
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photodynamics of potent antioxidants: ferulic and caffeic acids†
Michael D.
Horbury
,
Lewis A.
Baker
,
Wen-Dong
Quan
,
Simon E.
Greenough‡
* and
Vasilios G.
Stavros
*
Department of Chemistry, University of Warwick, Library Road, Coventry, CV4 7AL, UK. E-mail: v.stavros@warwick.ac.uk; ed.greenough@sheffield.ac.uk
First published on 7th June 2016
Abstract
The dynamics of ferulic acid (3-(4-hydroxy-3-methoxyphenyl)-2-propenoic acid) and caffeic acid (3-(3,4-dihydroxyphenyl)-2-propenoic acid) in acetonitrile, dioxane and water at pH 2.2 following photoexcitation to the first excited singlet state are reported. These hydroxycinnamic acids display both strong ultraviolet absorption and potent antioxidant activity, making them promising sunscreen components. Ferulic and caffeic acids have previously been shown to undergo trans–cis photoisomerization via irradiation studies, yet time-resolved measurements were unable to observe formation of the cis-isomer. In the present study, we are able to observe the formation of the cis-isomer as well as provide timescales of relaxation following initial photoexcitation.
Introduction
Ultraviolet (UV) radiation is a double-edged sword in biology. On the one hand it is required for the production of the vital vitamin D,1 on the other, UV light can induce dangerous photoreactions e.g. mutagenesis in DNA.2 This can lead to cancers of the skin, such as malignant melanoma,3–5 of which worldwide incidences are on the increase.6 While human skin has a natural photoprotective mechanism in the form of tanning, it is a delayed response during which photodamage can occur.7,8 Therefore, in order to complement the body's natural photoprotection, synthetic sunscreens have been developed.9,10 There have been controversies regarding the use of certain compounds as sunscreen agents in relation to the adverse dermatological effects they may cause.11–13 In particular, some reports have highlighted the possibility that certain photoexcited sunscreen molecules may produce harmful reactive oxygen species (ROS).14,15 This has led to the concept of including molecules with antioxidant properties within sunscreen products either to increase the photostability of sunscreen molecules, or to “mop up” any ROS.15–17A particularly potent set of antioxidants are hydroxycinnamic acids, which include: ferulic acid (3-(4-hydroxy-3-methoxyphenyl)-2-propenoic acid) and caffeic acid (3-(3,4-dihydroxyphenyl)-2-propenoic acid), shown in Fig. 1.18,19 These molecules also possess strong absorptions in the UVA (400–315 nm) and UVB (315–280 nm) regions of the electromagnetic spectrum; critical for use as absorbing species in sunscreens (see ESI†). Previous time-resolved pump–probe spectroscopy studies have been performed on hydroxycinnamic acids, though much of this work has been focused upon the archetypal p-coumaric acid (3-(4-hydroxyphenyl)-2-propenoic acid) as either the isolated molecule or a functionalized chromophore to mimic the photoactive yellow protein.20–27 In these studies p-coumaric acid was excited to the first excited electronic state (11ππ*, S1). The main relaxation pathway of the 11ππ* state was identified as a trans–cis photoisomerization resulting in the formation of the cis-photoproduct. A two-step mechanism was proposed for this photoisomerization. The initial step involved geometry relaxation out of the vertical Franck–Condon (vFC) region to a local minimum in the 11ππ* state and a solvent rearrangement. This was followed by excited state population traversing a barrier along the isomerization reaction coordinate, proceeding to an 11ππ*/S0 conical intersection (CI) (where S0 represents the electronic ground state) to either form the cis-isomer or return to the original trans-isomer.
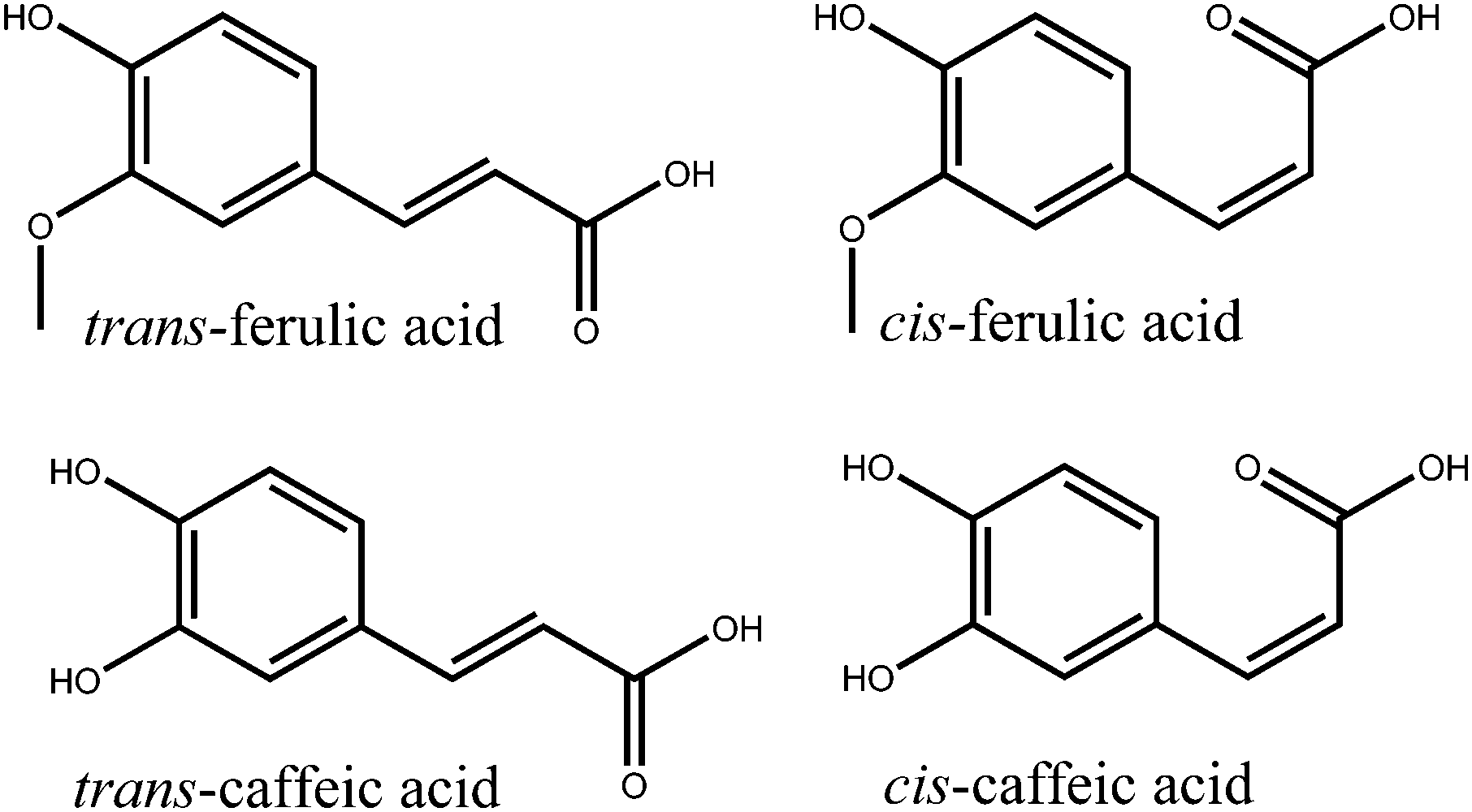 | ||
| Fig. 1 Structures of ferulic and caffeic acids in their trans and cis isomeric forms. | ||
