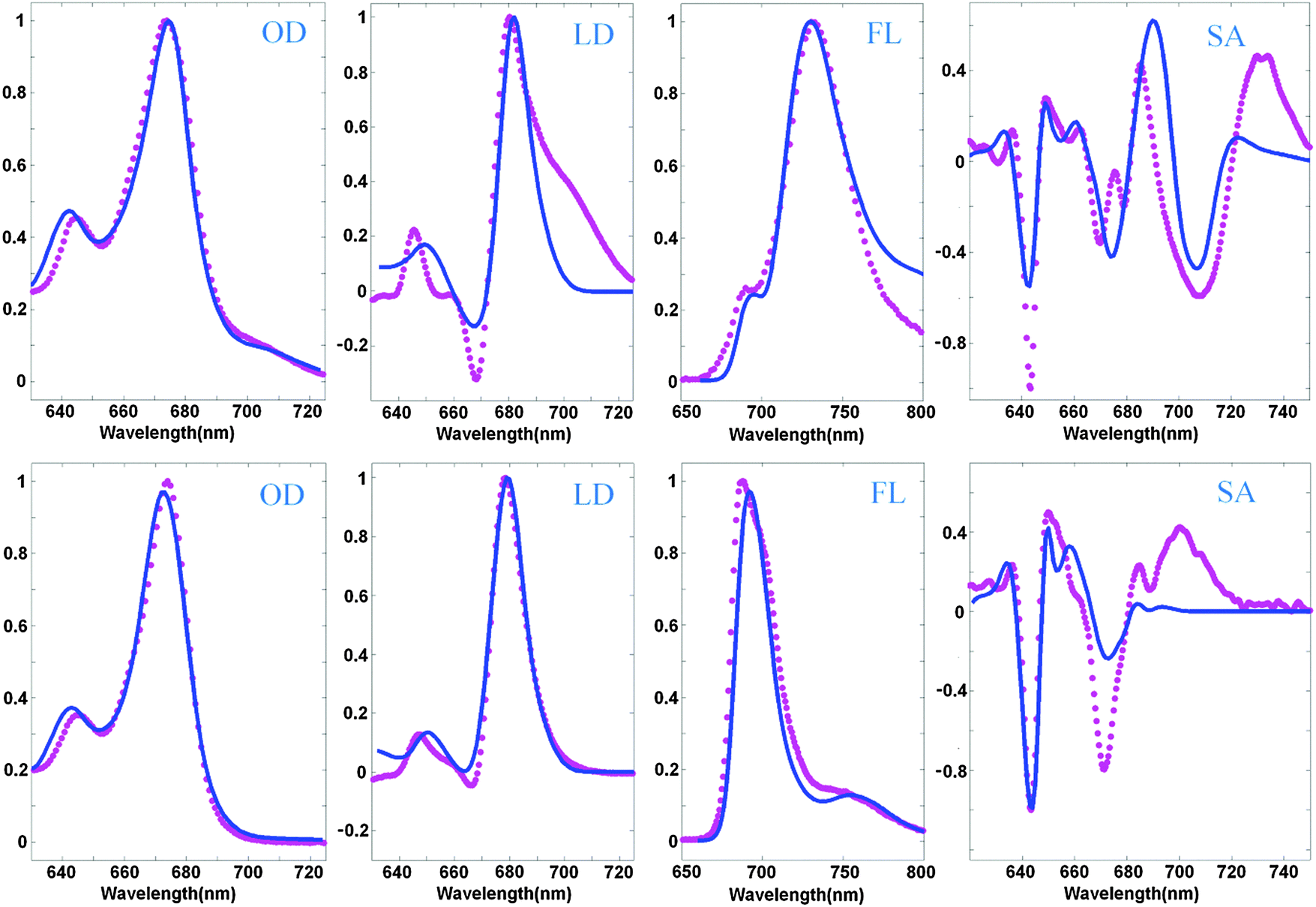 | ||
| Fig. 2 Simultaneous fit of the OD, LD, FL, and SA spectra of Lhca4 WT (top) and the NH mutant (bottom) at 77 K. Measured spectra (magenta points) are shown together with the calculated ones (blue lines). The OD, LD, and FL data are taken from ref. 20 and 22, and measured SA spectra are from ref. 39. The spectra are normalized to unity. Modeling is based on the high-resolution structure of Lhca4 and modified Redfield theory in a pure exciton basis. The exciton model includes 12 pigments (15 pigments from the structural data except 601, 617, and 618) and one CT state (corresponding to charge transfer between the 603 and 609 pigments). The site energies correspond to the E2 set from Table 2. | ||
The model assuming the presence of the a603–609 CT state and implying an exciton-type mixing of this CT state with the excited-state manifold allows a consistent explanation of the observed spectra, including the most important differences between the WT and NH samples. The red shoulder in OD, and broadening and red-shifting of the FL for the WT are reproduced at a quantitative level. In order to reproduce the weak 690 nm peak in the WT FL we suppose that some subpopulation (about 8%) of the WT complexes does not mix with the CT state. This assumption is in agreement with the character of the FL peak fluctuations observed for a single Lhca4 complex.40
The main peaks of the SA signal are reproduced as well, but it is difficult to reproduce the amplitude of the red-most 730 nm peak in the WT and the 700 nm peak in the NH spectrum. Notice that the normal ‘excitonic’ SA spectrum (without CT) should not contain any components in the red 690–730 nm region (as displayed by the calculated profile in Fig. 2). One can hypothesize that the red forms still present in NH may contain some CT state around 700 nm that is almost invisible in OD, but that gives a small shoulder in the FL and sizable contribution to the red region of the SA profile. For instance, one can suppose that some part of NH complexes still contains some charge separation within the a603–609 dimer, but this CT state is 3–5 times more weakly coupled to the excited states of the a603 and a609 pigments. Preliminary calculations show that such a model gives an additional 700 nm shoulder in FL and increases the Stark amplitude in the 680–700 nm region. Interestingly, similar features have been observed in the Stark fluorescence (SF) data for this mutant.39
The LD spectrum is satisfactorily reproduced in the absence of CT (i.e. for the NH mutant), but the WT LD contains a huge red tail that cannot be explained by our current model. Some borrowing of dipole strength by CT (needed to reproduce the amplitude of the red sub-band of OD at 690–720 nm) produces only a very weak red shoulder in the calculated LD. Trying to improve the fit of the red wing of the WT LD we always destroy the shape of the FL profiles. In principle, it is possible to shift a613 to the red (see configurations E5–7 in Table 2), producing a more localized state that contributes to the red tail of the LD together with the CT state (as shown in Fig. 3). But in the NH mutant (where CT disappears) this contribution remains, leading to a red shift of FL, and some extra red-tail in OD and LD (Fig. 3). Most probably the discrepancies in the LD fit are connected with the simplified character of the exciton-CT mixing in our model (see Discussion).
