
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtended charge accumulation in ruthenium–4H-imidazole-based black absorbers: a theoretical design concept†
Stephan
Kupfer
Institute of Physical Chemistry and Abbe Center of Photonics, Friedrich-Schiller-University Jena, Helmholtzweg 4, 07743 Jena, Germany. E-mail: stephan.kupfer@uni-jena.de
First published on 14th April 2016
Abstract
A theoretical-guided design concept aiming to achieve highly efficient unidirectional charge transfer and multi-charge separation upon successive photoexcitation for light-harvesting dyes in the scope of supramolecular photocatalysts is presented. Four 4H-imidazole–ruthenium(II) complexes incorporating a biimidazole-based electron-donating ligand sphere have been designed based on the well-known 4H-imidazole–ruthenium(II) polypyridyl dyes. The quantum chemical evaluation, performed at the density functional and time-dependent density functional level of theory, revealed extraordinary unidirectional charge transfer bands from the near-infrared to the ultraviolet region of the absorption spectrum upon multi-photoexcitation. Spectro-electrochemical simulations modeling photoexcited intermediates determined the outstanding multi-electron storage capacity for this novel class of black dyes. These remarkable photochemical and photophysical properties are found to be preserved upon site-specific protonation rendering 4H-imidazole–ruthenium(II) biimidazole dyes ideal for light-harvesting applications in the field of solar energy conversion.
1. Introduction
In recent years ruthenium(II) polypyridyl complexes have attracted enormous attention due to their outstanding (photo)chemical and (photo)physical properties, i.e., strong stability on heat, electricity and light coupled with broad absorption bands in the visible region, and redox and catalytic activity. Thus, possible applications of such complexes range from pharmaceuticals in phototherapy agents,1–7 light-emitting diodes,8–11 molecular sensors,12–16 semiconductors,17,18 and sensitizers in dye-sensitized solar cells19–22 (DSSCs) to supramolecular photocatalysts in the fields of water splitting and artificial photosynthesis.23–31Black absorbers based on ruthenium(II) polypyridyl complexes are widely applied in light-harvesting devices and photosensitizers due to their broad absorption bands. Such complexes, e.g. ruthenium(II) trisbipyridine and ruthenium(II) bisterpyridine-based dyes, typically present metal-to-ligand charge transfer (MLCT) bands centered below 550 nm. Several strategies have been pursued to enhance the absorption in the visible and near-infrared (NIR) regions in order to maximize overlap with the solar radiation spectrum.32,33 However, the improvement of light-induced (multi-)electron charge transfer processes and the stabilization of (multi-)charge-separated photoexcited intermediates are further major keystones in the field of supramolecular photocatalysis.34,35 Thus, dyes capable of donating and transferring several photoinduced charges in a unidirectional manner towards a catalytic center are of vital importance for light-driven hydrogen generation.34–39
One promising class of black absorbers to fulfil these pre-requirements are 4H-imidazole–ruthenium(II) polypyridyl complexes. Lately, we investigated the effects of (multi-)protonation,40,41 substitution with electron-withdrawing groups (EWGs) and electron-donating groups (EDGs) in the periphery of the 4H-imidazole ligand,40–42 anchoring on titanium dioxide nanoparticles43,44 and the influence of the polypyridyl ligand sphere, i.e. for terpyridine and bipyridine ligands,40,45 on their photophysical and photochemical properties by spectroscopic and computational methods. Furthermore, photoexcited relaxation channels42,46 have been assessed as well as their (multi-)photoelectron donor44 and storage capacities47,48 based on a spectro-electrochemical (SEC) approach. The absorption spectrum for this class of black absorbers was found to be dominated by mainly two intense bands: (i) a MLCT band (583 to 600 nm), with contributions towards both ligand spheres (4H-imidazole and polypyridyl), while (multi-)protonation leads to a bathochromic shift (from approximately 605 to 615 nm) accompanied by an enhanced unidirectionality of the underlying MLCT transition towards 4H-imidazole and, (ii) an intra-ligand (IL) band, correlated to a locally excited state of 4H-imidazole, at approximately 368–429 nm depending on the substitution pattern and the pH. Thus, these complexes feature two channels to accumulate photoexcited electrons on the 4H-imidazole ligand, via MLCT and IL states, which were further investigated by UV-vis and resonance Raman (RR-)SEC. The singly oxidized complex with an electron hole at ruthenium 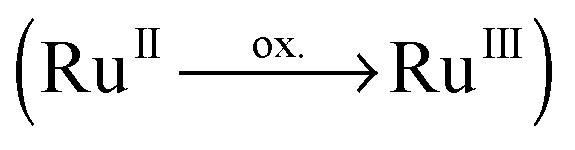 , mimicking the electronic structure upon MLCT excitation, was found to be capable of injecting a second electron into the low-lying π*-orbital of 4H-imidazole (π*im) via an IL centered state.44 Reductive SEC studies, aiming to estimate the multi-electron storage capacity, revealed that dual charge separation is closely related to the competitive population of a doubly reduced singlet (ground) state vs. a doubly reduced triplet (ground) state. For the singlet state, both excess charges are localized on 4H-imidazole, while the triplet state holds one charge on each ligand sphere.47,48 Thus, the key for multi-electron storage is to stabilize the doubly reduced singlet state with respect to the triplet analog. Such stabilization can be achieved by EWGs in the periphery of 4H-imidazole.