Ferulic acid has also been studied previously, at different pHs by van Grondelle and co-workers.28 In this study, ferulic acid was photoexcited to the 11ππ* state, before probing the excited state decay with transient electronic (UV-visible) absorption spectroscopy (TEAS). A global analysis of their data required two time constants at pH 7 and three time constants at pH 10.5 to fully describe the dynamics data. The first time constant was attributed to a solvent rearrangement and geometry relaxation out of the initially populated vFC region (in accordance to p-coumaric acid, vide supra), which manifested as a shift in the detected stimulated emission (SE). The second time constant was assigned to the decay of the excited state via an aborted isomerization as no cis-isomer was detected. This aborted isomerization in the propenoic acid “tail” of the molecule leads to a coupling between the 11ππ* state and the S0 by an 11ππ*/S0 CI. In contrast, static measurements of the effects of UV irradiation on both ferulic acid and caffeic acid have observed the formation of the cis-isomer following exposure to UV.29–32
Recent gas-phase ab initio calculations by Karsili and co-workers33 for ferulic and caffeic acids have suggested that photoisomerization occurs in the 21ππ* (S3) state after excitation to the 11ππ* state (this is in contrast to occurring on the 11ππ* as proposed previously)21–28 proceeding via an 21ππ*/S0 CI. Additionally, this work also highlighted a potential dissociation of the hydrogen atom on the phenolic hydroxyl group via tunnelling; this mechanism has been previously seen experimentally in the building blocks of ferulic and caffeic acids, notably guaiacol and catechol respectively.34,35 In light of these works, and the apparent need for a better understanding of the photodynamics of ferulic acid and caffeic acid as sunscreening agents, we use femtosecond (fs) TEAS to revisit the photodynamics of ferulic acid and provide new insight toward the hitherto unstudied caffeic acid, following photoexcitation to the 11ππ* state, in a range of solvent environments of varying polarity.
Experimental
The detailed experimental procedures pertaining to TEAS have been described elsewhere34,36 and are briefly summarized here. The pump pulse was generated using a commercially available optical parametric amplifier, (TOPAS-C, Spectra Physics). A femtosecond white light continuum (330–675 nm) probe pulse was formed through supercontinuum generation from 800 nm light in a 1 mm thick CaF2 window. The pump wavelength was set to 319 nm (3.88 eV) for ferulic acid and 314 nm (3.95 eV) for caffeic acid, corresponding to excitation to the 11ππ* state.33 The fluence of the pump beam was set between 1–2 mJ cm−2. The difference between the pump and probe polarizations was held at magic angle (54.7°) to negate anisotropic effects. Changes in the optical density (ΔOD) of the samples were calculated from probe intensities, collected using a spectrometer (Avantes, AvaSpec-ULS1650F). The ferulic acid (99%, Sigma Aldrich) and caffeic acid (98%, Sigma Aldrich) samples were made to a concentration of 3 mM in: buffer solutions at pH 2.2 (50 mM Glygly) with nano-pure water, acetonitrile (CH3CN, ≥99.9%, Sigma Aldrich) and dioxane (>99%, Fisher Chemical). The delivery system for the samples was a flow-through cell (Demountable Liquid Cell by Harrick Scientific Products Inc.) with a 100 μm path length to limit optical dispersion. The sample was circulated using a PTFE tubing peristaltic pump (Masterflex) recirculating sample from a 50 ml reservoir in order to provide each pulse with fresh sample.In addition to the transient absorption spectra (TAS – see Fig. 2), static difference absorption spectra, “ΔUV/vis spectra”, were collected. These were acquired by irradiating the solutions using an arc lamp (OBB, Tunable KiloArc) for 10 minutes. The desired irradiation wavelength was selected using a monochromator, providing light centred at the same wavelength as the pump wavelength in the TEAS measurements, with a 10 nm bandwidth. The “before” and “after” UV-vis spectra were collected using a UV/vis spectrometer (Cary 300, Agilent Technologies). To generate the ΔUV/vis spectra, the “before” spectra were subtracted from the “after” spectra and then normalized.
