
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An investigation of the effect of carbon support on ruthenium/carbon catalysts for lactic acid and butanone hydrogenation†
Daniel R.
Jones
a,
Sarwat
Iqbal
a,
Simon A.
Kondrat
a,
Giacomo M.
Lari
a,
Peter J.
Miedziak
a,
David J.
Morgan
 a,
Stewart F.
Parker
bc and
Graham J.
Hutchings
*a
a,
Stewart F.
Parker
bc and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, Main Building, Park place, Cardiff, CF10 3AT, UK. E-mail: hutch@cardiff.ac.uk
bISIS Facility, STFC Rutherford Appleton Laboratory, Chilton, Didcot, Oxon, OX11 0QX, UK
cUK Catalysis Hub, Research Complex at Harwell, STFC Rutherford Appleton Laboratory, Chilton, Didcot, Oxfordshire, OX11 0FA, UK
First published on 1st April 2016
Abstract
A series of ruthenium catalysts supported on two different carbons were tested for the hydrogenation of lactic acid to 1,2-propanediol and butanone to 2-butanol. The properties of the carbon supports were investigated by inelastic neutron scattering and correlated with the properties of the ruthenium deposited onto the carbons by wet impregnation or sol-immobilisation. It was noted that the rate of butanone hydrogenation was highly dependent on the carbon support, while no noticeable difference in rates was observed between different catalysts for the hydrogenation of lactic acid.
Introduction
Lactic acid is produced by the fermentation of glucose, a by-product of lignocellulose. The production of lactic acid is around 350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 t per year and worldwide growth is expected to be 12–15% per year.1 The chemistry and applications of lactic acid have previously been reviewed.2 Hydrogenation of lactic acid into 1,2-propanediol (a commodity chemical) is an industrially important reaction. 1,2-Propanediol (PDO) has a number of applications, mainly as a solvent for the production of polyester resins, drugs, cosmetics, food, de-icing fluid and antifreeze. Currently PDO is synthesized by the hydration of propene oxide using Cr based catalysts. This production route involving hydroperoxidation chemistry has many environmental problems because of the toxicity of Cr catalysts. Lactic acid hydrogenation provides a viable green alternative for the synthesis of PDO.3
000 t per year and worldwide growth is expected to be 12–15% per year.1 The chemistry and applications of lactic acid have previously been reviewed.2 Hydrogenation of lactic acid into 1,2-propanediol (a commodity chemical) is an industrially important reaction. 1,2-Propanediol (PDO) has a number of applications, mainly as a solvent for the production of polyester resins, drugs, cosmetics, food, de-icing fluid and antifreeze. Currently PDO is synthesized by the hydration of propene oxide using Cr based catalysts. This production route involving hydroperoxidation chemistry has many environmental problems because of the toxicity of Cr catalysts. Lactic acid hydrogenation provides a viable green alternative for the synthesis of PDO.3
The first catalytic hydrogenation of lactic acid was reported by Broadbent et al.4 using ruthenium black as a catalyst at 150 °C and 27 MPa hydrogen pressure. Since then, although various metal catalysts have been reported5,6 ruthenium based catalysts still remain the centre of interest due to their excellent hydrogenation activity.7–9 For example Zhang et al.10 have shown Ru/C to be an effective catalyst for the complete conversion of lactic acid into PDO at 100–170 °C at a hydrogen pressure of 7–14 MPa. Similarly, a MgO–NH2–Ru complex has been applied for the same reaction at 240 °C and a 100% yield of PDO was reported.11 An Ru-B–Al2O3 catalyst prepared by a reductant impregnation method has been reported to be an active catalyst for the hydrogenation of ethyl lactate yielding few by-products such as lactic acid and n-propanol.12 The same authors have also reported an effect of the addition of various promoters including Sn, Co, Fe and Zn and they found that Sn and Fe addition into a Ru-B–Al2O3 catalyst improved the activity and selectivity while Co and Zn decreased the conversion.13 In fact, from a broad perspective, ruthenium has been demonstrated to be highly active, even the pre-eminent metal, for the selective aqueous-phase hydrogenation of a wide range of bio-derived platform molecules.14,15
To synthesise PDO rather than propionic acid the formation of an alcohol by the hydrogenation of a carboxyl group without the removal of the α-hydroxyl group is required. Thermodynamic calculations have also supported the formation of propionic acid as a favoured reaction product as compared to 1,2-propanediol synthesis.16 Most of the reported studies have shown relatively severe reaction conditions for the hydrogenation of lactic acid and its esters because of the intrinsically low reactivity of the carboxylic groups adjacent to hydrogen.17 While the selective hydrogenation of bio-derived molecules, specifically that of lactic acid to 1,2-propane diol, has already been demonstrated with Ru/C, most studies focus on using the commercially available Ru/C catalyst. Recently we have shown, in a detailed study of Ru/C catalysts for this reaction,15 that there are some very important factors in the catalyst preparation, which can affect catalyst activity and selectivity. It was found that the catalytic performance was influenced by the choice of catalyst preparation method.
