
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the molecular character of periodic mesoporous organosilicates via photoluminescence of Lewis acid–base adducts†
Indre
Thiel
,
Alexey
Fedorov
,
Rene
Verel
,
Sergii
Yakunin
,
Maksym V.
Kovalenko
and
Christophe
Copéret
*
Department of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1–5, 8093 Zürich, Switzerland. E-mail: ccoperet@ethz.ch
First published on 9th May 2016
Abstract
Photoluminescence decay was used as a structure-sensitive method to compare the distribution of emitting sites in periodic mesoporous organosilicates (PMOs) to their respective molecular analogs. The observed close similarity of PL decays confirms the molecular nature of PMOs and high homogeneity of emitting sites.
Combining inorganic and organic fragments allows for the introduction of molecular properties into a functional hybrid material.1,2 Such materials can be rationally designed due to the modularity of the organic scaffold embedded in a stable inorganic matrix, typically silica.1,2 One approach relies on a co-condensation of an organosilane such as R–Si(OAlk)3 with tetraethoxysilane in the presence of a structure-directing agent, yielding mesostructured materials (usually, MCM or SBA-type) that incorporate organic groups (R) in their pores with high regularity.2 A related strategy utilizes an organic linker with two trialkoxysilyl groups [(AlkO)3Si–R–Si(OAlk)3] that can be condensed to give the so-called periodic mesoporous organosilicas.1 The latter high surface area hybrid materials feature organic functionalities uniformly distributed within the pores and the walls of an inorganic silica matrix.1 PMOs derived from aromatic precursors usually feature a 2D hexagonal framework of mesopores resulting in a long-range crystal order detected by XRD. These properties make PMOs attractive platforms for the post-synthetic modification towards molecularly defined heterogeneous catalysts.3,4
Phenylpyridine units have been previously utilized to form a PMO material (ppy-PMO, Scheme 1) and to subsequently immobilize several transition metal complexes.3d,e,5,6 In the case of the Cp*IrIII-ppy-PMO system, dynamic nuclear polarization surface enhanced NMR spectroscopy was used to differentiate between outer and inner layers of phenylpyridine ligands.5 Photoluminescence (PL) is another powerful structure-sensitive technique that was recently used to investigate the homogeneity of the environment of surface species.7 Encouraged by this result, we sought to extend the molecular level characterization of PMO materials by using PL. As solid molecular adducts of B(C6F5)3 and various O/N Lewis bases were reported to exhibit photoluminescence,8 we prepared related B(C6F5)3 adducts with PMO materials and compared their PL properties to respective molecular systems. Here, we demonstrate that PL decay of sites formed by B(C6F5)3 coordination to pyridine units of PMO materials is similar to the molecular analogues, which highlights the homogeneous environment and the molecular character of the organic units in PMO's.
 | ||
| Scheme 1 Synthesis of B-ppy-PMOpas, B-ppyMe-PMOpas and B-biph-PMOpas materials and their molecular models. | ||
We followed literature procedures to prepare ppy-PMO,3d,5 and also synthesized its previously unknown, sterically more bulky analogue ppyMe-PMO (Scheme 1). Material biph-PMO was prepared according to the literature9 as a nitrogen-free reference PMO (Scheme 1, see ESI† file for synthetic details). As B(C6F5)3 was anticipated to react with free surface silanols,10 the residual ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiOH groups of all three PMO materials were passivated using TMSBr (PMOpas), as evidenced by the disappearance of the OH band at 3720 cm−1 (Fig. S15 and 16, ESI†). All materials described herein exhibit a Type II isotherm in the N2 adsorption/desorption measurement, high surface areas and microporosity (pores < 2.4 nm by BJH analysis, Table 1). Incipient wetness impregnation of the three PMOpas with a toluene solution of B(C6F5)3 (0.2 equiv. B(C6F5)3 per nitrogen site of the PMOs) gave the respective B-PMOpas materials (Scheme 1).