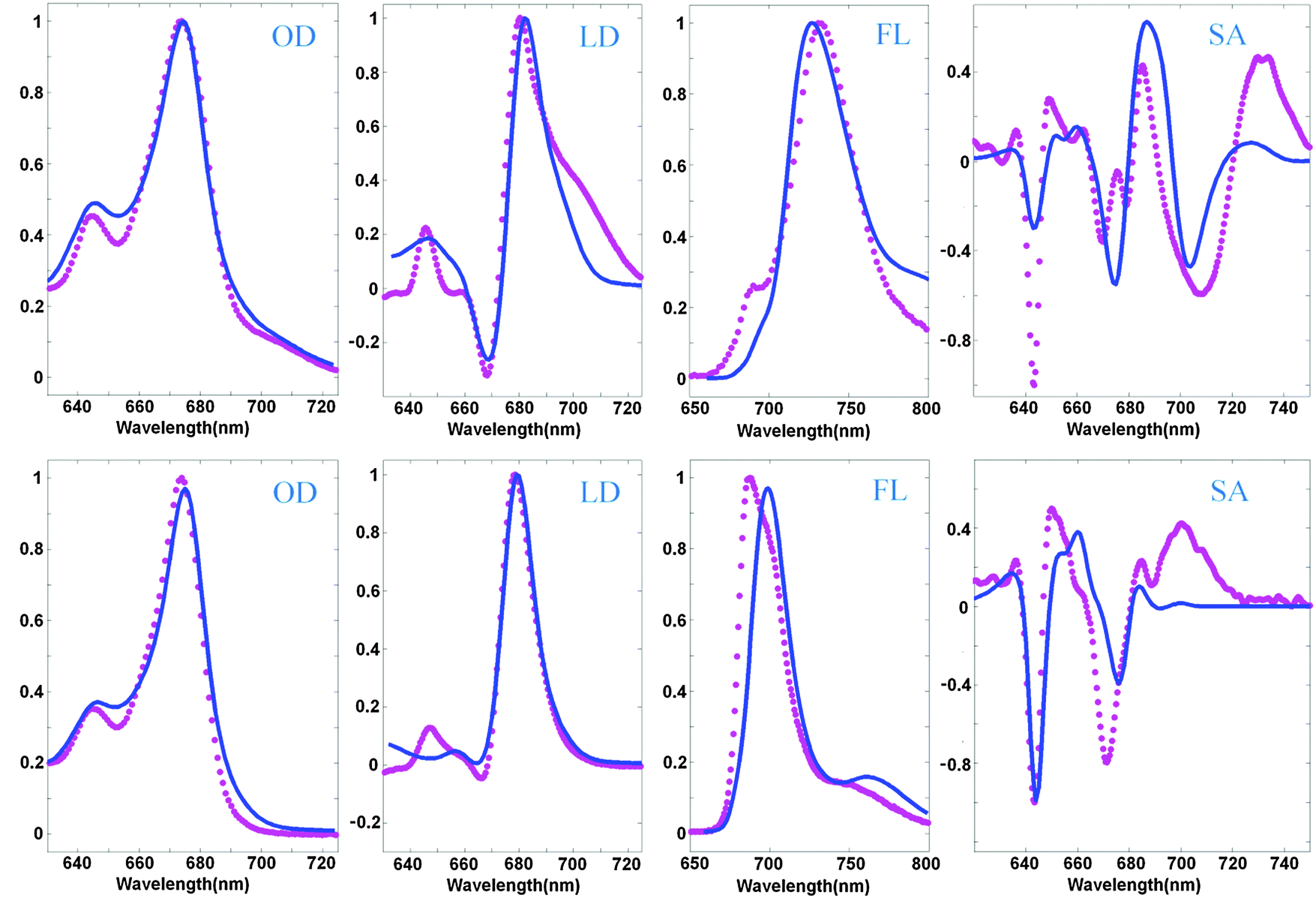
Besides the 12-state model described above we have also checked models including more pigments. We have found that including the extra pigments does not improve the fit. A decent quality of the fit can be obtained with the model that includes the Chl a617 pigment. Including all the three sites a601, a617, and b618 further decreases the quality of the fit.