In addition to the selective hydrogenation of carboxylic acids an enantioselective hydrogenation of ketones using heterogeneous catalysts is one of the most promising routes for the synthesis of various molecules which can be used in the pharmaceutical and fine chemicals industries. The enantioselective hydrogenation of butanone has been previously studied using chirally modified Ru18 and Ni based catalysts.19 We have therefore selected butanone to use as a comparison with lactic acid using the Ru/C catalysts. The reaction was chosen as a simple model system because of the similar molecular structure of both substrates (butanone and lactic acid), the higher activities observed for this hydrogenation and the reported 100% selectivity to butanol without the complication of the formation of side products. There are several reasons for the higher activity of 2-butanone hydrogenation compared to lactic acid hydrogenation. 2-Butanone hydrogenation is commonly catalysed by Ru/C catalysts because they exhibit the highest activity of all hydrogenation metals. The presence of water is important – it has been shown to interact with 2-butanone on Ru/C resulting in a lower activation energy than other typical catalytic hydrogenation reactions,20 such as lactic acid hydrogenation. However, the lower activity in lactic acid hydrogenation may well be due to a support effect. A study by Corma and co-workers on lactic acid hydrogenation showed that the use of Ru supported on TiO2 resulted in a three-fold increase in activity compared to the typical Ru/C catalyst.21 The activity increase was attributed to Ti3+ defect sites participating in the hydrogenation through stabilisation of the carbonyl group. This support effect is absent in our system, resulting in lower activity for lactic acid hydrogenation. When our results are taken into account, it could be suggested that the carbonyl group in 2-butanone is adequately stabilised by the solvent, but the carbonyl group in lactic acid requires greater stabilisation by the support.
In this work we have investigated, in greater depth, the effect of carbon support properties, as characterised by inelastic neutron spectroscopy and X-ray photoelectron spectroscopy, on the final Ru-supported catalyst. We tested the catalysts for the challenging hydrogenation of lactic acid to 1,2-propanediol and also the intrinsically more reactive hydrogenation of butanone to 2-butanol.
Experimental
Chemicals
Carbons (Cabot Vulcan XC72R and G60) were obtained from the Cabot Corporation and Sigma Aldrich respectively. Ru(NO)(NO3)3 solution (Sigma Aldrich, 1.5 wt% Ru in HNO3), RuCl3 (Sigma Aldrich, 45–55 wt% Ru) and Ru(acac)3 (Sigma Aldrich, 97%) were used as ruthenium precursors. Lactic acid (Sigma Aldrich, 98%) was used as received. Products (1,2-propanediol, n-propanol, 1,5-pentanediol, 1,4-pentanediol, 1,2-pentanediol) used as standards for calibration were of analytical purity and purchased from Sigma Aldrich and used as received.Catalyst preparation
Characterization
Results and discussion
INS spectroscopy is a complementary form of vibrational spectroscopy, where the scattering event is between a neutron and the atomic nucleus, thus the electronic nature of the material, conductor, semiconductor, or insulator, is irrelevant. The scattering intensity depends on the incoherent inelastic scattering cross section and the amplitude of vibration. For 1H both of these are large, consequently the scattered intensity is dominated by hydrogenous motion. Neutrons are highly penetrating, so the spectra are representative of the bulk rather than just the surface.The INS spectra of the carbons, normalised to 1 g of sample, are shown in Fig. 1. XC72R is ×2 ordinate expanded with respect to the G60 sample. This demonstrates that the samples have different hydrogen contents. Previous work25–27 would suggest that there is at least 10 times as much hydrogen in G60 and that the hydrogen content of XC72R approaches that of a pure graphitic carbon. To confirm this we have carried out CHN analysis, which is reported in Table 1. In agreement with the previous literature there is almost exactly 10 times as much hydrogen in the G60 carbon compared to the XC72R. This is supported by the INS data by comparison of the difference spectrum: [XC72R–G60] (which removes the hydrogen-related features) with that of graphite, Fig. 2, which are clearly very similar. The features around 1150 and 850 cm−1 are assigned6 to the in-plane and out-of-plane C–H bending modes respectively, of hydrogen terminating the graphene planes. The position and number of the out-of-plane modes reflects the local environment: whether there are isolated C–H or two or three adjacent C–H oscillators.6 There is a marked difference between the G60 sample and the XC72R carbon. The differences would suggest that there are mostly isolated C–H groups in XC72R, whereas there are mostly two or more adjacent C–H groups in G60. This is consistent with the relative hydrogen contents of the carbons. Potentially this difference in hydrogen content could alter the dispersion and oxidation state of the metal supported on the two respective carbon supports. Although many noble metal catalysts can be supported on carbons, supported ruthenium is particularly prevalent in the literature for the aqueous phase hydrogenation of various different bio-derived platform molecules.