, mimicking the electronic structure upon MLCT excitation, was found to be capable of injecting a second electron into the low-lying π*-orbital of 4H-imidazole (π*im) via an IL centered state.44 Reductive SEC studies, aiming to estimate the multi-electron storage capacity, revealed that dual charge separation is closely related to the competitive population of a doubly reduced singlet (ground) state vs. a doubly reduced triplet (ground) state. For the singlet state, both excess charges are localized on 4H-imidazole, while the triplet state holds one charge on each ligand sphere.47,48 Thus, the key for multi-electron storage is to stabilize the doubly reduced singlet state with respect to the triplet analog. Such stabilization can be achieved by EWGs in the periphery of 4H-imidazole.
Here we present a theory-guided design concept to improve the light-harvesting properties of 4H-imidazole–ruthenium-based black absorbers by increasing the unidirectionality of the CT, and to simultaneously enhance the multi-(photo)electron storage capacity, which is essential for the functionality in the scope of artificial photosynthesis devices. Structural modifications based on the chloro-η3-4,4′,4′′-tritert-butyl-2,2′:6′,2′′-terpyridine-η2-phenyl-4,5-(p-ethylcarboxyphenyl-imino)-imidazolate–ruthenium(II) (RuTerpy) reference system aim to replace the terpyridine and chlorine ligands by means of a new ligand sphere that: (i) does not contribute (significantly) to the bright MLCT, IL and intra-ligand CT (ILCT) states in the visible region enhancing the unidirectionality of the CT, (ii) features high-lying bonding orbitals donating electron density towards the ruthenium center to (partially) recover the initial ruthenium(II) species upon primary photoexcitation quenching charge recombination processes, and (iii) allows multi-photoinduced charge accumulation on 4H-imidazole due to its HOMO–LUMO (highest occupied molecular orbital–lowest unoccupied molecular orbital) gap. This schematic design concept is illustrated in Fig. 1a along with the molecular orbitals (MOs) involved in the electronic transition underlying the MLCT band of RuTerpy (Fig. 1b and d, dxy → π*im) and the charge-separated triplet state that is populated upon intersystem crossing and subsequent depletion along the excited state relaxation cascades (Fig. 1c and d, dxz, π*im).
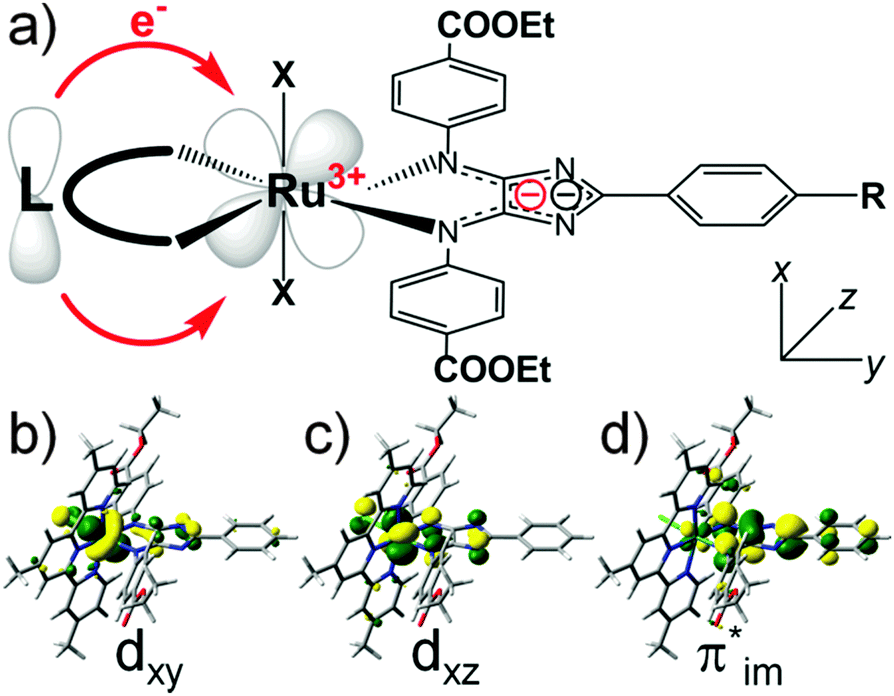 | ||
| Fig. 1 (a) Basic design concept employing an electron-donating ligand sphere capable of (partially) recovering the initial ruthenium(II) center upon primary photoexcitation. Frontier orbitals of the RuTerpy reference complex involved in the first bright MLCT transition, (b) dxy and (d) π*im, and the charge-separated triplet state, (c) dxz and (d) π*im. | ||
A promising class of ligands to realize these three objectives are bibenzimidazoles. The photoactivity and electrochemistry of bibenzimidazolyl complexes has been investigated for a long time.49–53 Bibenzimidazole ligands incorporated in e.g. ruthenium(II) bipyridyl and ruthenium(II) phenanthrolyl complexes (and related systems) function as non-electron-accepting ligands and thus do not participate in low-lying excited states such as MLCT and IL states.15,54–59 Hence, unidirectional CT can be achieved upon photoexcitation, however, the (photo)physical and (photo)chemical properties of ruthenium(II) bibenzimidazolyl, and structurally-related complexes, are known to be highly pH-dependent, i.e., with protonation of the non-coordinating nitrogen atoms of bibenzimidazole.15,56,58,60,61 Such pH-dependency is of interest for applications in molecular sensors but it is undesired in the scope of molecular photocatalysis. Several attempts have been proposed to overcome this shortcoming by protecting the non-coordinating nitrogen atoms with phenylene or 1,2-ethanidyl groups.59,62