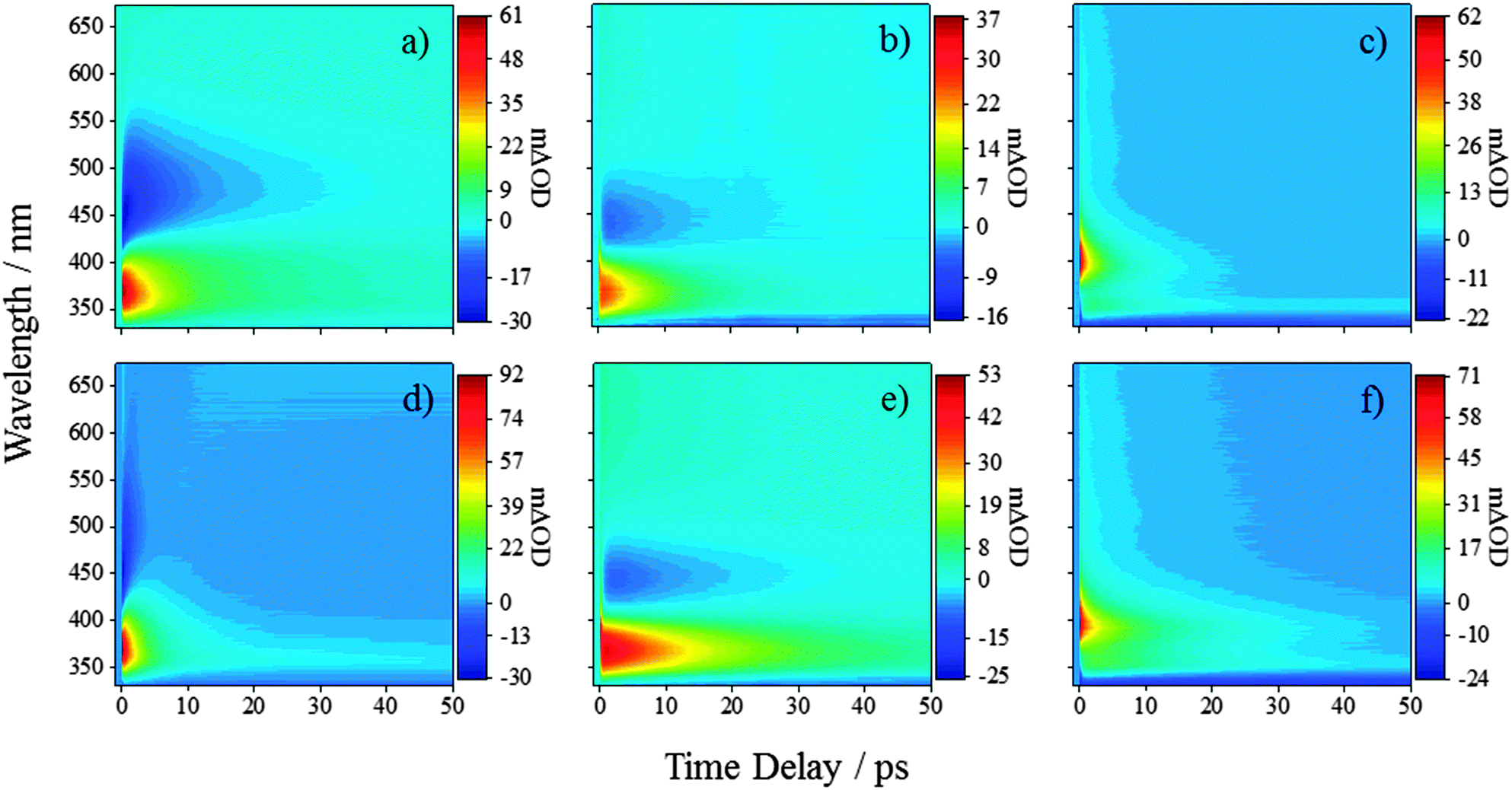 | ||
| Fig. 2 TAS of ferulic acid in (a) water at pH 2.2, (b) CH3CN, and (c) dioxane after excitation using a pump pulse of 319 nm. TAS of caffeic acid in (d) water at pH 2.2, (e) CH3CN, and (f) dioxane after excitation using a pump pulse of 314 nm. | ||
Results
We will first consider the TAS of ferulic and caffeic acids in water at pH 2.2, shown in Fig. 2a and d, respectively (other pHs are provided in the ESI†). At this pH, the carboxyl group is protonated in >99% of the molecules (see ESI† for details). After the initial excitation to the 11ππ* state, the TAS possess three dominant features. The first of these is a large excited state absorption (ESA) centred at 375 nm for ferulic acid and 370 nm for caffeic acid. The second is an SE signal (negative-going) spanning from ∼425–575 nm for both acids. Finally, a broad absorption is present in the region of ∼550–675 nm. As the large ESA and SE decay away, a negative-going signal at the blue edge of the spectra is uncovered (<350 nm) alongside the positive-going signal (>350 nm). These features, along with a broad (and rising) absorption in the red, persist out to the maximum experimental time delay of 500 ps (see Fig. 3a and d, black traces). To further our grasp of the environmental effects, we also performed TEAS on ferulic and caffeic acids in CH3CN, a polar aprotic environment (εr = 37.5). These TAS, presented in Fig. 2b and e respectively, are similar to those acquired in aqueous solution and once again decay to reveal similar features seen in water at pH 2.2, however with a notable absence of the broad (and rising) absorption in the red (Fig. 3b and e, black traces).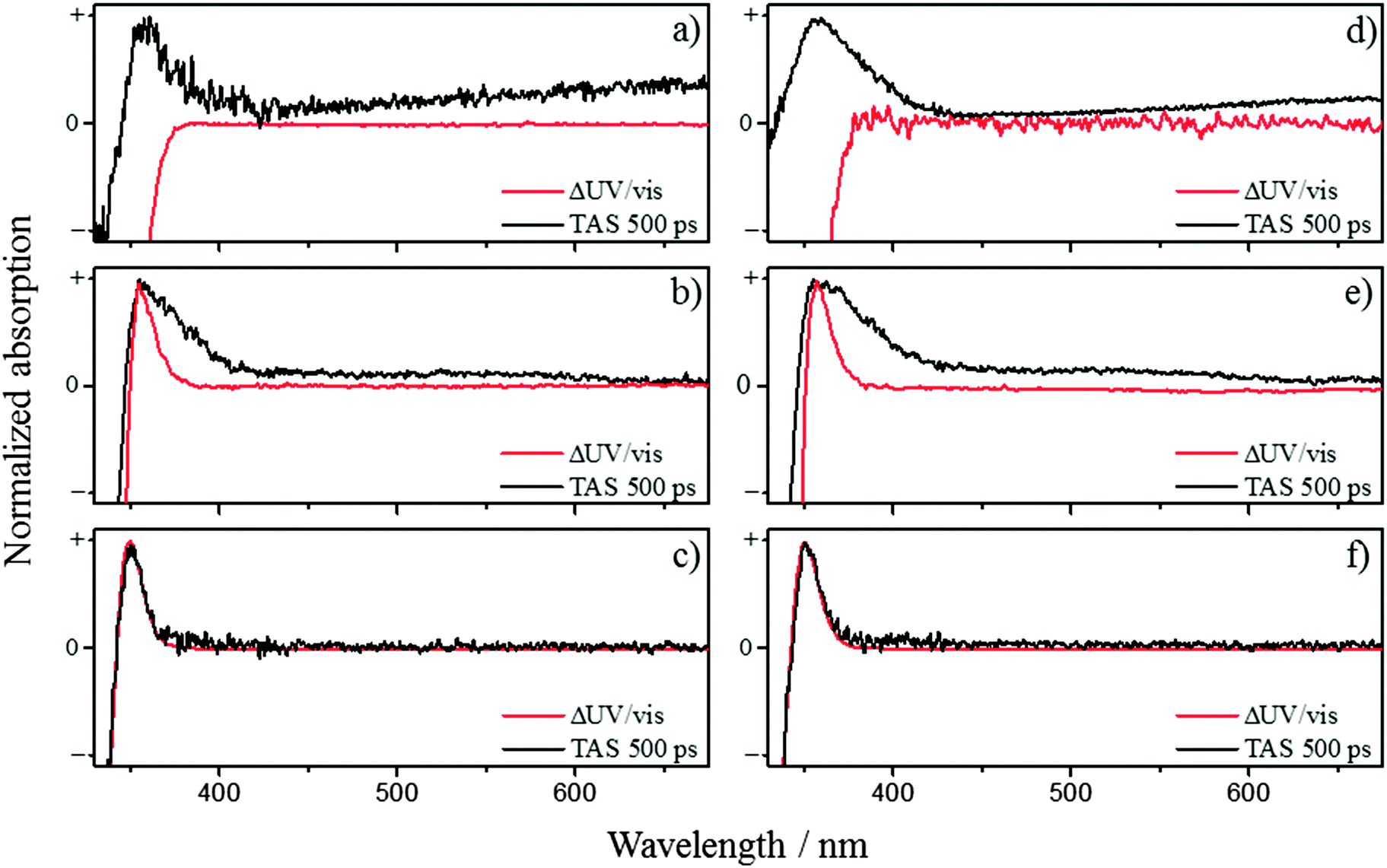 | ||
| Fig. 3 The 500 ps TAS overlaid with ΔUV/vis spectra for ferulic acid in (a) water at pH 2.2, (b) CH3CN, and (c) dioxane. Similarly, for caffeic acid in (d) water at pH 2.2, (e) CH3CN, and (f) dioxane. | ||
Finally, both acids were studied in dioxane (εr = 2.3), serving to closely mimic the gas-phase calculations owing to the weakly perturbing environment offered by this solvent. The TAS for ferulic and caffeic acids, Fig. 2c and f respectively, display a notable difference to the corresponding TAS in water at pH 2.2 and CH3CN; no SE can be seen in the TAS recorded in dioxane (likely masked by the large ESA). In addition, an initial large absorption is present, now at ∼400 nm in both acids (cf. ∼370 nm for TAS in pH 2.2 and CH3CN). As this peak decays, an absorption peak at ∼350 nm for both ferulic and caffeic acid is revealed (Fig. 3c and f, black traces), in addition to a negative-going signal (<345 nm), the cause of which is discussed below.
To model the observed dynamics, a global fitting technique (see ESI† for further details)37,38 using the sum of five exponentials, convoluted with a Gaussian representing our instrument response (∼80 fs), is deployed. One of the exponentials represents the instrument response which includes signal attributed to the solvent- and glass-only dynamics (<50 fs). The second exponential represents a baseline offset, which we attribute to long-lived species generated following photoexcitation, such as radical species and/or cis photoproduct (vide infra). This persists beyond the temporal window of our measurements (>500 ps). The remaining three exponentials are used to recover the time constants of the observed dynamics in ferulic and caffeic acids and are labelled as τ1, τ2 and τ3, as shown in Table 1.