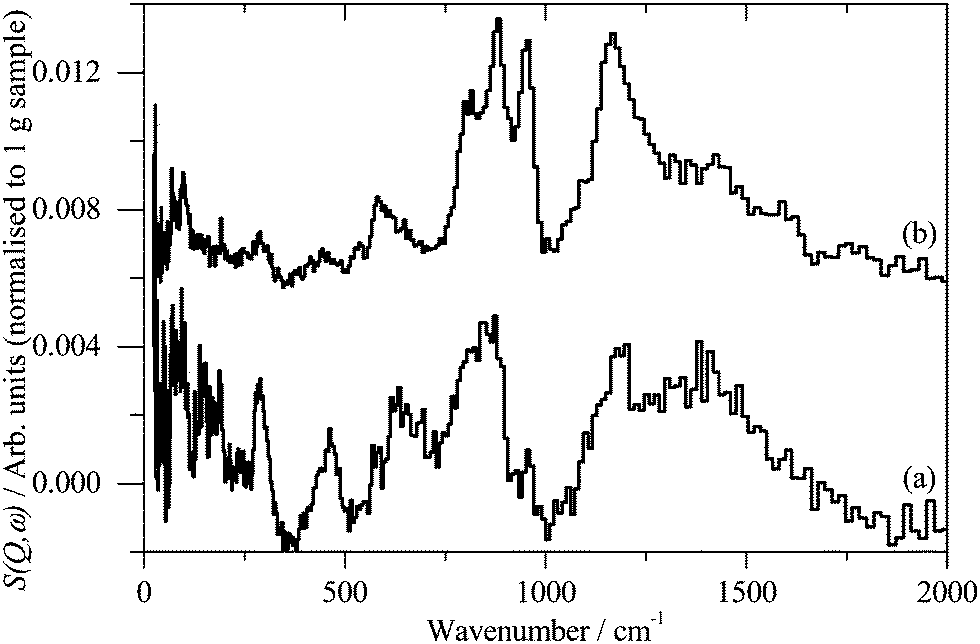 | ||
| Fig. 1 Inelastic neutron scattering spectrum of (a). XC72R, (b). G-60. (a) Is ×2 ordinate expanded relative to (b). | ||
| Carbon support | Element (%wt/wt) | ||
|---|---|---|---|
| Carbon | Hydrogen | Nitrogen | |
| G60 | 86.76 | 1.15 | 0.26 |
| XC72 | 98.64 | 0.14 | 0.46 |
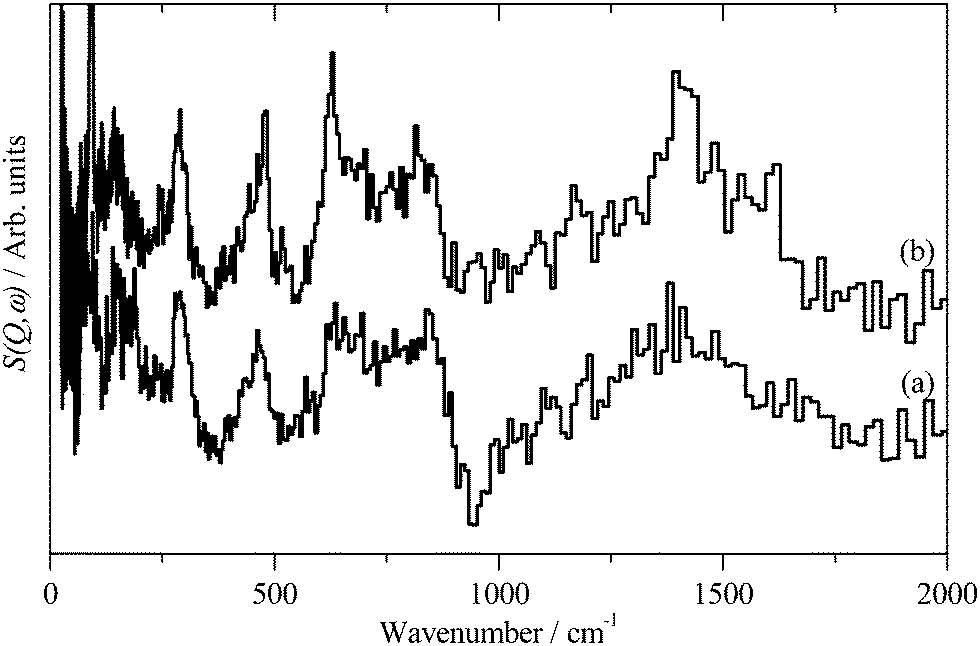 | ||
| Fig. 2 (a) Inelastic neutron subtracted spectra of: [XC72R–G-60] to emphasise the non-hydrogen features, (b) graphite. | ||
Two series of 1 wt% Ru/C catalysts were prepared using various ruthenium precursors on the two different types of carbon, with a variation of catalyst preparation methods. TEM analysis was carried out on the catalysts to try to determine the metal particle size, however it was difficult to identify any metal particles, this is likely to be because the metal particles are too small to be detected by this instrument and due to the amorphous nature of the carbon supports. In our previous studies we have reported aberration corrected TEM and shown that the ruthenium particles are below the expected detection limit of conventional TEM.28 To confirm that ruthenium was present on the catalysts we performed EDX mapping on the catalysts prepared by the sol-immobilisation prepared catalysts. For both the chloride and nitrate ruthenium precursors, ruthenium was detected over the whole area of the carbon supports suggesting the ruthenium is well dispersed over the carbon, as previously reported, the EDX maps are shown in the ESI,† Fig. S1 and S2.