In this contribution we present a priori quantum chemical evaluation of the photophysical and photochemical properties of four 4H-imidazole–ruthenium(II)–bi(benz)imidazolyl complexes (complexes Ru1–Ru4 in Fig. 2) upon multi-photoexcitation. On the one hand, Ru1 and Ru2 are designed according to the concept shown in Fig. 1a, where 4H-imidazole and the bibenzimidazole/biimidazole ligands reside in the same plane (xy) to allow for direct interaction of their π-systems with the dxy donor orbital of ruthenium, involved in the lowest bright MLCT excitation towards 4H-imidazole (MOs in Fig. 1b and d). On the other hand, the related bisbibenzimidazole and bisbiimidazole complexes, Ru3 and Ru4, aim to stabilize the charge-separated triplet state by improving the overlap of the bi(benz)imidazole π-orbitals with the dxz and dyz orbitals of ruthenium. Furthermore, the (multi-)electron donor and (multi-)electron storage capacities as well as the impact of the pH on the photophysical and photochemical properties will be studied.
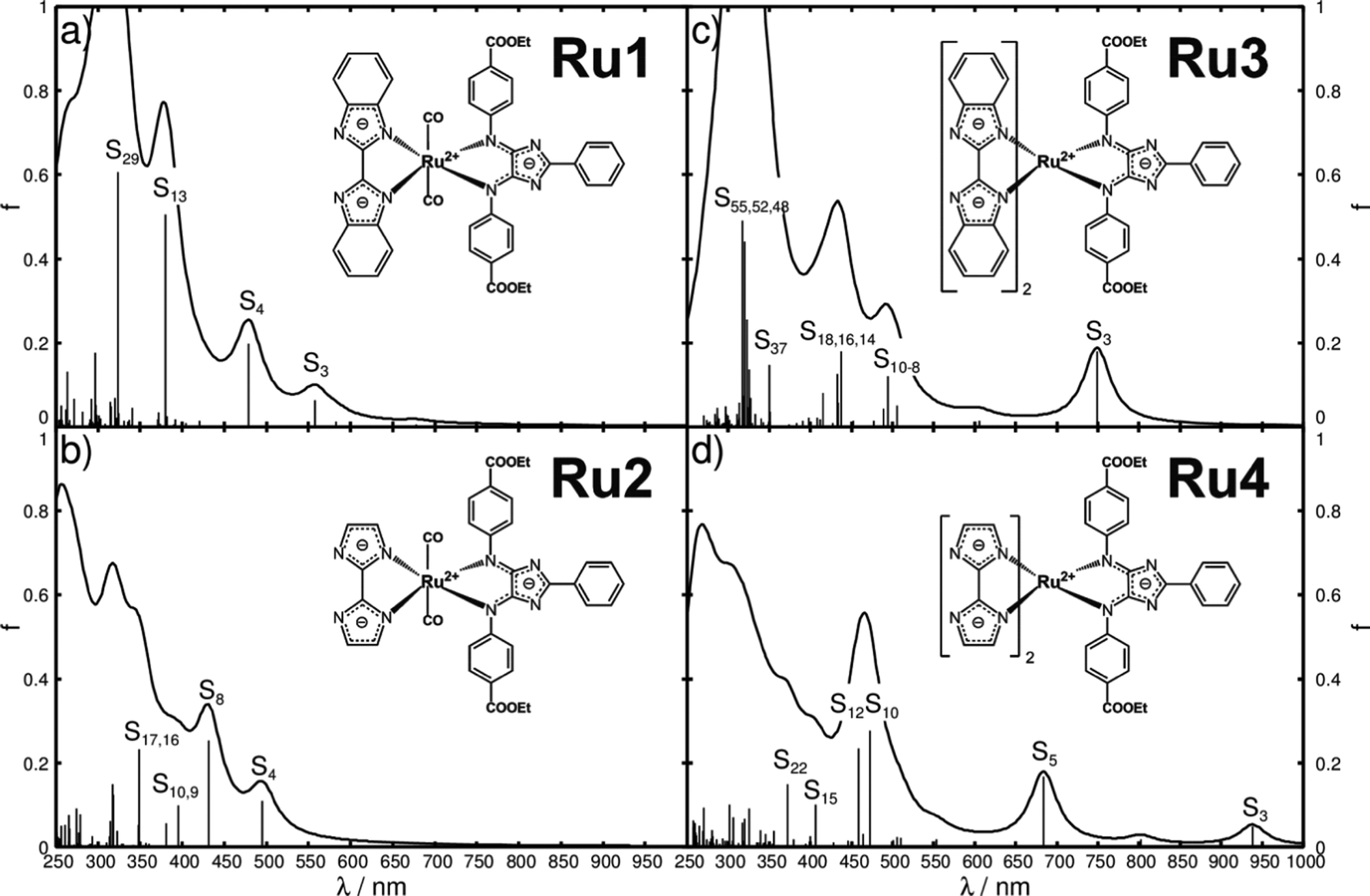 | ||
| Fig. 2 Molecular structures and simulated UV-vis absorption spectra for the primary photoactivation of Ru1 (a), Ru2 (b), Ru3 (c) and Ru4 (d) in acetonitrile. The calculated oscillator strengths are represented by black bars, and a Lorentzian function with a half-width of 20 nm is employed to broaden the transitions. | ||
2. Computational details
The structural and electronic data for complexes Ru1, Ru2, Ru3 and Ru4 were obtained from quantum chemical calculations performed with the Gaussian 09 program.63 While the complexes Ru1–Ru3 have been exclusively studied in their non-reduced singlet form, Ru4 has been furthermore investigated in several redox states, namely: (i) the singly oxidized doublet, (ii) the singly reduced doublet and (iii) the doubly reduced singlet and triplet. In addition, protonation effects have been taken into account for Ru4, while single protonation at three positions has been considered: (i) at one non-bonding nitrogen atom of the biimidazole, (ii) at one non-bonding nitrogen atom of the 4H-imidazole and (iii) at one carbonyl group of the ester substituent. Furthermore, the non-reduced triplet species of Ru4 was studied as well as its three aforementioned singly protonated species. The equilibrium geometries of the electronic ground state were calculated by means of density functional theory (DFT) with the XC functional B3LYP64,65 for each redox state of each compound, respectively. The 6-31G(d) double-ζ basis set66 was employed for all main group elements. The 28-electron relativistic core potential MWB67 was applied with its basis set for the ruthenium atom, that is, 4s, 4p, 4d and 5s electrons are treated explicitly, while the first three inner shells are described by the core pseudo-potential. A subsequent vibrational analysis carried out for each fully optimized structure verified that the obtained structures correspond to minima of the ground state potential energy surface (PES).Excited state properties, such as excitation energies and oscillator strengths, have been calculated at the time-dependent DFT (TDDFT) level of theory within the adiabatic approximation with the same XC functional, pseudo-potential and basis set. Several joint experimental–theoretical studies on structurally-related polypyridyl–ruthenium–4H-imidazole complexes proved that this computational protocol enables an accurate prediction of ground and excited state properties with respect to experimental data, e.g. UV-vis absorption, RR spectra and excited state relaxation pathways, as well as (spectro-)electrochemical properties.40,42,45,47 As shown in ref. 40 the B3LYP functional provides a balanced description of the MLCT (metal-to-ligand charge transfer), LLCT (ligand-to-ligand intra-ligand charge transfer), ILCT (intra-ligand charge transfer) and LMCT (ligand-to-metal charge transfer) states as well as of the local ππ* states of the respective ligand spheres for ruthenium–4H-imidazole complexes. The absorption spectra of Ru1–Ru4 were simulated by means of the 100 lowest excited states of the respective ground state multiplicity. The effects of the interaction with a solvent (acetonitrile, ε = 35.688, n = 1.344) on the geometry, frequencies, excitation energies and transition dipole moments were taken into account by the integral equation formalism of the polarizable continuum model.68
3. Results
In order to estimate the multi-photon light-harvesting capacity for this class of novel black absorbers, we apply quantum chemical simulations on complexes Ru1–Ru4 to study their photophysical properties. Subsequently, their multi-photoelectron donor and acceptor capacities are evaluated by means of a SEC approach. Additionally, the pH-dependency upon site-specific protonation and its impact on the photophysical and photochemical properties is assessed.3.1 UV-vis absorption for primary and secondary photoexcitation
The preliminary step to evaluate dyes and their eligibility as light-harvesting units in DSSCs or as photosensitizers in light-driven supramolecular catalysis is to obtain insight into their UV-vis absorption spectra. Thus, the UV-vis spectra of Ru1, Ru2, Ru3 and Ru4, depicted in Fig. 2a–d, have been simulated by means of TDDFT with the B3LYP functional.Detailed information with respect to the optimized equilibrium geometries, excited state properties and MOs involved in the leading transitions are collected in Tables S1–S3 of the ESI.† The quantum chemical simulations on Ru1, featuring one bibenzimidazole and two carbonyl ligands, revealed that the visible region is dominated by one medium bright IL state of 4H-imidazole (S3) and one bright LLCT state from bibenzimidazole towards 4H-imidazole (S4) at 2.22 (557 nm) and 2.59 eV (479 nm), respectively. The UV region features one additional bright 4H-imidazole-centered IL state (S13) at 3.26 eV (380 nm) and one IL state of the bibenzimidazole ligand (S29) at 3.84 eV (324 nm). Surprisingly, only minor contributions of MLCT character are estimated for Ru1, which is correlated to the bulky bibenzimidazole ligand distorting the structure of the entire complex (α1, α2 and the butterfly angle αBF in Table S1 of the ESI†). Detailed information with respect to excited state properties, charge density differences (CDDs) and the MOs involved in the leading transitions of Ru1 (and Ru2–Ru4) are collected in Tables S2–S4 (ESI†).