| τ 1/fs | τ 2/ps | τ 3/ps | ||
|---|---|---|---|---|
| Ferulic acid | pH 2.2 | 406 ± 32 | 2.60 ± 0.19 | 15.4 ± 0.9 |
| CH3CN |
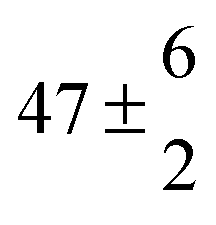
|
0.45 ± 0.04 | 8.9 ± 0.3 | |
| Dioxane |
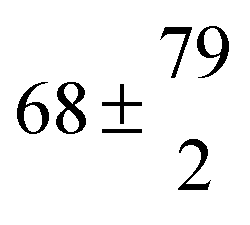
|
0.97 ± 0.13 | 6.3 ± 0.4 | |
| Caffeic acid | pH 2.2 | 279 ± 58 | 1.05 ± 0.13 | 4.8 ± 0.4 |
| CH3CN | 78 ± 6 | 0.52 ± 0.04 | 16.7 ± 0.5 | |
| Dioxane |
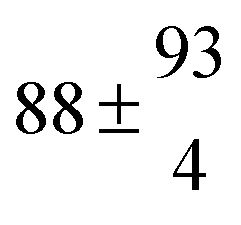
|
1.15 ± 0.12 | 14.1 ± 0.7 |
Discussion
We now discuss the three extracted time constants and the dynamical processes we attribute them to. We reiterate that in the previous study by van Grondelle and co-workers on ferulic acid,22,28 the authors used two time constants at pH 7 to sufficiently model their transient data at similar excitation wavelengths (318 nm here versus their 319 nm); here two time constants were insufficient to fully model our TAS (including our pH 7.4, see ESI†) which is likely due to the enhanced time-resolution of the present measurements. This, we believe, has enabled us to effectively separate the first time constant into two components (τ1 and τ2) leaving the third time constant (τ3) in excellent agreement with van Grondelle and co-workers measurements. Considering τ1, we attribute this time constant to the population flux from the vFC region, induced by a geometry relaxation, as well as any solvent rearrangement, lowering the energy of the initially excited 11ππ* state. This is in agreement with the abovementioned studies, where similar changes to the features occurred within this timeframe.22,28 We also recognise that with CH3CN and dioxane that this value is similar to the instrument response and time-zero artefacts will undoubtedly contribute to this time constant.Regarding the time-constants τ2 and τ3, there are two potential decay mechanisms that may account for these. We discuss each of these individually. The first, which we designate mechanism 1, is based on previous time-resolved studies on hydroxycinnamic acids, which proposed that the excited state decays along the initially populated 11ππ* state, whereby excited state depopulation is mediated via a 11ππ*/S0 CI along the trans–cis isomerization coordinate.22,26,28 If this model is also responsible for the observed dynamics in ferulic and caffeic acid, the additional time-constant τ2, is likely to correspond to the spectral displacement in the TAS, which reflects excited state population evolving along the isomerization coordinate. The decay of the ESA, as well as the concurrent decay in the SE seen in water at pH 2.2 and CH3CN, represents the flow of excited state flux through a 11ππ*/S0 CI leading to the reformation of the initial trans-ground state or the cis-isomer photoproduct. This process would then be reflected by the time-constant τ3. A schematic of this decay model is shown in Fig. 4a.