All of these catalysts were tested for lactic acid conversion and their data in terms of turn over numbers (TON) are shown in Fig. 3. It was noted that the activities of all wet impregnation catalysts were comparable, for both types of carbon support and the catalyst precursors. The activity of the sol immobilisation catalysts prepared with the nitrate precursor was also found to be comparable to the wet impregnation catalysts. Only the catalysts prepared by sol-immobilisation using the chloride Ru precursor were noticeably different in activity, with TON values ca. 3 times greater than those noted for other catalysts. However, there was still no significant difference (within error) between the catalysts supported on the two different carbons. Selectivity towards 1,2-propanediol was 100% for all catalysts investigated.
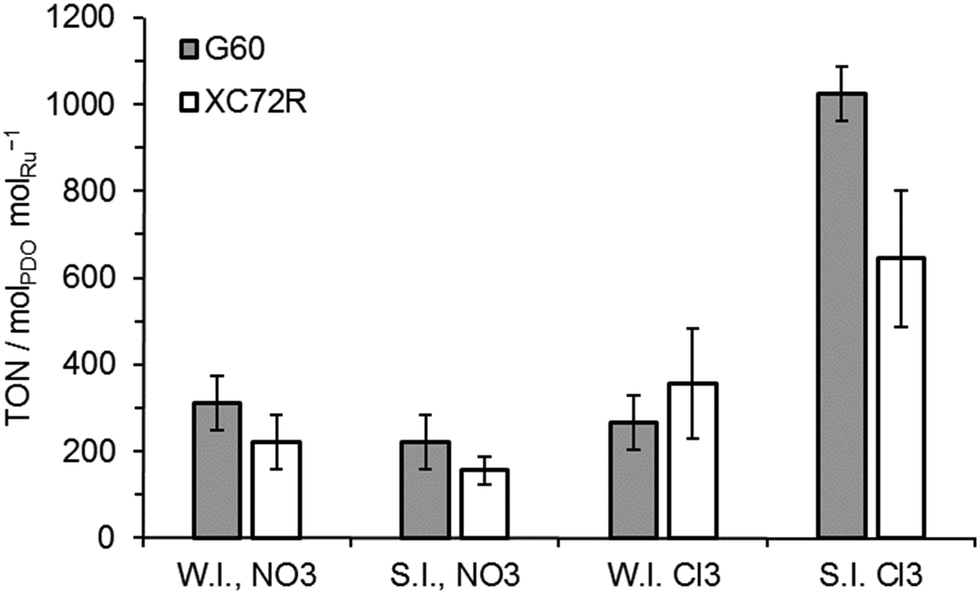 | ||
| Fig. 3 The effect of different carbons, precursors, and preparation methods on Ru/C catalysts for lactic acid hydrogenation. WI = wet impregnation; SI = sol immobilisation; NO3 = Ru(NO)(NO3)3 precursor; Cl3 = RuCl3 precursor. Reaction conditions: 125 °C, 35 bar H2, 2.5 h, 5 wt% lactic acid in H2O, 0.1 g of catalyst. | ||
The apparent invariance of activity of the catalysts for lactic acid hydrogenation was not observed in the hydrogenation of the ketone, butanone, to 2-butanol (Fig. 4). For this intrinsically more reactive substrate the observed TONs of catalysts prepared with the G60 carbon support were consistently higher than those of catalysts supported on XC72R. No significant variation between preparation techniques was observed.
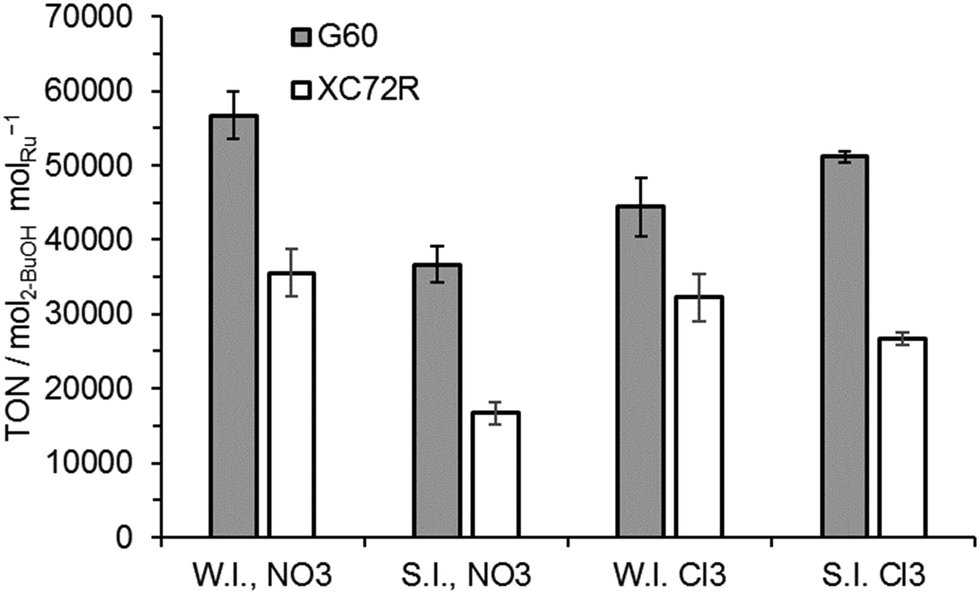 | ||
| Fig. 4 The effect of different carbons, precursors, and preparation methods on Ru/C catalysts for 2-butanone hydrogenation. WI = wet impregnation; SI = sol immobilisation; NO3 = Ru(NO)(NO3)3 precursor; Cl3 = RuCl3 precursor. Reaction conditions: 110 °C, 1 bar H2, 0.5 h, 5 wt% 2-butanone in H2O, 0.05 g of catalyst. | ||
Surface area data for all the catalysts is reported in Table 2. The surface area of the two carbon supports was noticeably different with XC72R having a much lower surface area of 220 m2 g−1 compared to 680 m2 g−1 for the G60 carbon. It was noted that the surface areas were not significantly influenced by the Ru deposition for any catalyst. The higher activity of the G60 catalysts for butanone hydrogenation could be associated with a higher dispersion of Ru, facilitated by the high surface area of this support. However, this potentially improved Ru dispersion did not have a noticeable effect on lactic acid hydrogenation rates.