To overcome this steric limitation, bibenzimidazole was reduced to biimidazole from Ru1 to Ru2 (detailed structural data shown in Table S1, ESI†). As predicted, the first absorption band of Ru2 in the visible region given by the S4 state at 2.50 eV (495 nm) (Fig. 2b) is now of MLCT character towards 4H-imidazole, while the character of the 4H-imidazole IL band, S8, at 2.87 eV (431 nm) is not affected significantly. Analogous to Ru1, the UV region of Ru2 is dominated by bright IL states of both ligand spheres. Generally, a bathochromic shift of the absorption features is predicted by the quantum chemical evaluation from Ru1 to Ru2, which is intrinsically connected to the reduction of the π-system from bibenzimidazole in Ru1 to biimidazole in Ru2.
In order to combine an extended π-system for the ligands with minimization of the steric hindrance, Ru1, a bisbibenzimidazole sphere, is introduced in Ru3, where all three ligands are orientated perpendicular to each other to minimize steric hindrance. The resulting absorption spectrum, illustrated in Fig. 2c, is substantially shifted to higher wavelengths, and thus exhibits increased overlap with the solar radiation spectrum. The bright MLCT state to 4H-imidazole (S3) is stabilized to 1.66 eV (749 nm), further bright 4H-imidazole-centered IL states, i.e. S9 and S16, as well as (medium) bright MLCT states, S10, S14 and S18, which dominate the visible region of the absorption spectrum of Ru3. However, the S14, S16 and S18 states show pronounced contributions of MLCT transitions towards the bisbibenzimidazole ligand sphere, which leads to a decrease of unidirectionality of the CT (CDDs in Table S4, ESI†). Analogous to Ru1 and Ru2, intense IL states of 4H-imidazole (S37) and bisbibenzimidazole (S48, S51, S52 and S55) are calculated in the UV region.
Ru4 features a bisbiimidazole ligand sphere to decrease the CT to bisbibenzimidazole. The simulated UV-vis spectrum, shown in Fig. 2d, features bright and medium bright MLCT and IL states, exclusively directed towards 4H-imidazole. Detailed information with respect to the bright excited states of Ru4 is given in Table 1, while the respective CDDs between the respective excited singlet state and the singlet ground state are illustrated in Fig. 3; the MOs involved in the underlying transitions are collected in Table S3 of the ESI.†