Notice that a617 is located close to the a603–609 dimer, but there is no strong exciton mixing between them (interaction energies with a603 and a609 in the dipole–dipole approximation are 11 and 37 cm−1, respectively). As a result, the presence of a617 does not change significantly the exciton structure in the red region, in particular it does not improve the fit of the red tail of the LD. On the other hand, the center-to-center distance between a617 and a609 (1.02 nm) is comparable with the intradimer a603–609 spacing (0.89 nm), possibly allowing for the presence of one additional CT state, i.e. with a charge separation between the a617 and a609. Although the a617 pigment is not present in monomeric complexes used in this study, it can be present in dimeric complexes (Lhca4–Lhca1), where it might contribute to the red spectral forms due to the formation of additional CT states.20
Knowledge of the site energies extracted from the fit makes it possible to explore the structure of the exciton and mixed exciton–CT states of the Lhca4 complex. Here we will do this using the site energies corresponding to the E2 set from Table 2. In Fig. 4 we show the calculated OD spectrum averaged over disorder together with its 13 exciton components (emerging from mixing of the 12 excited states and one CT state). For each of these exciton components we show a distribution of the dipole strengths calculated for 2500 realizations of the disorder. The dipole strengths are shown as a function of the wavelength of the pure electronic zero-phonon transitions (for each exciton level in every realization of the disorder). Note that the zero-phonon lines (ZPLs) (i.e. spectral components corresponding to zero-phonon transitions) are shifted to the red with respect to the absorption maxima of the exciton components. For each realization we count the exciton states in increasing order of energy, i.e. from k = 1 to 13. Participation of the pigments in these states (averaged over disorder) is shown in Fig. 5. Notice that the pigments can contribute differently to certain exciton states for different realizations, so that the participation of many pigments in any state k does not always reflect a delocalization, but may contain statistical averaging (meaning that the k-th state can be localized at different pigments for different realizations of the disorder).
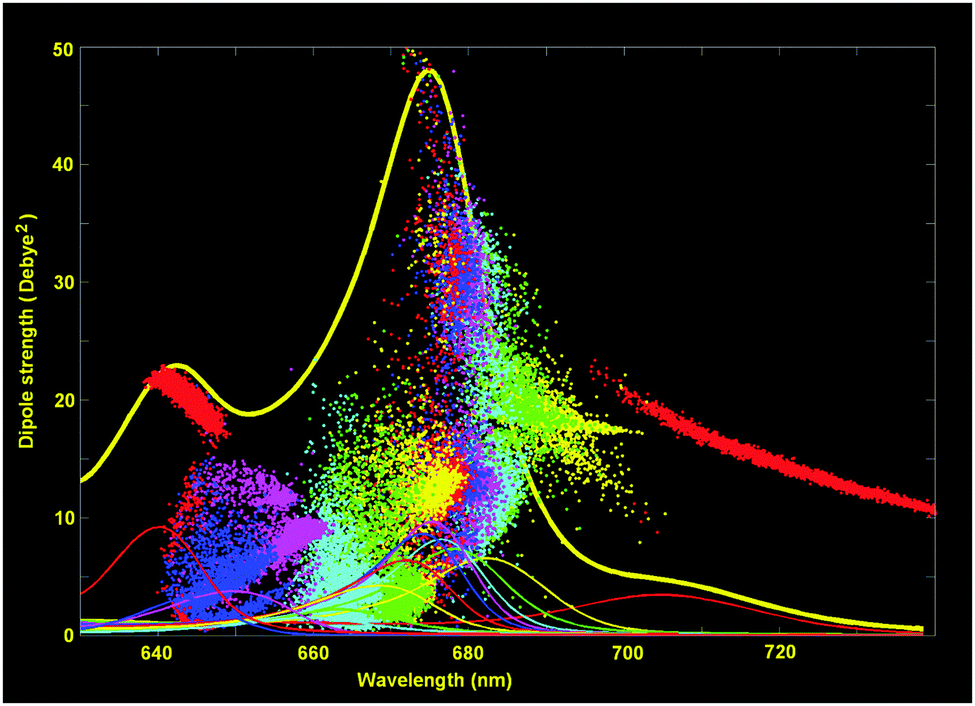 | ||
| Fig. 4 Exciton structure of the WT Lhca4 absorption. The calculated OD spectrum (thick yellow line) is shown together with the 13 exciton components averaged over disorder (thin lines). The points show the distribution of the dipole strengths vs. ZPL positions for 2500 realizations of the disorder (dipole strengths for different exciton components are shown in the same colors as the absorption profiles of the corresponding components). The site energies correspond to the E2 set from Table 2. | ||
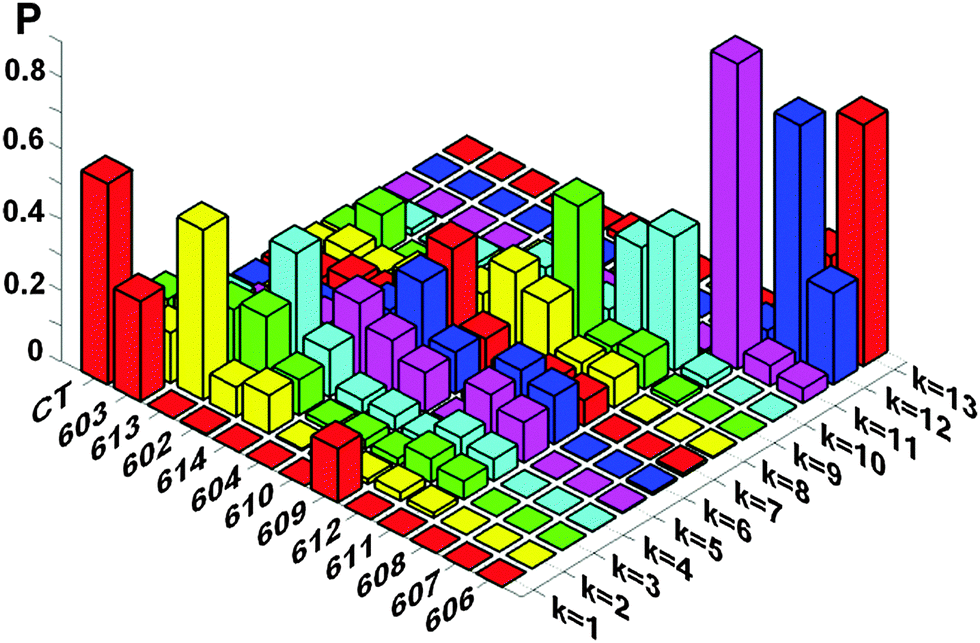 | ||
| Fig. 5 Participation of the pigments (and CT diabatic state) in the exciton states of Lhca4. The exciton states are counted in increasing order of energy (their colors are the same as in Fig. 4). Participation of the pigments in these states is averaged over disorder. Calculation is done with the same site energies as in Fig. 4, i.e. with the E2 set from Table 2. | ||
The lowest k = 1 state (shown by red in Fig. 4 and 5) is determined by the CT state mixed with a603 and (to less extend) with a609. The dipole strength borrowed by the CT state from the excited states is 10–20 D2 (depending on the position of the CT state). The averaged dipole strength of the lowest state is about 14 D2, corresponding to 0.87 of the monomeric dipole strength for Chl a (16 D2). This means that the CT state is really strongly mixed with the a603–609 dimer borrowing on average 44% of its total dipole strength.