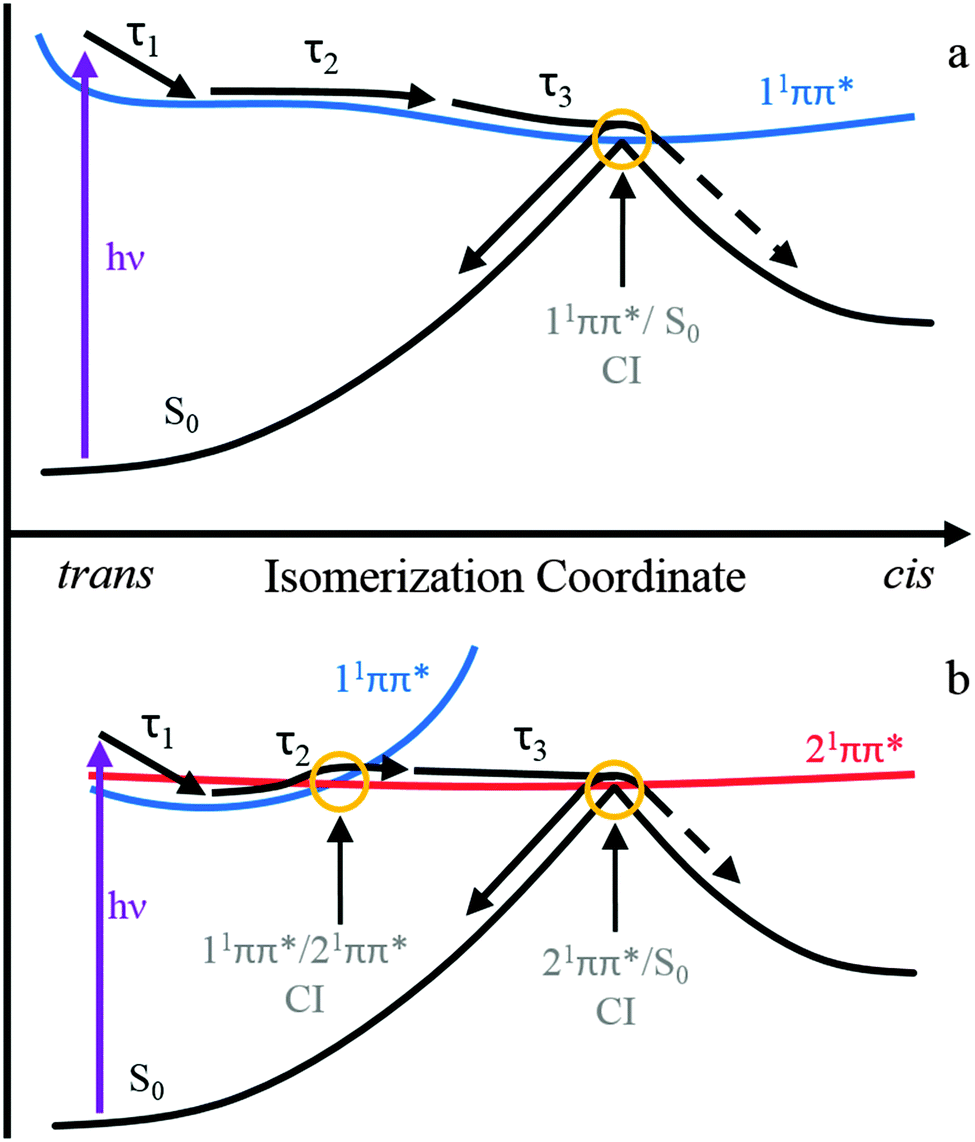 | ||
| Fig. 4 A representative schematic of the two proposed relaxation mechanisms along the trans–cis isomerization coordinate. The top panel (a, mechanism 1) represents the isomerization occurring along the 11ππ* state as proposed previously for hydroxycinnamic acids.22,26,28 The bottom panel (b, mechanism 2) is based upon recently calculated potential energy surfaces along the isomerization coordinate and involves both the 11ππ* and 21ππ* states.33 | ||
The second potential decay mechanism 2, is in accordance to the most recent gas-phase ab initio calculations on ferulic and caffeic acids, that suggest that the photoisomerization occurs on a second electronically excited state.33 The potential energy surfaces (PESs) in these theoretical studies identified the state mediating the isomerization as the 21ππ* state. Fig. 4b shows a schematic of the relevant PESs along the isomerization coordinate and the relaxation mechanism. The spectral displacement in the ESA, and SE seen in water at pH 2.2 and CH3CN, corresponding to the time-constant τ2, could be due to population of the 21ππ* excited state, following a non-adiabatic transition via a 11ππ*/21ππ* CI. These ab initio calculations also predict a second CI of interest between the 21ππ* and electronic ground state, 21ππ*/S0, which lies along the isomerization coordinate between trans and cis isomers.33 Upon reaching the CI, excited state flux is able to progress onto either the trans or cis ground state. The disappearance of the ESA, and in pH 2.2 and CH3CN the concomitant decay of the SE feature, are attributed to the depopulation of the 21ππ* state, via a 21ππ*/S0 CI, and results in the time-constant τ3. One caveat is in order here: these calculations suggest that this pathway is ∼8500 cm−1 above the photoexcitation wavelengths used in the present experiments. However, these are gas-phase calculations and the presence of solvent will inevitably alter the 11ππ* to 21ππ* state crossing pathways, as suggested in other studies of similar molecules.39
Two points are worthy of discussion here. Firstly, exploring the change, if any, in the anisotropy of the SE may provide insight into the underlying mechanism in operation, i.e., 11ππ* → S0 (mechanism 1) or 11ππ* → 21ππ* → S0 (mechanism 2); one would anticipate a change in anisotropy when traversing from the 11ππ* to the 21ππ* state (mechanism 2), given any differences in the transition dipole moments (TDMs) for 11ππ* → S0 and 21ππ* → S0. Preliminary experiments investigating the change in anisotropy in the SE (for ferulic acid in CH3CN and H2O, data not shown) before τ2 and after τ2 however were inconclusive, which may be a direct result of similarities in the TDMs between 11ππ* → S0 and 21ππ* → S0, sensitivity in our measurements or both. Secondly, the global fitting technique applied here has a limitation in that all of the exponentials used in the fit begin at zero time-delay. Such non-sequential kinetic modelling means that each time-constant reported will have some contribution from dynamical processes represented by shorter and/or longer time-constants, inevitably impacting assignment of a given time-constant to a particular dynamical process.