SiOH groups of all three PMO materials were passivated using TMSBr (PMOpas), as evidenced by the disappearance of the OH band at 3720 cm−1 (Fig. S15 and 16, ESI†). All materials described herein exhibit a Type II isotherm in the N2 adsorption/desorption measurement, high surface areas and microporosity (pores < 2.4 nm by BJH analysis, Table 1). Incipient wetness impregnation of the three PMOpas with a toluene solution of B(C6F5)3 (0.2 equiv. B(C6F5)3 per nitrogen site of the PMOs) gave the respective B-PMOpas materials (Scheme 1).
| Organic linker | Surface area [m2 g−1] | ||
|---|---|---|---|
| PMO | PMOpas | B-PMOpas | |
| ppy | 634 | 436 | 175 |
| ppyMe | 976 | 853 | 372 |
| biph | 776 | 728 | 176 |
The 1H magic angle spinning (MAS) NMR spectrum of ppyMe-PMOpas has signals at δ 2.0 and 6.9 ppm (Fig. S12, ESI†) that are assigned to aliphatic and aromatic protons of the ppyMe moiety, respectively, and the 13C cross-polarization CP-MAS NMR of ppyMe-PMOpas features aliphatic and aromatic peaks as well (Fig. S13, ESI†). The 29Si CP-MAS spectrum of ppyMe-PMOpas shows expected signals of T2 and T3-sites at −69.3 and −78.6 ppm, respectively (Fig. S14, ESI†). 11B NMR spectra provided insight into the binding of B(C6F5)3 in B-PMOpas materials. Free, non-interacting B(C6F5)3 gives a broad signal with δiso = 58 ppm (ηCS = 0.03 and Cq = 4.3 MHz) due to the quadrupolar boron nucleus and the high symmetry of B(C6F5)3 (Fig. S18, ESI†). A priori, B(C6F5)3 can form adducts with O sites of silica as well as N sites of the phenylpyridine moiety.11 Coordination of a Lewis base (O or N) to B(C6F5)3 results in an upfield shift of the boron signal in the 11B NMR.12 All impregnated B-PMOpas materials contain no free B(C6F5)3. 2D 11B multiple quantum (MQ-MAS) experiments allow to distinguish between B–O and B–N interactions in B-PMOpas materials (Fig. 1). The signal at −2 ppm is ascribed to a B–O surface adduct as it is present in all three materials including nitrogen-free B-biph-PMOpas. This boron chemical shift is also close to that of molecular B(C6F5)3–O-adducts in solution.13 Impregnated pyridine-containing materials feature an additional signal centred around −4 ppm. This signal is attributed to the B(C6F5)3–Py adduct since the molecular B(C6F5)3–lutidine adduct in solution has a chemical shift at −3.9 ppm.14 Solid state NMR data of the molecular adduct of B(C6F5)3 and phenylpyridine also corroborates this assignment (Fig. S19, ESI†).
 | ||
| Fig. 1 11B ssNMR MQ-MAS spectra of B-ppy-PMOpas (A), B-ppyMe-PMOpas (B) and B-biph-PMOpas (C). | ||
While instructive in distinguishing between the N/O boron adducts, the 11B NMR only reveals one, relatively broad signal for a B–N interaction, which is not conclusive in terms of the broadness of the B–N sites distribution. We therefore set about to investigate the photoluminescence properties of B-PMOpas materials. Previously, coordination of B(C6F5)3 to the pyridine unit of the light emitting polymer was reported to lead to a red shift in the emission spectra, longer excited state lifetimes as well as higher quantum yields.8b A recent study reported on the photoluminescence properties of solid adducts of B(C6F5)3 with various acetophenone derivatives as well.8a
DRIFT UV-VIS spectra show a red shift upon impregnation of our PMOpas materials with B(C6F5)3 (Fig. S21, ESI†). The photoluminescence spectra of all PMOpas obtained after excitation at 350 nm display no emission. Interestingly, both molecular models of the B-PMOpas adducts with alkoxy silyl moieties on the aromatic linkers (ppySi and ppySiMe, Scheme 1) show no photoluminescence as well, in contrast to the simple B(C6F5)3 adducts with ppy and ppyMe. The siloxane moieties are likely causing different relaxation pathways and/or compete for boron coordination, thereby suppressing the photoluminescence from the Lewis B–N adduct. No luminescence was observed for the biph-derived materials, pointing towards the necessity of a B–N interaction for photoluminescence. In the case of ppy and ppyMe systems, the molecular boron adduct as well as the impregnated material show photoluminescence. For the molecular adducts the maximum of emission is at 355 nm and 363 nm, while for the impregnated materials a red shift to 418 and 420 nm, respectively, is observed (Fig. 2A).