| Carbon | Ru precursor | Preparation method | Surface area (m2 g−1) |
|---|---|---|---|
| G60 | Ru(NO)(NO3)3 | WI | 650 |
| SI | 630 | ||
| RuCl3 | WI | 670 | |
| SI | 610 | ||
| XC72R | Ru(NO)(NO3)3 | WI | 220 |
| SI | 200 | ||
| RuCl3 | WI | 210 | |
| SI | 200 | ||
XRD analysis (Fig. 5) was performed in order to gain information on the active phases present on the catalyst and on the average crystallite size. No reflections of Ru or RuO2 were observed in the catalysts prepared with G60 as the support or XC72R catalysts prepared with the Ru nitrate precursor. This is not surprising given the low metal loading of 1 wt% Ru used and the potential for highly dispersed Ru. Small broad reflections at 2θ = 34° and 54° associated with RuO2 were observed for the catalysts prepared from RuCl3 with the XC72R carbon. These reflections correspond to those of RuO2 physical mixed with the carbon support. The presence of observable RuO2 reflections in the XC72R catalysts suggests that Ru is less well supported on these catalysts and is also present as an oxide, as opposed to reduced Ru0. However, the low metal loadings and poor resolution of Ru species from XRD, requires alternative characterisation techniques to provide greater information on the nature of the deposited Ru species.
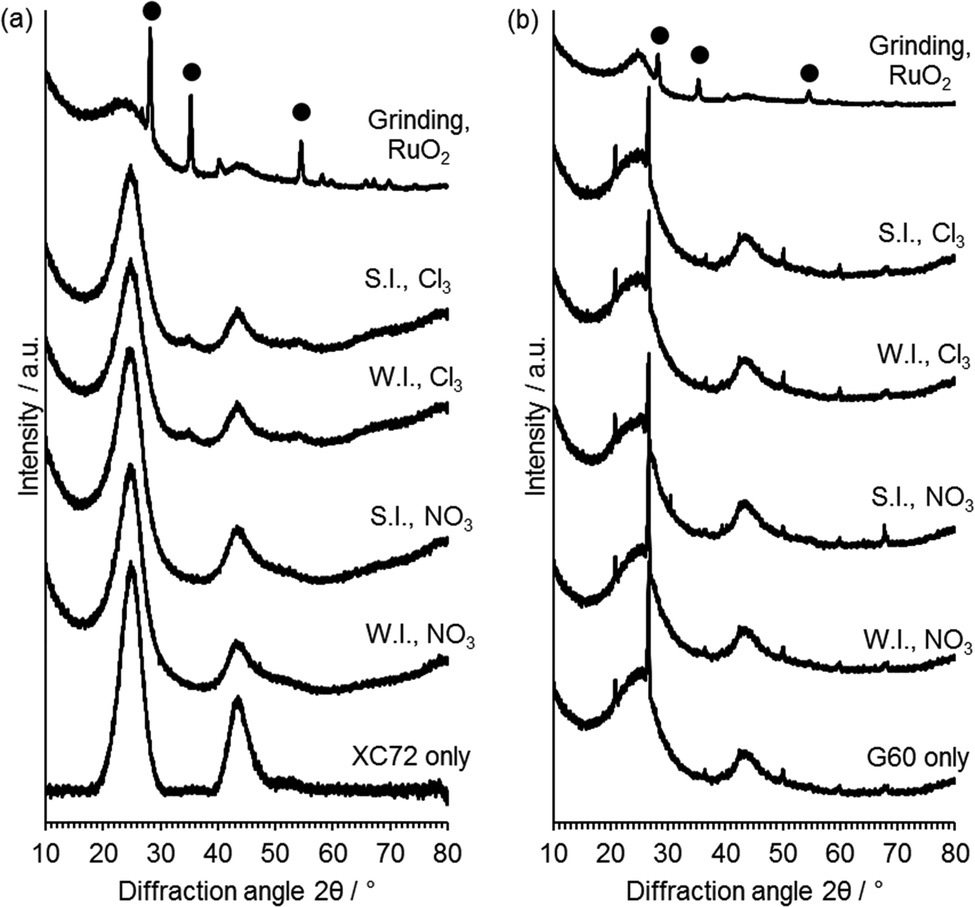 | ||
| Fig. 5 XRD patterns for 1 wt% Ru/C catalysts prepared using (a) XC72 carbon and (b) G60 carbon. ● RuO2. WI = wet impregnation; SI = sol immobilisation; NO3 = Ru(NO)(NO3)3 precursor; Cl3 = RuCl3 precursor. | ||
TPR was used to gain insight into the oxidation state and speciation of Ru in the catalysts (Fig. 6). Three principle reduction peaks were observed in most catalysts. The attribution of the peaks is still not clear but are usually described as follows. The lower temperature peaks are said to be either due to reduction of ruthenium oxide to the metal or to the reduction of Ru(IV) or Ru(III) to Ru(II), while the high temperature one is said to be due to the reduction of surface groups of the carbon support or to the reduction of Ru(II) to the metal.29,30 The catalysts prepared by the wet impregnation method on G60 and XC72R using the nitrosyl nitrate precursor have shown two reduction signals which can be linked with the reduction of ruthenium oxide (Ru (III)) along with a broad reduction signal of carbon. The catalyst prepared by the sol immobilization method on XC72R using a chloride precursor showed one very intense reduction signal at 107 °C and one weak signal at a higher temperature (210 °C). This catalyst presented a lower catalytic activity compared with the catalyst prepared on G60 carbon, both for lactic acid and butanone hydrogenation. On the other hand the catalyst prepared by the sol immobilization