It is clear that the solvent environment (including pH 7.4, 11 and 14, see ESI† for details) has an effect upon the values of τ1, τ2 and τ3, yet no trend is apparent. We make no attempt to reconcile these differences (due to their highly complex nature), only to say that we believe that these variances are primarily caused by electrostatic solvent influences upon the energies of the relevant electronic states. Furthermore, calculations33 have shown the possibility that the 11nπ* (S2) state crosses both 11ππ* and 21ππ* states. It is therefore conceivable that some of the excited state population is funneled into the 11nπ* state and becomes trapped. Thus it is apparent that further theoretical studies are a necessary to fully understand the effects of the solvent on the electronic (and geometric) structure of these molecules.
After the decay of the ESA (and the SE in pH 2.2 and CH3CN) several features are still present within the TAS and persist out to the longest available time delay (500 ps). By comparing these features to the ΔUV/vis spectra, we can determine if the features are caused by the cis-photoproduct as expected from our proposed models. The ΔUV/vis spectra will only show changes to the absorption of the sample caused by one-photon absorption (owing to the light source) and non-transient species. Fig. 3 show the comparison between the ΔUV/vis spectra and 500 ps TAS collected in ferulic and caffeic acids. In water at pH 2.2, the TAS at long time delays (Fig. 3a (ferulic acid) and Fig. 3d (caffeic acid)) display broad absorptions in the red, akin to that of the ubiquitous solvated electron.40 The absorption feature in the blue-end of the spectrum also matches closely to the phenolic radical absorption, which has been proposed to be formed through dissociation via a tunnelling mechanism, along the O–H coordinate of the phenolic moiety, following one-photon absorption.33–35,41 However these absorptions show a quadratic dependence upon pump fluence in the power dependence measurements (see ESI†). The phenolic radical (along with the solvated electron) of p-coumaric acid has been shown to be generated by a two-photon resonant ionization process in aqueous solutions with the pump pulse.26 Furthermore, these features are absent in solvent and buffer only scans (see ESI†) and therefore strongly imply they originate from ferulic and caffeic acid. Therefore, in keeping with previous literature, we confidently assign these two absorptions to the radical and solvated electron generated via two-photon ionization, recognising this is an artefact of our measurements and will very unlikely occur when these molecules are exposed to solar UV. Unfortunately, the presence of this two-photon driven process means that it will mask any spectral signature of other transient species which includes the cis-isomer photoproduct. We once again note that previous studies have observed the formation of the cis-isomer after UV exposure29–32 and evidence for the cis-isomer is present in our ΔUV/vis spectra. It is worth noting that, in the ΔUV/vis spectra of caffeic acid in water at pH 11 and 14 (see ESI,† Fig. S17), the photoproduct absorption is heavily red-shifted and is no-longer convoluted with the radical absorption. Even with the spectral separation of the photoproduct and radical absorption, no photoproduct is observed in the corresponding TAS. This may be a result of a poor quantum yield for photoproduct formation, making its detection very difficult.
In the case of CH3CN as the solvent, the absorption peak in the TAS shown in Fig. 3b (ferulic acid) and Fig. 3e (caffeic acid), overlays well with the peak in the ΔUV/vis spectrum for both ferulic and caffeic acids. However, a discrepancy between the two is still present, making it difficult to determine whether the cis-isomer is actually being formed. The peak in the ΔUV/vis spectrum is significantly narrower than the absorption peak in the TAS which also possesses a long, sloped shoulder in the red end of the absorption. Furthermore the broadness of this peak is akin to that of the phenolic radical for both ferulic and caffeic acid.41 In order to determine if this feature is the radical absorption, formed through two-photon ionization (as above), we once again performed power dependence measurements (see ESI†). These measurements demonstrated that different spectral regions in the TAS displayed differing dependences upon pump power. Integrating the signal at the peak of the absorption in the ΔUV/vis spectrum of ferulic acid, shows a linear dependence, while caffeic acid is near-linear. It is worth noting that the peak maximum in the ΔUV/vis spectrum is in the same spectral location as the peak maximum in the TAS. Yet when we integrate signal where no absorption is present in the ΔUV/vis spectrum, both ferulic and caffeic acid display a power dependence of ∼1.6. Therefore, we propose that the long-lived absorption is a convolution between the phenolic radical generated by two-photon ionization, akin to the pH 2.2 measurements (vide supra), and the absorption from the cis-isomer photoproduct.