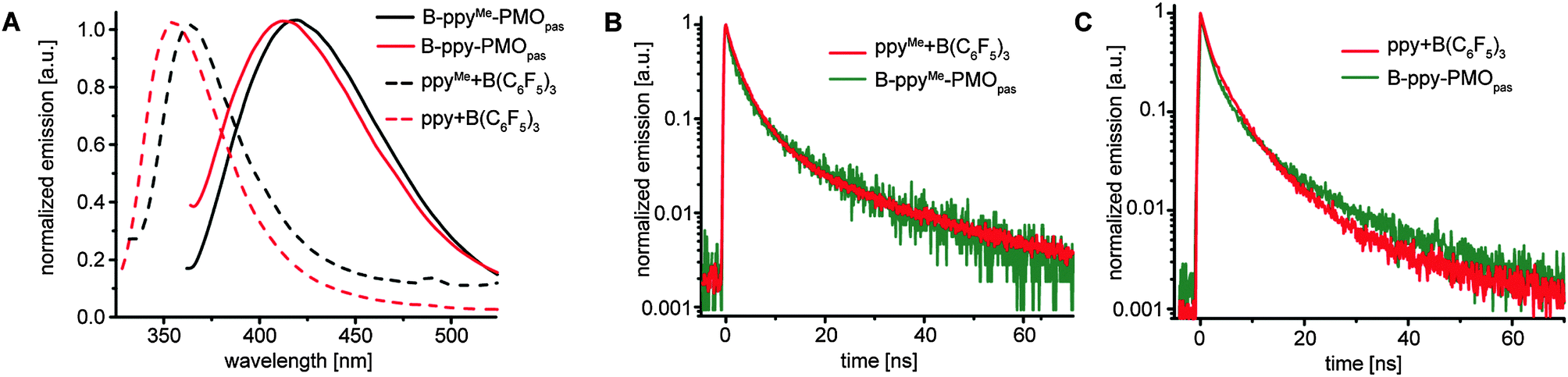 | ||
| Fig. 2 (A) PL spectra of ppyMe- and ppy-derivatives; ppy-B(C6F5)3 and ppyMe-B(C6F5)3 were excited at 300 nm wavelength, B-ppy-PMOpas and B-ppyMe-PMOpas were excited at 350 nm wavelength; (B) PL decay spectra of ppyMe-derivatives; (C) PL decay spectra of ppy-derivatives. | ||
Photoluminescence decay of the emitting samples described above features a bi-exponential mode (Fig. 2B, C and Table 2). The differences in the fitting parameters between molecular adducts (entries 1, 2) and impregnated materials (entries 3, 4) are relatively small and most likely not attributed to a larger distribution of sites in B-PMOpas, but caused by the presence of a greater number of deactivation pathways in the solid state that result in the quenching of photoluminescence. Note that a bi-exponential mode was also observed for the PL decay of molecular adducts of B(C6F5)3 with aromatic ketones.8a
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2)
exp(−t/τ1) + A2exp(−t/τ2)
| Entry | Sample | A 1 | τ 1 [ns] | A 2 | τ 2 [ns] |
|---|---|---|---|---|---|
| 1 | ppy-B(C6F5)3 | 0.53 | 1.2 | 0.46 | 5.0 |
| 2 | ppyMe-B(C6F5)3 | 0.76 | 1.9 | 0.21 | 8.2 |
| 3 | B-ppy-PMOpas | 0.75 | 1.2 | 0.27 | 6.5 |
| 4 | B-ppyMe-PMOpas | 0.70 | 1.1 | 0.29 | 7.0 |
Conclusions
We reported that PMOs with phenylpyridine moieties impregnated with B(C6F5)3 feature distinct signals for B–N and B–O interactions in 11B ssNMR MQ-MAS spectra. The B–N Lewis acid–base pair provides photoluminescence in the solid state while the B–O pair does not. The comparison of photoluminescence decay featured by molecular adducts in the solid state with respective impregnated materials demonstrates the molecular nature of highly homogeneous surface sites in PMOs.Acknowledgements
I. T. and A. F. thank the German Academic Exchange Service (DAAD) and the Holcim Stiftung for a postdoctoral and a habilitation fellowship, respectively. The authors acknowledge ScopeM at ETH Zürich for the use of their electron microscopy facilities.Notes and references
- (a) F. Hoffmann and M. Froba, Chem. Soc. Rev., 2011, 40, 608–620 RSC; (b) N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789–800 RSC; (c) P. Van Der Voort, D. Esquivel, E. De Canck, F. Goethals, I. Van Driessche and F. J. Romero-Salguero, Chem. Soc. Rev., 2013, 42, 3913–3955 RSC; (d) The sol–gel handbook: synthesis, characterization and applications, ed. D. Levy and M. Zayat, Wiley-VCH, Weinheim, 2015 Search PubMed; (e) C. Sanchez, C. Boissière, D. Grosso, C. Laberty and L. Nicole, Chem. Mater., 2008, 20, 682–737 CrossRef CAS; (f) R. J. P. Corriu, J. J. E. Moreau, P. Thepot and M. W. C. Man, Chem. Mater., 1992, 4, 1217–1224 CrossRef CAS.