method on G60 using chloride precursor showed a slight reduction in signal at low temperature (∼105 °C) and two weak signals at slightly higher temperature. Clearly, the catalyst was reduced at lower temperature and showed the highest catalytic activity for both reactions. Quantification of hydrogen consumption was found to be problematic as mass spectrometry of the TPR effluent gas (Fig. S3, ESI†) revealed that methane, from Ru catalysed methanation of the carbon supports, was produced concurrently with water from RuOx reduction. The temperature of RuOx reduction was found to be a simpler and more reliable measure of catalyst reducibility. Grinding both of the carbons with ruthenium oxide showed reduction at higher temperatures (188 °C, and 190 °C) and did not show any catalytic activity for the hydrogenation reactions. Therefore, we conclude that the interaction of Ru with the support effects the degree of Ru species reducibility and this has an important role in catalytic activity for butanone hydrogenation.
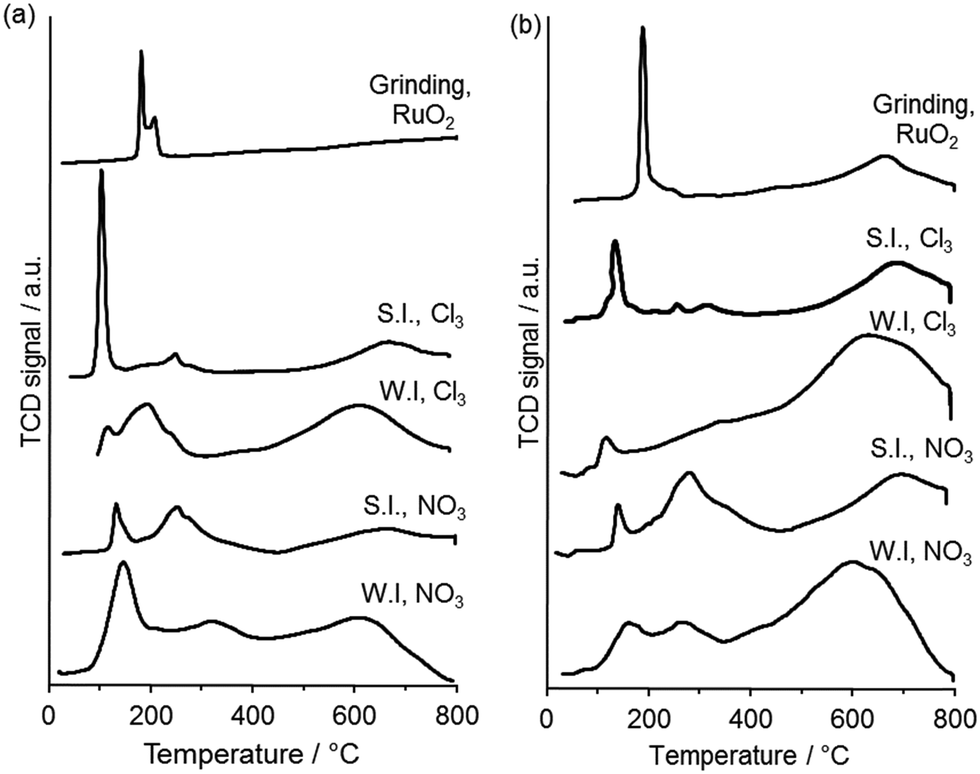 | ||
| Fig. 6 TPR profiles for 1 wt% Ru/C catalysts prepared using (a) XC72 carbon and (b) G60 carbon. WI = wet impregnation; SI = sol immobilisation; NO3 = Ru(NO)(NO3)3 precursor; Cl3 = RuCl3 precursor. | ||
The XPS data (Table 3) for the XC72R and G60 samples clearly indicates a distinct influence of the support on the type of Ru species formed. It is evident that G60 typically yields both Ru(0) and RuO2 species with the exception of the RuCl3 sol which reveals a single RuO2 state. This suggests that the G60 carbon facilitates the partial reduction of RuO2 species, while all the Ru was present as an oxide for the XC72R supported catalysts. The small shifts in energy (ca. 0.3 eV) for both Ru(0) and RuO2 species may be attributed to, or be a combination of, particle-size effects31 or the level of hydration of the ruthenium oxide, which has been shown to affect both binding energy and peak width.32 In accordance with the activity data presented, it suggests that RuO2, or its hydrated form is, or at least the precursor to, the catalytically active species. Conclusive determination of as to whether it is the pure oxide or its hydrated form is difficult to ascertain from the XPS analysis as RuO2 has a high affinity for water, even under UHV conditions.33 Analysis of the catalysts after both lactic acid and butanone hydrogenation, reveals C (1s) and O (1s) core-levels similar to those for the fresh respective carbon supports. The XPS does reveal the sol for the RuCl3 prepared XC72R catalysts is lost during reaction, although the ruthenium remains. The most notable changes is observed for the G60 catalyst, wherein no metallic ruthenium is observed to remain after reaction.