We close by discussing the data obtained in dioxane, given in Fig. 3c (ferulic acid) and Fig. 3f (caffeic acid). Firstly, we note that there is a lack of SE in the TAS unlike water and CH3CN. We believe this is caused by either a spectral shift or reduced probability of the emission causing it to be masked by the ESA. Regarding the photoproduct, we see excellent agreement between the 500 ps TAS and ΔUV/vis spectrum, providing strong evidence that in dioxane, the cis-isomer is being formed. Adding to this, the power dependency measurements show that this peak has a linear dependence on power. Importantly, if this peak was indeed caused by the radical absorption (following one-photon absorption), it would be absent in the ΔUV/vis spectrum as the radical is not expected to be present in solution on a second to a minute timescale that these static measurements are carried out. This is in contrast to van Grondelle and co-workers where no cis-isomer is seen, however their measurements are only performed in water at pH 7 and 10.5 and, like the aqueous measurements presented here, struggle with convolution with the phenolic radical absorption generated through two-photon absorption.28
It is worth mentioning as a closing statement here that while we have determined when the cis-isomer is present or not, we are unable to determine the extent to which it is being formed. Therefore, it is apparent that further studies are needed that are more sensitive to the structural differences between the two isomers. Measurements employing time-resolved vibrational spectroscopy would certainly be warranted.
Conclusions
To conclude, the 11ππ* excited state relaxation dynamics of ferulic and caffeic acids, following UV photoexcitation, have been studied in a range of solvent environments using transient electronic absorption spectroscopy. Two possible relaxation mechanisms (1 and 2) are proposed, based on previous work on hydroxycinnamic acids and recent ab initio calculations on the present systems. Both involve a geometry relaxation out of the initial Franck–Condon region as well as solvent rearrangement with the time constant τ1. Mechanism 1 then involves the evolution along the trans–cis isomerisation coordinate on the 11ππ* state on the timescale of τ2, followed by internal conversion to S0 mediated by a 11ππ*/S0 CI. This occurs on a timescale of τ3. Mechanism 2 on the other hand involves internal conversion from the 11ππ* to the 21ππ* state, via a 11ππ*/21ππ* CI, described by τ2 and subsequent evolution along the trans–cis isomerisation coordinate on the 21ππ* state and internal conversion onto S0, mediated by a 21ππ*/S0 CI. This is described by τ3. For both mechanisms, at the 11ππ*/S0 or 21ππ*/S0 CI, the excited species can either reform the original trans-ground state or lead to a cis-photoproduct.Finally, we return to briefly assess the sunscreening capabilities of ferulic and caffeic acids, in light of the present findings. Both display a photostability induced by the molecules' ability to quickly deactivate the excess electronic energy, very likely via an isomerization pathway, leading to the reformation of the initial ground state or the cis-isomer. It is expected that the cis-isomer (the major photoproduct) will display a similar excited state relaxation pathway, providing comparable levels of photoprotection as the trans-isomer, thereby completing the cycle of photoprotection. Naturally further studies are clearly warranted to validate this hypothesis and are on-going in our research group.
Acknowledgements
The authors are grateful to Prof. Peter J. Sadler for the use the KiloArc and Cary 300, and Ms Faye Monk for experimental assistance. We thank Dr Tolga Karsili for helpful discussions and reading of the manuscript, along with Dr Gareth Roberts. M. D. H. thanks the University of Warwick for EPSRC doctoral training awards. L. A. B. and W. D. Q. thank the EPSRC for providing studentships under grant number EP/F500378/1, through the Molecular Organisation and Assembly in Cells Doctoral Training Centre. S. E. G. thanks the Warwick Institute of Advanced Studies, for postdoctoral funding. V. G. S. thanks the EPSRC for an equipment grant (EP/J007153) and the Royal Society for a University Research Fellowship.Notes and references
- M. F. Holick, J. Cell. Biochem., 2003, 88, 296–307 CrossRef CAS PubMed.
- G. P. Pfeifer, Y.-H. You and A. Besaratinia, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2005, 571, 19–31 CrossRef CAS PubMed.
- J. P. Ortonne, Br. J. Dermatol., 2002, 146, 7–10 CrossRef CAS PubMed.
- R. S. Mason and J. Reichrath, Adv. Anticancer Agents Med. Chem., 2013, 13, 83–97 CrossRef CAS.
- S. González, M. Fernández-Lorente and Y. Gilaberte-Calzada, Clin. Dermatol., 2008, 26, 614–626 CrossRef PubMed.
- V. G. Stavros, Nat. Chem., 2014, 6, 955–956 CrossRef CAS PubMed.
- M. Brenner and V. J. Hearing, Photochem. Photobiol., 2008, 84, 539–549 CrossRef CAS PubMed.
- N. Agar and A. R. Young, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2005, 571, 121–132 CrossRef CAS PubMed.
- S. Forestier, J. Am. Acad. Dermatol., 2008, 58, S133–S138 CrossRef PubMed.
- F. Urbach, J. Photochem. Photobiol., B, 2001, 64, 99–104 CrossRef CAS.
- M. Loden, H. Beitner, H. Gonzalez, D. W. Edström, U. Åkerström, J. Austad, I. Buraczewska-Norin, M. Matsson and H. C. Wulf, Br. J. Dermatol., 2011, 165, 255–262 CrossRef CAS PubMed.
- M. E. Burnett and S. Q. Wang, Photodermatol., Photoimmunol. Photomed., 2011, 27, 58–67 CrossRef CAS PubMed.
- K. Skotarczak, A. Osmola-Mańkowska, M. Lodyga, A. Polańska, M. Mazur and Z. Adamski, Eur. Rev. Med. Pharmacol. Sci., 2015, 19, 98–112 CAS.
- N. Serpone, A. Salinaro, A. V. Emeline, S. Horikoshi, H. Hidaka and J. Zhao, Photochem. Photobiol. Sci., 2002, 1, 970–981 CAS.
- S. Afonso, K. Horita, J. P. Sousa e Silva, I. F. Almeida, M. H. Amaral, P. A. Lobão, P. C. Costa, M. S. Miranda, J. C. G. E. da Silva and J. M. S. Lobo, J. Photochem. Photobiol., B, 2014, 140, 36–40 CrossRef CAS PubMed.