- (a) M. P. Conley, C. Copéret and C. Thieuleux, ACS Catal., 2014, 1458–1469 CrossRef CAS; (b) A. Mehdi, C. Reye and R. Corriu, Chem. Soc. Rev., 2011, 40, 563–574 RSC.
- (a) Q. Yang, J. Liu, L. Zhang and C. Li, J. Mater. Chem., 2009, 19, 1945–1955 RSC; (b) M. Waki, Y. Maegawa, K. Hara, Y. Goto, S. Shirai, Y. Yamada, N. Mizoshita, T. Tani, W.-J. Chun, S. Muratsugu, M. Tada, A. Fukuoka and S. Inagaki, J. Am. Chem. Soc., 2014, 136, 4003–4011 CrossRef CAS PubMed; (c) P. Wang, X. Liu, J. Yang, Y. Yang, L. Zhang, Q. Yang and C. Li, J. Mater. Chem., 2009, 19, 8009–8014 RSC; (d) M. Waki, N. Mizoshita, T. Tani and S. Inagaki, Angew. Chem., Int. Ed., 2011, 50, 11667–11671 CrossRef CAS PubMed; (e) Y. Maegawa and S. Inagaki, Dalton Trans., 2015, 44, 13007–13016 RSC; (f) N. Mizoshita and S. Inagaki, Angew. Chem., Int. Ed., 2015, 54, 11999–12003 CrossRef CAS PubMed; (g) T. Seki, K. McEleney and C. M. Crudden, Chem. Commun., 2012, 48, 6369–6371 RSC.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed.
- W. R. Grüning, A. J. Rossini, A. Zagdoun, D. Gajan, A. Lesage, L. Emsley and C. Coperet, Phys. Chem. Chem. Phys., 2013, 15, 13270–13274 RSC.
- W. R. Grüning, G. Siddiqi, O. V. Safonova and C. Copéret, Adv. Synth. Catal., 2014, 356, 673–679 CrossRef.
- G. Lapadula, A. Bourdolle, F. Allouche, M. P. Conley, I. del Rosal, L. Maron, W. W. Lukens, Y. Guyot, C. Andraud, S. Brasselet, C. Copéret, O. Maury and R. A. Andersen, Chem. Mater., 2014, 26, 1062–1073 CrossRef CAS.
- (a) M. M. Hansmann, A. López-Andarias, E. Rettenmeier, C. Egler-Lucas, F. Rominger, A. S. K. Hashmi and C. Romero-Nieto, Angew. Chem., Int. Ed., 2016, 55, 1196–1199 CrossRef CAS PubMed; (b) P. Zalar, Z. B. Henson, G. C. Welch, G. C. Bazan and T.-Q. Nguyen, Angew. Chem., Int. Ed., 2012, 51, 7495–7498 CrossRef CAS PubMed.
- M. P. Kapoor, Q. Yang and S. Inagaki, J. Am. Chem. Soc., 2002, 124, 15176–15177 CrossRef CAS PubMed.
- Y.-J. Wanglee, J. Hu, R. E. White, M.-Y. Lee, S. M. Stewart, P. Perrotin and S. L. Scott, J. Am. Chem. Soc., 2011, 134, 355–366 CrossRef PubMed.
- T. Onak, Organoborane Chemistry, Academic Press, 1975, pp. 136–163 Search PubMed.
- H. Nöth and B. Wrackmeyer, in Nuclear Magnetic Resonance Spectroscopy of Boron Compounds, ed. H. Nöth and B. Wrackmeyer, Springer Berlin Heidelberg, Berlin, Heidelberg, 1978, pp. 74–101 Search PubMed.
- M. S. Oderinde and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 9834–9837 CrossRef CAS PubMed.
- (a) A. Karkamkar, K. Parab, D. M. Camaioni, D. Neiner, H. Cho, T. K. Nielsen and T. Autrey, Dalton Trans., 2013, 42, 615–619 RSC; (b) S. J. Geier and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 3476–3477 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and physico-chemical characterization. See DOI: 10.1039/c6cp02176j |
| This journal is © the Owner Societies 2016 |