| State | Transition | Weight (%) | E e (eV) | λ (nm) | f | 〈s2〉 |
|---|---|---|---|---|---|---|
| Primary photoexcitation (singlet, S0 geometry) | ||||||
| S3 | dxy + πbim(199) → π*im(201) (MLCT) | 77 | 1.32 | 938 | 0.049 | 0.00 |
| dxy + πbim(197) → π*im(201) (MLCT) | 9 | |||||
| dyz + πbim(200) → π*im(201) (MLCT) | 9 | |||||
| S5 | dxy + πbim(197) → π*im(201) (MLCT) | 87 | 1.81 | 684 | 0.168 | 0.00 |
| S10 | πim(195) → π*im(201) (ILCT) | 71 | 2.63 | 472 | 0.278 | 0.00 |
| dxy + πbim(199) → π*im(203) (MLCT) | 23 | |||||
| S12 | dxy + πbim(199) → π*im(203) (MLCT) | 64 | 2.70 | 459 | 0.233 | 0.00 |
| πim(195) → π*im(201) (ILCT) | 24 | |||||
| S15 | dxy + πbim(197) → π*im(203) (MLCT) | 90 | 3.05 | 406 | 0.101 | 0.00 |
| S22 | πdelocal(191) → π*im(201) (CT) | 89 | 3.33 | 372 | 0.149 | 0.00 |
| Secondary photoexcitation (triplet, T1 geometry) | ||||||
| T3 | πbim(199β) → dxz(200β) (LMCT) | 74 | 1.23 | 1005 | 0.010 | 2.03 |
| dyz(196β) → dxz(200β) (MC) | 11 | |||||
| T4 | πbim(198β) → dxz(200β) (LMCT) | 64 | 1.33 | 934 | 0.016 | 2.03 |
| dxy(197β) → dxz(200β) (MC) | 14 | |||||
| dyz(196β) → dxz(200β) (MC) | 11 | |||||
| T6 | πbim(198β) → π*im(201β) (LLCT) | 49 | 1.68 | 739 | 0.017 | 2.04 |
| πim(195β) → dxz(200β) (LMCT) | 13 | |||||
| πbim(199β) → π*im(201β) (LLCT) | 10 | |||||
| dxy(197β) → π*im(201β) (MLCT) | 10 | |||||
| T8 | π*im(201α) → π*im(202α) (ILCT) | 84 | 1.90 | 651 | 0.243 | 2.08 |
| T9 | π*im(201α) → π*im(203α) (IL) | 56 | 1.99 | 623 | 0.039 | 2.06 |
| πim(195β) → dxz(200β) (LMCT) | 18 | |||||
| T10 | dyz(196β) → π*im(201β) (MLCT) | 47 | 2.10 | 591 | 0.020 | 2.04 |
| dxy(197β) → π*im(201β) (MLCT) | 33 | |||||
| T11 | π*im(201α) → π*im(203α) (IL) | 29 | 2.13 | 581 | 0.191 | 2.04 |
| dyz(196β) → π*im(201β) (MLCT) | 26 | |||||
| dxy(197β) → π*im(201β) (MLCT) | 23 | |||||
| T12 | πim(195β) → π*im(201β) (ILCT) | 89 | 2.27 | 546 | 0.340 | 2.04 |
| T13 | π*im(201α) → π*im(204α) (IL) | 58 | 2.70 | 459 | 0.031 | 2.39 |
| πim(194β) → dxz(200β) (LMCT) | 14 | |||||
| T14 | πim(194β) → dxz(200β) (LMCT) | 76 | 2.75 | 450 | 0.132 | 2.12 |
| π*im(201α) → π*im(204α) (IL) | 9 | |||||
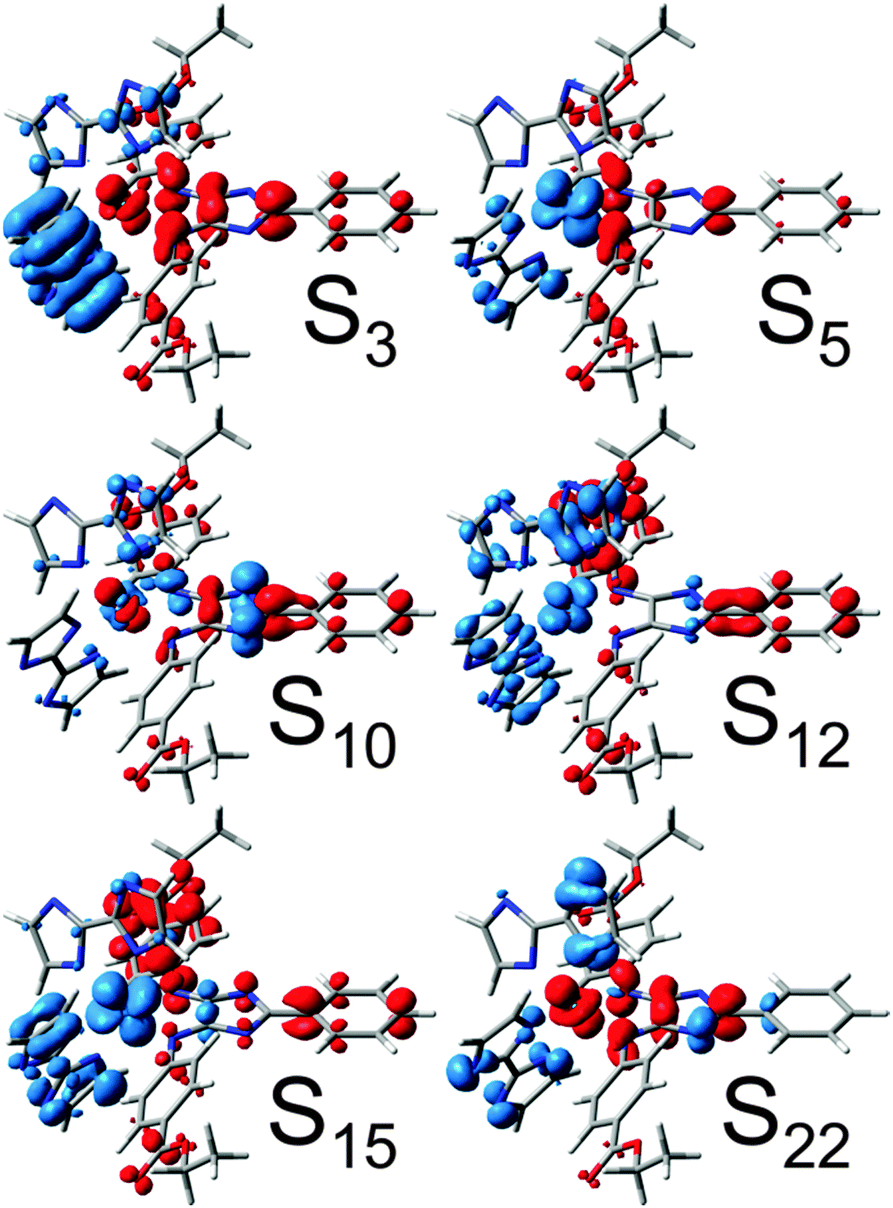 | ||