- M. S. Matsui, A. Hsia, J. D. Miller, K. Hanneman, H. Scull, K. D. Cooper and E. Baron, J. Invest. Dermatol. Symp. Proc., 2009, 14, 56–59 CrossRef CAS PubMed.
- A. Saija, A. Tomaino, D. Trombetta, A. De Pasquale, N. Uccella, T. Barbuzzi, D. Paolino and F. Bonina, Int. J. Pharm., 2000, 199, 39–47 CrossRef CAS PubMed.
- H. Kikuzaki, M. Hisamoto, K. Hirose, K. Akiyama and H. Taniguchi, J. Agric. Food Chem., 2002, 50, 2161–2168 CrossRef CAS PubMed.
- K. Sevgi, B. Tepe and C. Sarikurkcu, Food Chem. Toxicol., 2015, 77, 12–21 CrossRef CAS PubMed.
- A. Espagne, D. H. Paik, P. Changenet-Barret, M. M. Martin and A. H. Zewail, ChemPhysChem, 2006, 7, 1717–1726 CrossRef CAS PubMed.
- N. Mataga, H. Chosrowjan, S. Taniguchi, N. Hamada, F. Tokunaga, Y. Imamoto and M. Kataoka, Phys. Chem. Chem. Phys., 2003, 5, 2454–2460 RSC.
- H. Kuramochi, S. Takeuchi and T. Tahara, J. Phys. Chem. Lett., 2012, 3, 2025–2029 CrossRef CAS.
- K. Heyne, O. F. Mohammed, A. Usman, J. Dreyer, E. T. J. Nibbering and M. A. Cusanovich, J. Am. Chem. Soc., 2005, 127, 18100–18106 CrossRef CAS PubMed.
- A. Espagne, P. Changenet-Barret, P. Plaza and M. M. Martin, J. Phys. Chem. A, 2006, 110, 3393–3404 CrossRef CAS PubMed.
- G. Groenhof, M. Bouxin-Cademartory, B. Hess, S. P. De Visser, H. J. C. Berendsen, M. Olivucci, A. E. Mark and M. A. Robb, J. Am. Chem. Soc., 2004, 126, 4228–4233 CrossRef CAS PubMed.
- D. S. Larsen, I. H. M. van Stokkum, M. Vengris, M. A. van der Horst, F. L. de Weerd, K. J. Hellingwerf and R. van Grondelle, Biophys. J., 2004, 87, 1858–1872 CrossRef CAS PubMed.
- P. Changenet-Barret, A. Espagne, S. Charier, J.-B. Baudin, L. Jullien, P. Plaza, K. J. Hellingwerf and M. M. Martin, Photochem. Photobiol. Sci., 2004, 3, 823–829 CAS.
- M. Vengris, D. S. Larsen, M. A. van der Horst, O. F. A. Larsen, K. J. Hellingwerf and R. van Grondelle, J. Phys. Chem. B, 2005, 109, 4197–4208 CrossRef CAS PubMed.
- J. S. Challice and A. H. Williams, J. Chromatogr. A, 1966, 21, 357–362 CrossRef CAS PubMed.
- R. D. Hartley and E. C. Jones, J. Chromatogr. A, 1975, 107, 213–218 CrossRef CAS.
- T. W. Fenton, M. M. Mueller and D. R. Clandinin, J. Chromatogr. A, 1978, 152, 517–522 CrossRef CAS.
- G. Kahnt, Phytochemistry, 1967, 6, 755–758 CrossRef CAS.
- T. N. V. Karsili, B. Marchetti, M. N. R. Ashfold and W. Domcke, J. Phys. Chem. A, 2014, 118, 11999–12010 CrossRef CAS PubMed.
- S. E. Greenough, M. D. Horbury, J. O. F. Thompson, G. M. Roberts, T. N. V. Karsili, B. Marchetti, D. Townsend and V. G. Stavros, Phys. Chem. Chem. Phys., 2014, 16, 16187–16195 RSC.
- M. D. Horbury, L. A. Baker, W.-D. Quan, J. D. Young, M. Staniforth, S. E. Greenough and V. G. Stavros, J. Phys. Chem. A, 2015, 119, 11989–11996 CrossRef CAS PubMed.
- S. E. Greenough, G. M. Roberts, N. A. Smith, M. D. Horbury, R. G. McKinlay, J. M. Żurek, M. J. Paterson, P. J. Sadler and V. G. Stavros, Phys. Chem. Chem. Phys., 2014, 16, 19141–19155 RSC.
- A. S. Chatterley, C. W. West, V. G. Stavros and J. R. R. Verlet, Chem. Sci., 2014, 5, 3963–3975 RSC.
- L. A. Baker, M. D. Horbury, S. E. Greenough, P. M. Coulter, T. N. V. Karsili, G. M. Roberts, A. J. Orr-Ewing, M. N. R. Ashfold and V. G. Stavros, J. Phys. Chem. Lett., 2015, 6, 1363–1368 CrossRef CAS PubMed.
- L. A. Baker, M. D. Horbury, S. E. Greenough, F. Allais, P. S. Walsh, S. Habershon and V. G. Stavros, J. Phys. Chem. Lett., 2016, 7, 56–61 CrossRef CAS PubMed.
- J. Peon, G. C. Hess, J.-M. L. Pecourt, T. Yuzawa and B. Kohler, J. Phys. Chem. A, 1999, 103, 2460–2466 CrossRef CAS.
- S. Foley, S. Navaratnam, D. J. McGarvey, E. J. Land, T. G. Truscott and C. A. Rice-Evans, Free Radical Biol. Med., 1999, 26, 1202–1208 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: UV/visible absorption spectra, global fitting error analysis, residuals and decay associated spectra, power dependence measurement and experimental results on ferulic and caffeic acid at additional pH levels. See DOI: 10.1039/c6cp01595f |
| ‡ Current address: Department of Chemistry, University of Sheffield, Sheffield, S3 7HF, UK. |
| This journal is © the Owner Societies 2016 |