The second state (k = 2, yellow) is predominantly localized on a613. The next state (k = 3, green) may contain (depending on the realization of the disorder) contributions from a613 or a602. The k = 4 state (cyan) is mostly determined by a602. Notice that depending on the disorder the a602 and a613 sites can be coherently mixed with a603 and a614, respectively. It means that the k = 2–4 states can be localized (at a613 or a602), but also may contain contributions from delocalized configurations. That is why these states are characterized by a big spread of dipole strengths, i.e. varying from 8 to 36 D2 in different realizations as shown in Fig. 4. A similar (or even bigger) spread of dipole strengths is observed for the next set of states (i.e. k = 5–7 states shown in magenta, blue, and red) that are determined by the delocalized lower exciton state of the a610–611–612 trimer. These delocalized contributions are statistically superimposed with more localized states, corresponding to the higher exciton level of the a613–614 dimer (predominantly localized at a614), and with the monomeric a604 state.
The next group of states (k = 8–10, shown in yellow, green and cyan) corresponds to the higher exciton levels of the stromal-side Chl a clusters, i.e. the a610–611–612 trimer and the higher level of the a602–603–609 trimer, localized at a609. In contrast to the superradiant lower states of these clusters, the higher states are weakly allowed (with a dipole strength of less than 5 D2 in most of the realizations).
The higher states k = 11–13 are determined by the three b-sites, i.e. a monomeric b608 on the stromal side (k = 11, magenta), and the luminal-side dimer b606–607 (k = 12–13 states, shown in blue and red). The energy difference between the b606 and b607 is comparable with the disorder value, meaning that in realization with a big energy gap the excited states can be localized, thus giving dipole strengths close to the monomeric value (i.e. 10.2 D2 for Chl b). In realization with a small gap between the two sites we obtain delocalization within the b606–607 dimer with the weakly allowed k = 12 and superradiant k = 13 state (with dipole strengths around 20 D2 in the 640–650 region).
More information about contributions of the pigments to the exciton states can be obtained by calculating the density of exciton states Dk together with the distribution of the pigment participations Dn (see ESI†). Distribution Dn shows the participation of the n-th pigment to the exciton states (summed over all states) as a function of the excited-state energy. This function defined in ref. 41 was originally denoted as ‘the exciton state pigment distribution’.
Fig. 6a shows the participation of the diabatic states from the strongly coupled block a602–603–609–CT that includes three excited states mixed with the CT state. The a603 distribution (green) displays three maxima reflecting its mixing with the a609 and CT states. The CT distribution (blue) has two maxima due to its mixing with a603. The blue-shifted a609 site is mixed to a lesser extent with the a603 and CT states, as reflected by relatively weak maxima near 685 and 720 nm (red curve). The a602 site (magenta) is predominantly localized (due to relatively weak coupling with a603). Comparing the pigment participations with the density of exciton states, we conclude that the states k = 4 and 9 are determined mostly by contributions from a602 and a609, respectively, whereas the states k = 1 and 3 are delocalized and contain a mixing of the a603, a608, and CT state (as we have also shown in Fig. 5).