| Fig. 3 CDDs for bright singlet excitations contributing to the UV-vis absorption spectrum of Ru4. CT takes place from blue to red. | ||
The first medium bright MLCT state (S3) is observed in the NIR region at merely 1.32 eV (938 nm), and further bright MLCT states (S5, S12 and S15) are found at 1.81, 2.70 and 3.05 eV (684, 472 and 406 nm), respectively. A closer look into the MOs involved in the MLCT transitions reveals that the dxy, dxz and dyz orbitals of ruthenium are highly mixed with the π-system of the biimidazole ligands (MOs in Table S3, ESI†), which leads to a highly unidirectional CT from the biimidazoles and ruthenium towards the 4H-imidazole. In addition, bright 4H-imidazole-centered IL states, namely S10 and S22, are calculated at 2.63 and 3.33 eV (472 and 372 nm). Hence, Ru4 is a promising black absorber capable of transferring photoexcited electrons unidirectionally to the 4H-imidazole ligand via MLCT and IL states ranging from the NIR to the UV region. However, photosensitizers for hydrogen-evolving supramolecular photocatalysts need to supply at least two photoelectrons. Therefore, the next step is to investigate the absorption spectrum of the photoexcited Ru4, which will be denoted as secondary photoexcitation. Upon primary photoexcitation to the bright singlet excited states, ultra-fast inter-system crossing to the triplet manifold is assumed to occur, followed by population of the triplet ground state via excited state relaxation channels. Therefore, the triplet ground state (within the Frank–Condon region of the primary photoexcitation) was optimized and used as the initial structure for the secondary photoexcitation within triplet multiplicity, and structural data on the optimized triplet are collected in Table S5 (ESI†). In accordance with the reference complex RuTerpy (recall MOs in Fig. 1c and d), the triplet ground state of Ru4 features an electron hole in the dxz orbital, while the photoexcited electron resides in the lowest anti-bonding orbital of the 4H-imidazole (MOs dxz(200β) and π*im(201α) in Table S6, ESI†). The resulting absorption spectrum for the secondary excitation as well as the primary absorption spectrum are depicted in Fig. 4b, while detailed information with respect to the underlying excited states are collected in Table 1.