| Carbon | Ru precursor | Preparation method | Ru(3d5/2) (eV) | Ru(3p3/2) (eV) | Ru speciesb |
|---|---|---|---|---|---|
| a n/d = not determined as the concentration was too low. b Assignments made against binding energies determined for bulk reference samples. | |||||
| XC72R | Ru(NO)(NO3)3 | WI | 281.2 | 463.5 | RuO2/RuO2·xH2O |
| SI | n/da | 464.2 | RuO2/RuO2·xH2O | ||
| RuCl3 | WI | 281.3 | 463.7 | RuO2/RuO2·xH2O | |
| SI | 281.5 | 463.6 | RuO2/RuO2·xH2O | ||
| G60 | Ru(NO)(NO3)3 | WI | 280.4 (21%) and 281.2 (79%) | 462.8 | Ru(0) and RuO2/RuO2·xH2O |
| SI | 280.4 (22%) and 281.3 (78%) | 463.7 and 462.0 | Ru(0) and RuO2/RuO2xH2O | ||
| RuCl3 | WI | 280.1 (45%) and 281.0 (46%) | 462.2 and 464.0 | Ru(0) and RuO2/RuO2·xH2O | |
| SI | 281.4 | 463.7 | RuO2/RuO2·xH2O | ||
INS showed a distinct difference in the nature and concentration of hydrogen species on the two different carbon supports. XPS, XRD and TPR all show that the G60 carbon with the higher hydrogen concentration resulted in a greater degree of reducibility of the supported RuOx species. This clearly had an effect on the activity of the catalysts for butanone hydrogenation, which can be rationalised by attributing the active site of this reaction to being metallic Ru0. Higher TON is observed for the Ru/G60 carbon catalysts as they are more reducible. The lack of difference in TON for lactic acid hydrogenation between the two supported materials suggests that other factors than just metallic Ru0 contribute to activity. This could be that a synergistic effect between Lewis acidic RuOx and Ru0 is required for activity, as noted for furfural hydrogenation by Vlachos et al.34 or there is an importance of a substrate–carbon support interaction unrelated to C–H surface functionality.
Conclusions
We have investigated and reported three important parameters in the preparation of supported Ru catalysts for the hydrogenation of lactic acid and butanone, namely; effect of different types of activated carbon (G60 and XC72R), choice of ruthenium precursors, and various catalyst preparation methods. The two carbon supports were shown to have different surface hydrogen groups and surface areas, according to INS and BET analysis respectively. The higher concentration of C–H bonds in G60 explain the presence of partially reduced Ru species, according to XPS. While the isolated C–H network present in XC72R favoured the deposition of RuO2 species. For butanone hydrogenation the G60 supported catalysts were found to be more active than the XC72R supported catalysts. This was attributed to the observed partially reduced Ru species. However, the different Ru speciation did not significantly affect the rates of lactic acid hydrogenation with all catalysts having comparable TON for this reaction.Acknowledgements
The UK Catalysis Hub is kindly thanked for resources and support provided via our membership of the UK Catalysis Hub Consortium and funded by EPSRC (grants EP/K014706/1, EP/K014668/1, EP/K014854/1EP/K014714/1 and EP/M013219/1). We thank the STFC Rutherford Appleton Laboratory for access to neutron beam facilities on TOSCA.References
- K. L. Wasewar, A. A. Yawalkar, J. A. Moulijn and V. G. Pangarkar, Ind. Eng. Chem. Res., 2004, 43, 5969–5982 CrossRef CAS.
- R. Datta, S.-P. Tsai, P. Bonsignore, S.-H. Moon and J. R. Frank, FEMS Microbiol. Rev., 1995, 16, 221–231 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- H. S. Broadbent, G. C. Campbell, W. J. Bartley and J. H. Johnson, J. Org. Chem., 1959, 24, 1847–1854 CrossRef CAS.
- R. D. Cortright, M. Sanchez-Castillo and J. A. Dumesic, Appl. Catal., B, 2002, 39, 353–359 CrossRef CAS.