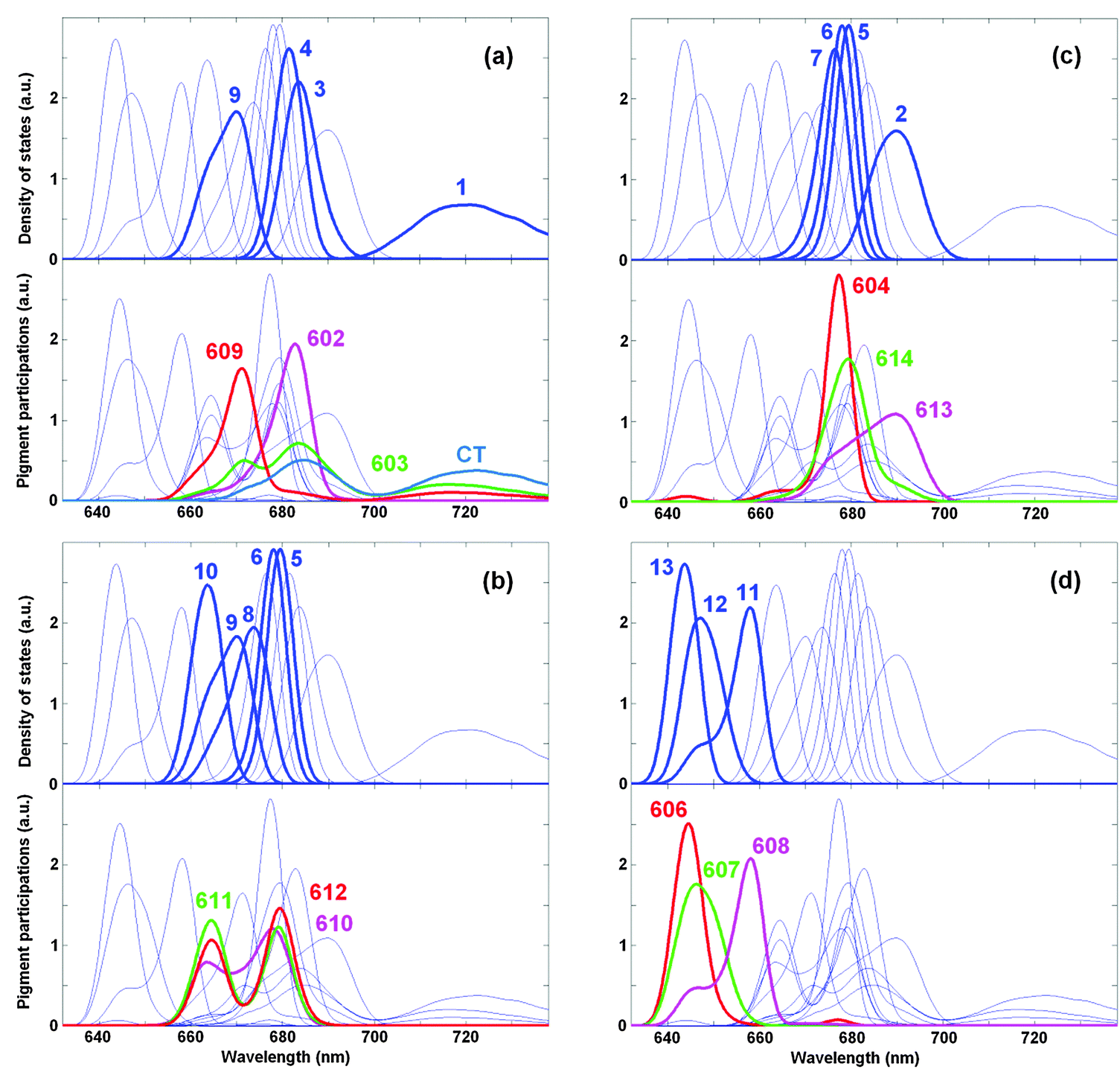 | ||
| Fig. 6 Density of exciton states Dk and distribution of the pigment participations Dn for the WT Lhca4. Calculation is done with the same site energies as in Fig. 4 and 5. Functions Dk and Dn are shown in arbitrary (but the same for Dk and Dn) units. The four frames (a–d) highlight the distributions of different groups of pigments (shown by thick red, magenta, green, and blue lines). The exciton states with predominant contributions from these pigments are highlighted by thick blue lines (numbering of the exciton states k = 1–13 is the same as in Fig. 4 and 5). Thin blue lines show the Dk distributions for all the states and the Dn distributions for all the pigments for a reference. | ||
The pigments from the trimer a610–611–612 give a distribution with two maxima due to strong coupling between a611 and a612 superimposed with some coherent admixture of a610 (Fig. 6b). These pigments contribute to the states k = 5, 6, 8, 9, and 10 (depending on the disorder).
Fig. 6c shows the participation of the Chls a from the luminal side, including the localized a604 site and the dimer a613–614. Notice that the intra-dimer coupling is relatively strong (see Table 1), but the red-shifting of a613 breaks the mixing of the two sites. As a result, instead of a distribution with two maxima (corresponding to the strongly mixed limit) we observe only weak shoulders in the a613 and a614 distributions. The a613 contributes mostly to the k = 2 state, whereas a614 and a604 contribute to the states k = 5–7 depending on the disorder.
In the b-region there are three sites, i.e. b606–607–608 participating in the states k = 10, 11, and 12, respectively. Notice that the distributions of the b606 and b607 participations are closer to each other than the distributions of the k = 12 and 13 states, reflecting some exciton mixing within the b606–607 dimer with a moderate coupling.
Discussion and conclusions
The most intriguing feature of the Lhca4 complex is connected with the CT character of the lowest absorption band, where the degree of the exciton–CT mixing is probably the largest among all known antenna complexes and reaction centers (RCs). Consequently, the main challenge in explaining the functioning of the complex is the choice of an adequate physical model for the coupling between excited states and CT states.The exciton–CT mixing was studied theoretically for bacterial RCs,42–46 for the PSI-core,47 and for the isolated RC of photosystem II (PSII-RC).35 In order to reproduce the details of the lineshape, a quite sophisticated treatment is needed, where a nontrivial interplay between the electronic and nuclear degrees of freedom is included explicitly.45,46 But these models are restricted to linear spectral responses. In a more simple approach, based on modified Redfield theory, the exciton–CT mixing is treated in the basis of pure electronic states, and the resulting eigenstates are then dressed by phonon lineshapes (including broadening and reorganization shifts depending on the wavefunctions of the mixed eigenstates).35 Such an approach is convenient in the calculation of steady-state (SA) and time-resolved nonlinear responses (transient absorption (TA), two-dimensional electronic spectroscopy (2DES)). In a series of recent papers this approach was successfully used in modeling relaxation and charge separation dynamics in PSII-RC.48–51 The same approach is used in the present study of Lhca4, where the exciton–CT mixing produces the same features as in the PSII-RC (red tail of the absorption, FL shift/broadening, and increased SA peaks on the red). However, in the case of Lhca4, all these effects are much more pronounced.
The degree of exciton–CT mixing in Lhca4 is really huge. Thus, the red-most CT state borrows on average 44% of the total dipole strength of the a603–609 dimer, i.e. 14 D2, which is 0.87 of the monomeric Chl a dipole strength. This is close to the result of fluorescence studies of Lhca4, yielding an emitting dipole strength for the red state of 0.64–0.84 (in monomeric units), suggesting a strong mixing between the excited and CT states.52 For comparison, in the PSII-RC the lowest CT state borrows about 4.9 D2 corresponding to an emitting dipole strength of 0.3 in monomeric units.35
Another manifestation of the exciton–CT mixing is the enhanced phonon coupling of the red state. To account for the spectral properties of the red state we have to assume a 3.2 times increased Huang-Rhys factor for the CT state, i.e. S = 2.7. This value is close to the result of hole-burning studies, giving S = 2.9, which is the largest Huang-Rhys factor found for any photosynthetic antenna system.19
Our modeling confirms a commonly used assumption that the CT states are more strongly coupled to the environmental degrees of freedom due to an additional Coulomb term (appearing due to the huge static dipole interacting with the surrounding charged environment).35,45–47,53 In our fit the coupling to phonons and the disorder value for CT state are increased by a factor of 3.2 and 1.5, respectively. Without such scaling it is impossible to explain the red shift and broadening of FL and the shape of SA.
The most frustrating result of our modeling is connected with the discrepancy in fitting the red tail of the LD spectrum in the presence of the CT state. This means that although we can reproduce the dipole strength of the red state (necessary to explain its isotropic absorption and emitting properties), we still cannot correctly describe the observed LD on the red. For some configurations it is possible to obtain the LD shoulder with the same relative amplitude as in OD, but this amplitude is still smaller than the measured one (in addition, the other LD bands are not well fitted in this case). Generally, the difference in the LD spectrum with and without the CT state is given by the details of the mixing within the a602–603–609–CT cluster with some small contribution from the a610–611–612 sites. Mixing in a pure electronic basis (that we are dealing with in the modified Redfield picture) produces a superposition of the ZPLs with the weights given by the couplings and energy gaps between the states. For example, the CT state will be mixed mostly with the a603 ZPL that is closer than the a609 ZPL (as shown in Fig. 4). A more realistic picture may include a mixing of the electron-vibrational states with different displacements along the nuclear coordinates (an example of such a picture was developed recently for a reduced model of the PSII-RC54). In this case the mixing of vibrational sublevels of lower states with ZPLs of higher states can strongly change the exciton structure (changing the delocalization of the states and producing additional states by splitting of ZPLs). For example, the vibrational satellites of the CT state (that are in quasi-resonance with the higher sites, including a602, 603, 609, 610, 612) can dramatically increase the character of mixing of the CT state with these states, thus changing the whole LD and, in particular changing (increasing) the relative amplitude of LD in the red. In the near future we will further develop this model to include complex structures as Lhca4.
Besides the simplified approach to the exciton-CT mixing, our results can be affected by other shortcomings of the modified Redfield theory (connected with secular and Markovian approximations and perturbative treatment of the off-diagonal system-bath coupling). The secular approximation results in too fast transfer rates between weakly coupled isoenergetic states, as was demonstrated by comparing with the exact hierarchical equation of motion (HEOM) method.55 We have also seen that the lineshapes for a real antenna systems are different in the modified Redfield and HEOM.55 Recently it was shown that a non-Markovian theory for the off-diagonal fluctuations, termed complex time dependent Redfield theory,56 gives more accurate lineshapes (noticeably closer to the exact HEOM shapes) than the modified Redfield. Obviously, a nonperturbative approach like HEOM is the best solution to the problem, but in contrast to Redfield this method is suitable only for small systems with a simple spectral density. Notice in this respect that recently a nonperturbative stochastic path integral method has been developed,57 where an arbitrary spectral density can be used. As a good alternative to numerically expensive nonperturbative methods the hybrid cumulant expansion method has been proposed,58 although perturbative, but still capable of generating reliable spectra and transfer rates.
In this paper we did not model the excitation dynamics of the complex. Obviously, for a large part these dynamics are expected to be very similar to those of LHCII. For example one can expect the same sub-ps Chl b-Chl a conversion, the same dynamics on the lumenal side, and similar (about 10 ps) times of equilibration between the stromal-side and luminal-side pigments.38 Population of the red-most CT state is expected to be fast due to strong exciton-CT mixing (implying fast exciton-type relaxation from the excited states). But equilibration in the red region will also contain the slow components, because the two lowest states (k = 1 and 2) are localized on the pigments from the stromal (a603–CT) and lumenal (a613) sides, respectively. Notice in this respect that earlier experimental studies revealed a slow (5 ps) component involving red pigments.59–61 According to present modeling we can assign this component to transfer from the k = 2 state (localized at a613) to the red-most k = 1 a603–CT state.
Another fundamental difference between Lhca4 and LHCII is connected with the localization of excitation after equilibrium. In Lhca4 the a610–611–612 trimer is no longer the reddest (and most populated), in contrast to LHCII. The equilibrated excitations are now localized at a603/CT and a613, i.e. at the opposite (to the a610–611–612 trimer) side, that is closer to the PSI core. The most populated a603/CT state is located close to the linker Chls within LHCI.
We conclude that the Lhca4 complex is a nice example of how the exciton–CT mixing can be used to control the spectral properties of plant LHCs in order to optimize light-harvesting by creating directed energy transfers.