- M. A. N. Santiago, M. A. Sanchez-Castillo, R. D. Cortright and J. A. Dumesic, J. Catal., 2000, 193, 16–28 CrossRef CAS.
- R. L. Augustine, Catal. Today, 1997, 37, 419–440 CrossRef CAS.
- M. J. Mendes, O. A. A. Santos, E. Jordao and A. M. Silva, Appl. Catal., A, 2001, 217, 253–262 CrossRef CAS.
- K. Tahara, H. Tsuji, H. Kimura, T. Okazaki, Y. Itoi, S. Nishiyama, S. Tsuruya and M. Masai, Catal. Today, 1996, 28, 267–272 CrossRef CAS.
- Z. Zhang, J. E. Jackson and D. J. Miller, Appl. Catal., A, 2001, 219, 89–98 CrossRef CAS.
- B.-W. Mao, Z.-Z. Cai, M.-Y. Huang and Y.-Y. Jiang, Polym. Adv. Technol., 2003, 14, 278–281 CrossRef CAS.
- G. Luo, S. Yan, M. Qiao, J. Zhuang and K. Fan, Appl. Catal., A, 2004, 275, 95–102 CrossRef CAS.
- G. Luo, S. Yan, M. Qiao and K. Fan, J. Mol. Catal. A: Chem., 2005, 230, 69–77 CrossRef CAS.
- C. Michel and P. Gallezot, ACS Catal., 2015, 5, 4130–4132 CrossRef CAS.
- S. Iqbal, S. A. Kondrat, D. R. Jones, D. C. Schoenmakers, J. K. Edwards, L. Lu, B. R. Yeo, P. P. Wells, E. K. Gibson, D. J. Morgan, C. J. Kiely and G. J. Hutchings, ACS Catal., 2015, 5, 5047–5059 CrossRef CAS.
- P. Maki-Arvela, I. L. Simakova, T. Salmi and D. Y. Murzin, Chem. Rev., 2014, 114, 1909–1971 CrossRef CAS PubMed.
- M. A. Dasari, P.-P. Kiatsimkul, W. R. Sutterlin and G. J. Suppes, Appl. Catal., A, 2005, 281, 225–231 CrossRef CAS.
- H. Wan, A. Vitter, R. V. Chaudhari and B. Subramaniam, J. Catal., 2014, 309, 174–184 CrossRef CAS.
- Z. Lou, X. Chen, L. Tian, M. Qiao, K. Fan, H. He, X. Zhang and B. Zong, J. Mol. Catal. A: Chem., 2010, 326, 113–120 CrossRef CAS.
- H. Wan, A. Vitter, R. V. Chaudhari and B. Subramaniam, J. Catal., 2014, 309, 174–184 CrossRef CAS.
- A. Primo, P. Concepcion and A. Corma, Chem. Commun., 2011, 47, 3613–3615 RSC.
- P. C. H. P. Mitchell, S. F. Parker, A. J. Ramirez-Cuesta and J. Tomkinson, Vibrational spectroscopy with neutrons, with applications in chemistry, biology, materials science and catalysis, World Scientific, Singapore, 2005 Search PubMed.
- D. Colognesi, M. Celli, F. Cilloco, R. J. Newport, S. F. Parker, V. Rossi-Albertini, F. Sacchetti, J. Tomkinson and M. Zoppi, Appl. Phys. A: Mater. Sci. Process., 2002, 74, S64–S66 CrossRef CAS.
- http://www.isis.stfc.ac.uk/ .
- P. Albers, K. Seibold, G. Prescher, B. Freund, S. F. Parker, J. Tomkinson, D. K. Ross and F. Fillaux, Carbon, 1999, 37, 437–444 CrossRef CAS.
- P. Albers, A. Karl, J. Mathias, D. K. Ross and S. F. Parker, Carbon, 2001, 39, 1663–1676 CrossRef CAS.
- P. W. Albers, J. Pietsch, J. Krauter and S. F. Parker, Phys. Chem. Chem. Phys., 2003, 5, 1941–1949 RSC.
- S. Iqbal, S. A. Kondrat, D. R. Jones, D. C. Schoenmakers, J. K. Edwards, L. Lu, B. R. Yeo, P. P. Wells, E. K. Gibson, D. J. Morgan, C. J. Kiely and G. J. Hutchings, ACS Catal., 2015, 5, 5047–5059 CrossRef CAS.
- I. Rossetti, N. Pernicone and L. Forni, Appl. Catal., A, 2003, 248, 97–103 CrossRef CAS.
- A. M. Hengne, N. S. Biradar and C. V. Rode, Catal. Lett., 2012, 142, 779–787 CrossRef CAS.
- M. Baron, O. Bondarchuk, D. Stacchiola, S. Shaikhutdinov and H. J. Freund, J. Phys. Chem. C, 2009, 113, 6042–6049 CAS.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- A. Foelske, O. Barbieri, M. Hahn and R. Kötz, Electrochem. Solid-State Lett., 2006, 9, A268–A272 CrossRef CAS.
- P. Panagiotopoulou, N. Martin and D. G. Vlachos, J. Mol. Catal. A: Chem., 2014, 392, 223–228 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp01311b |
| This journal is © the Owner Societies 2016 |