Acknowledgements
V. N. was supported by the Russian Foundation for Basic Research (Grant No. 15-04-02136) and by the visitors grant from the Dutch Foundation for Basic Research (NWO, Grant No. 040.11.428). I. P. and R. C. are supported by the the BioSolar Cells open innovation consortium, financed by the Dutch Ministry of Economic affairs, Agriculture and Innovation, by the Foundation for Fundamental research on Matter (FOM) and by the European Research Council via the ERC consolidator grant ASAP (281341). Md. W., E. R. and R. v. G. were supported by the VU University Amsterdam, the Laserlab-Europe Consortium, the TOP grant (700.58.305) from the Foundation of Chemical Sciences part of NWO, the advanced investigator grant (267333, PHOTPROT) from the European Research Council. E. R. and R. v. G. were also supported by the EU FP7 project PAPETS (GA 323901). R. v. G. gratefully acknowledges his Academy Professor grant from the Netherlands Royal Academy of Sciences (KNAW) and his support from the Canadian Institute for Advanced Research (CIFAR).References
- B. Gobets and R. van Grondelle, Biochim. Biophys. Acta, 2001, 1057, 80–99 CrossRef
.
- R. Croce and H. van Amerongen, Photosynth. Res., 2013, 116, 153–166 CrossRef CAS PubMed
- N. Nelson and W. Junge, Annu. Rev. Biochem., 2015, 84, 659–683 CrossRef CAS PubMed
- A. Ben Shem, F. Frolow and N. Nelson, Nature, 2003, 426, 630–635 CrossRef CAS PubMed
- E. Wientjes and R. Croce, Biochem. J., 2011, 433, 477–485 CrossRef CAS PubMed
- S. Jansson, Trends Plant Sci., 1999, 4, 236–240 CrossRef PubMed
- X. Qin, M. Suga, T. Kuang and J.-R. Shen, Science, 2015, 348, 989–995 CrossRef CAS PubMed
- Y. Mazor, A. Borovikova and N. Nelson, eLife, 2015, 4, e07433 Search PubMed
- B. Gobets, J. T. M. Kennis, J. A. Ihalainen, M. Brazzoli, R. Croce, L. H. M. van Stokkum, R. Bassi, J. P. Dekker, H. van Amerongen, G. R. Fleming and R. van Grondelle, J. Phys. Chem. B, 2001, 105, 10132–10139 CrossRef CAS
-
N. V. Karapetyan, E. Schlodder, R. van Grondelle and J. P. Dekker, in Photosystem I: the light-driven plastocyanin ferredoxin oxidoreductase, Advances in photosynthesis and respiration, ed. J. H. Golbeck, Springer, Dordrecht, 2006, vol. 24, pp. 177–192 Search PubMed
- L. O. Palsson, C. Flemming, B. Gobets, R. van Grondelle, J. P. Dekker and E. Schlodder, Biophys. J., 1998, 74, 2611–2622 CrossRef CAS PubMed
- E. Engelmann, G. Zucchelli, A. P. Casazza, D. Brogioli, F. M. Garlaschi and R. C. Jennings, Biochemistry, 2006, 45, 6947–6955 CrossRef CAS PubMed
- E. Wientjes, H. van Amerongen and R. Croce, Biochim. Biophys. Acta, 2013, 1827, 420–426 CrossRef CAS PubMed
- R. C. Jennings, G. Zucchelli, R. Croce and F. M. Garlaschi, Biochim. Biophys. Acta, Bioenerg., 2003, 1557, 91–98 CrossRef CAS
- R. Croce, G. Zucchelli, F. M. Garlaschi and R. C. Jennings, Biochemistry, 1998, 37, 17355–17360 CrossRef CAS PubMed
- V. H. R. Schmid, K. V. Cammarata, B. U. Bruns and G. W. Schmidt, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 7667–7672 CrossRef CAS
- H. Zhang, H. M. Goodman and S. Jansson, Plant Physiol., 1997, 115, 1525–1531 CAS
- S. Castelletti, T. Morosinotto, B. Robert, S. Caffarri, R. Bassi and R. Croce, Biochemistry, 2003, 42, 4226–4234 CrossRef CAS PubMed
- J. A. Ihalainen, M. Ratsep, P. E. Jensen, H. V. Scheller, R. Croce, R. Bassi, J. E. I. Korppi-Tommola and A. Freiberg, J. Phys. Chem. B, 2003, 107, 9086–9093 CrossRef CAS
- R. Croce, A. Chojnicka, T. Morosinotto, J. A. Ihalainen, F. van Mourik, J. P. Dekker, R. Bassi and R. van Grondelle, Biophys. J., 2007, 93, 2418–2428 CrossRef CAS PubMed
- E. Romero, M. Mozzo, I. H. M. van Stokkum, J. P. Dekker, R. van Grondelle and R. Croce, Biophys. J., 2009, 96, L35–L37 CrossRef CAS PubMed
- T. Morosinotto, J. Breton, R. Bassi and R. Croce, J. Biol. Chem., 2003, 278, 49223–49229 CrossRef CAS PubMed
- T. Morosinotto, M. Mozzo, R. Bassi and R. Croce, J. Biol. Chem., 2005, 280, 20612–20619 CrossRef CAS PubMed
- E. Wientjes, G. Roest and R. Croce, Biochim. Biophys. Acta, 2012, 1817, 711–717 CrossRef CAS PubMed
- Z. Liu, H. Yan, K. Wang, T. Kuang, J. Zhang, L. Gui, X. An and W. Chang, Nature, 2004, 428, 287–292 CrossRef CAS PubMed
- X. Pan, M. Li, T. Wan, L. Wang, C. Jia, Z. Hou, X. Zhao, J. Zhang and W. Chang, Nat. Struct. Mol. Biol., 2011, 18, 309–315 CAS
- V. I. Novoderezhkin, M. A. Palacios, H. van Amerongen and R. van Grondelle, J. Phys. Chem. B, 2005, 109, 10493–10504 CrossRef CAS PubMed
- J. S. Frähmcke and P. J. Walla, Chem. Phys. Lett., 2006, 430, 397–403 CrossRef
- F. Müh, M. E. Madjet and T. Renger, J. Phys. Chem. B, 2010, 114, 13517 CrossRef PubMed
- F. Müh, M. E. Madjet and T. Renger, Photosynth. Res., 2012, 111, 87–101 CrossRef PubMed
- J. Chmeliov, W. P. Bricker, C. Lo, E. Jouin, L. Valkunas, A. V. Ruban and C. D. P. Duffy, Phys. Chem. Chem. Phys., 2015, 17, 15857–15867 RSC
- R. Croce, Science, 2015, 348, 970–971 CrossRef CAS PubMed
- R. N. Frese, M. Germano, F. L. de Weerd, I. H. M. van Stokkum, A. Y. Shkuropatov, V. A. Shuvalov, H. J. van Gorkom, R. van Grondelle and J. P. Dekker, Biochemistry, 2003, 42, 9205–9213 CrossRef CAS PubMed
- M. A. Palacios, R. N. Frese, C. C. Gradinaru, I. H. M. van Stokkum, L. L. Premvardhan, P. Horton, A. V. Ruban, R. van Grondelle and H. van Amerongen, Biochim. Biophys. Acta, 2003, 1605, 83–95 CrossRef CAS
- V. I. Novoderezhkin, J. P. Dekker and R. van Grondelle, Biophys. J., 2007, 93, 1293–1311 CrossRef CAS PubMed
- O. J. G. Somsen, V. Chernyak, R. N. Frese, R. van Grondelle and S. Mukamel, J. Phys. Chem. B, 1998, 102, 8893–8908 CrossRef CAS
- V. I. Novoderezhkin, M. A. Palacios, H. van Amerongen and R. van Grondelle, J. Phys. Chem. B, 2004, 108, 10363–10375 CrossRef CAS
- V. I. Novoderezhkin, A. Marin and R. van Grondelle, Phys. Chem. Chem. Phys., 2011, 13, 17093–17103 RSC
- Md. Wahadoszamen, unpublished results.
- T. P. J. Krüger, E. Wientjes, R. Croce and R. van Grondelle, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13516–13521 CrossRef PubMed
- G. Raszewski, W. Saenger and T. Renger, Biophys. J., 2005, 88, 986–998 CrossRef CAS PubMed
- A. Warshel and W. W. Parson, J. Am. Chem. Soc., 1987, 109, 6143–6152 CrossRef CAS
- W. W. Parson and A. Warshel, J. Am. Chem. Soc., 1987, 109, 6152–6163 CrossRef CAS
- G. Small, Chem. Phys., 1995, 197, 239–257 CrossRef CAS
- T. Renger, Phys. Rev. Lett., 2004, 93, 188101 CrossRef PubMed
- T. Mančal, L. Valkunas and G. R. Fleming, Chem. Phys. Lett., 2006, 432, 301–305 CrossRef
- S. Vaitekonis, G. Trinkunas and L. Valkunas, Photosynth. Res., 2005, 86, 185–201 CrossRef CAS PubMed
- V. I. Novoderezhkin, E. Romero, J. P. Dekker and R. van Grondelle, ChemPhysChem, 2011, 12, 681–688 CrossRef CAS PubMed
- K. L. M. Lewis, F. D. Fuller, J. A. Myers, C. F. Yocum, S. Mukamel, D. Abramavicius and J. P. Ogilvie, J. Phys. Chem. A, 2013, 117, 34–41 CrossRef CAS PubMed
- A. Gelzinis, L. Valkunas, F. D. Fuller, J. P. Ogilvie, S. Mukamel and D. Abramavicius, New J. Phys., 2013, 15, 075013 CrossRef
- E. Romero, R. Augulis, V. I. Novoderezhkin, M. Ferretti, J. Thieme, D. Zigmantas and R. van Grondelle, Nat. Phys., 2014, 10, 676–682 CrossRef CAS PubMed
- J. A. Ihalainen, R. Croce, T. Morosinotto, I. H. M. van Stokkum, R. Bassi, J. P. Dekker and R. van Grondelle, J. Phys. Chem. B, 2005, 109, 21150–21158 CrossRef CAS PubMed
- L. M. C. Barter, J. R. Durrant and D. R. Klug, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 946–951 CrossRef CAS PubMed
- V. I. Novoderezhkin, E. Romero and R. van Grondelle, Phys. Chem. Chem. Phys., 2015, 17, 30828–30841 RSC
- V. Novoderezhkin and R. van Grondelle, J. Phys. Chem. B, 2013, 117, 11076–11090 CrossRef CAS PubMed
- A. Gelzinis, D. Abramavicius and L. Valkunas, J. Chem. Phys., 2015, 142, 154107 CrossRef PubMed
- J. M. Moix, J. Ma and J. Cao, J. Chem. Phys., 2015, 142, 094108 CrossRef PubMed
- J. Ma, J. Moix and J. Cao, J. Chem. Phys., 2015, 142, 094107 CrossRef PubMed
- A. N. Melkozernov, S. Lin, V. H. R. Schmid, H. Paulsen, G. W. Schmidt and R. E. Blankenship, FEBS Lett., 2000, 471, 89–92 CrossRef CAS PubMed
- E. Wientjes, I. H. M. van Stokkum, H. van Amerongen and R. Croce, Biophys. J., 2011, 100, 1372–1380 CrossRef CAS PubMed
- E. Wientjes, I. H. M. van Stokkum, H. van Amerongen and R. Croce, Biophys. J., 2011, 101, 745–754 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp02225a |
| This journal is © the Owner Societies 2016 |