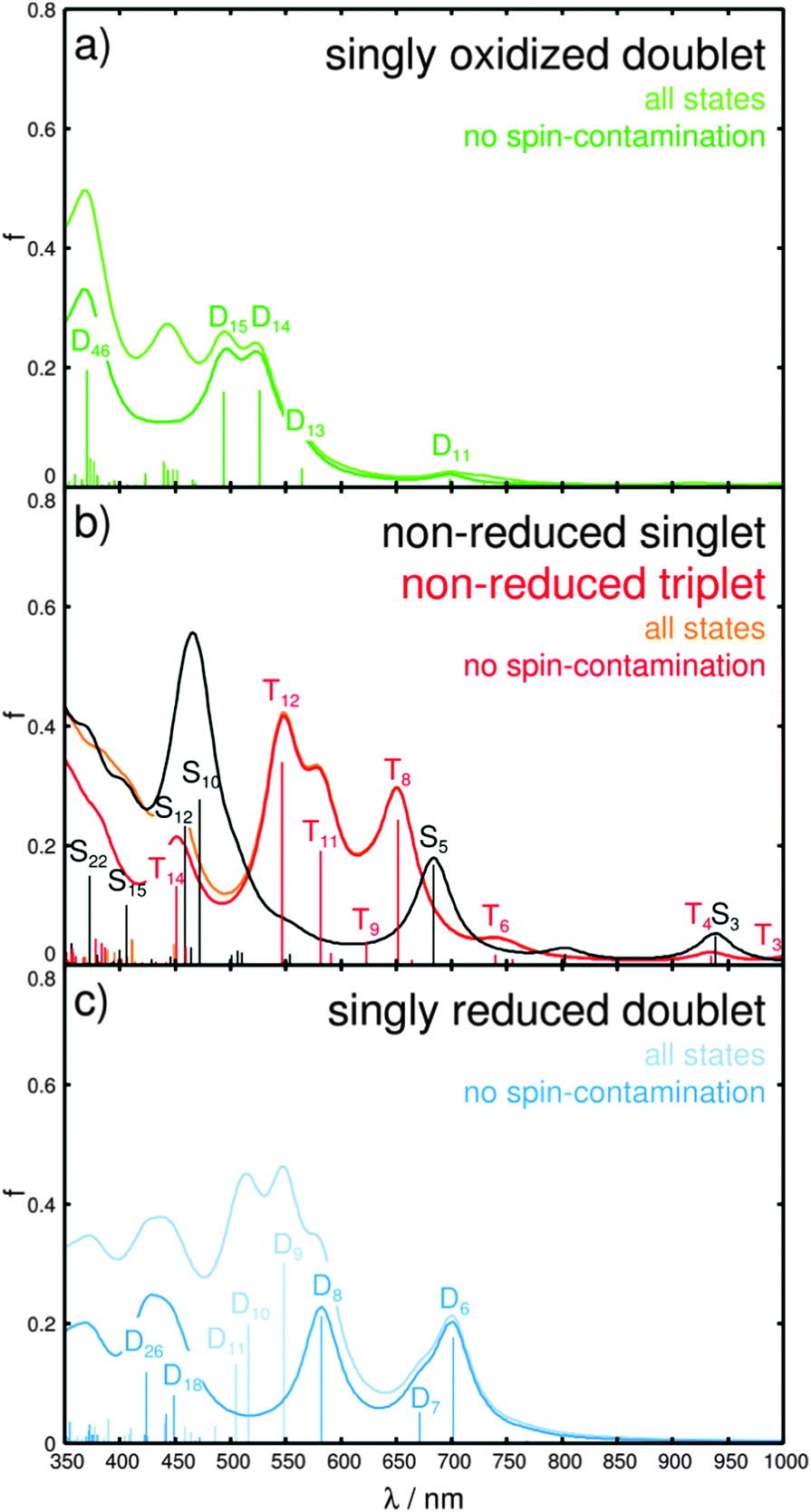 | ||
| Fig. 4 (a and c) Simulated UV-vis spectra of the singly oxidized (green) and singly reduced (blue) doublet species of Ru4, respectively; (b) absorption spectrum upon primary (singlet) and secondary (triplet-to-triplet) photoexcitation in black and red. The calculated oscillator strengths are represented by bars, and Lorentzian functions with a half-width of 20 nm are employed to broaden the transitions. For open-shell systems (doublet and triplet) all states as well as those states within a threshold for spin-contamination of 〈s2〉 ≤ s(s + 1) + 0.75 are given (see respective color code). | ||
The respective CDDs for the triplet excitations are depicted in Fig. 5, while the MOs involved in the leading transitions are shown in Table S6 (ESI†). The visible range of the absorption spectrum upon secondary excitation is dominated by mainly four states, T8, T11, T12 and T14, at 1.90 (651 nm), 2.13 (581 nm), 2.27 (546 nm) and 2.75 eV (450 nm). The bright excitations into T8 and T12 are 4H-imidazole-centered ILCT states, while T11 features mixed MLCT/ILCT character towards 4H-imidazole. However, the bright T14 is mainly related to a LMCT state from 4H-imidazole to the electron hole at ruthenium (MOs πim(194β) and dxz(200β) in Table S6, ESI†) leading to charge recombination. Further medium bright LMCT states are calculated in the NIR region, i.e. T3 and T4 at 1.23 (1005 nm) and 1.33 eV (934 nm). These LMCT states are not correlated to charge recombination, since the CT occurs from the π-system of the biimidazole ligands towards the dxz orbital (MOs πbim(198β), πbim(199β) and dxz(200β) in Table S6, ESI†). Thus, the (singly) charge-separated triplet species of Ru4 could transfer a second photoelectron via a secondary absorption process into the low-lying anti-bonding orbitals of the 4H-imidazole ligand, which leads in consequence to a doubly charge-separated state. However, the possibility of such a secondary absorption process needs to be further investigated by means of time-resolved spectroscopy. The population of the doubly charge-separated states is found to be dependent on the excitation wavelength. While photoexcitation from the NIR region to approximately 500 nm leads mainly to a doubly charge-separated species with two excess charges localized on the 4H-imidazole ligand (T8, T11 and T12), excitation in the blue region (T14 at 450 nm) is partially associated with an undesired charge recombination process.
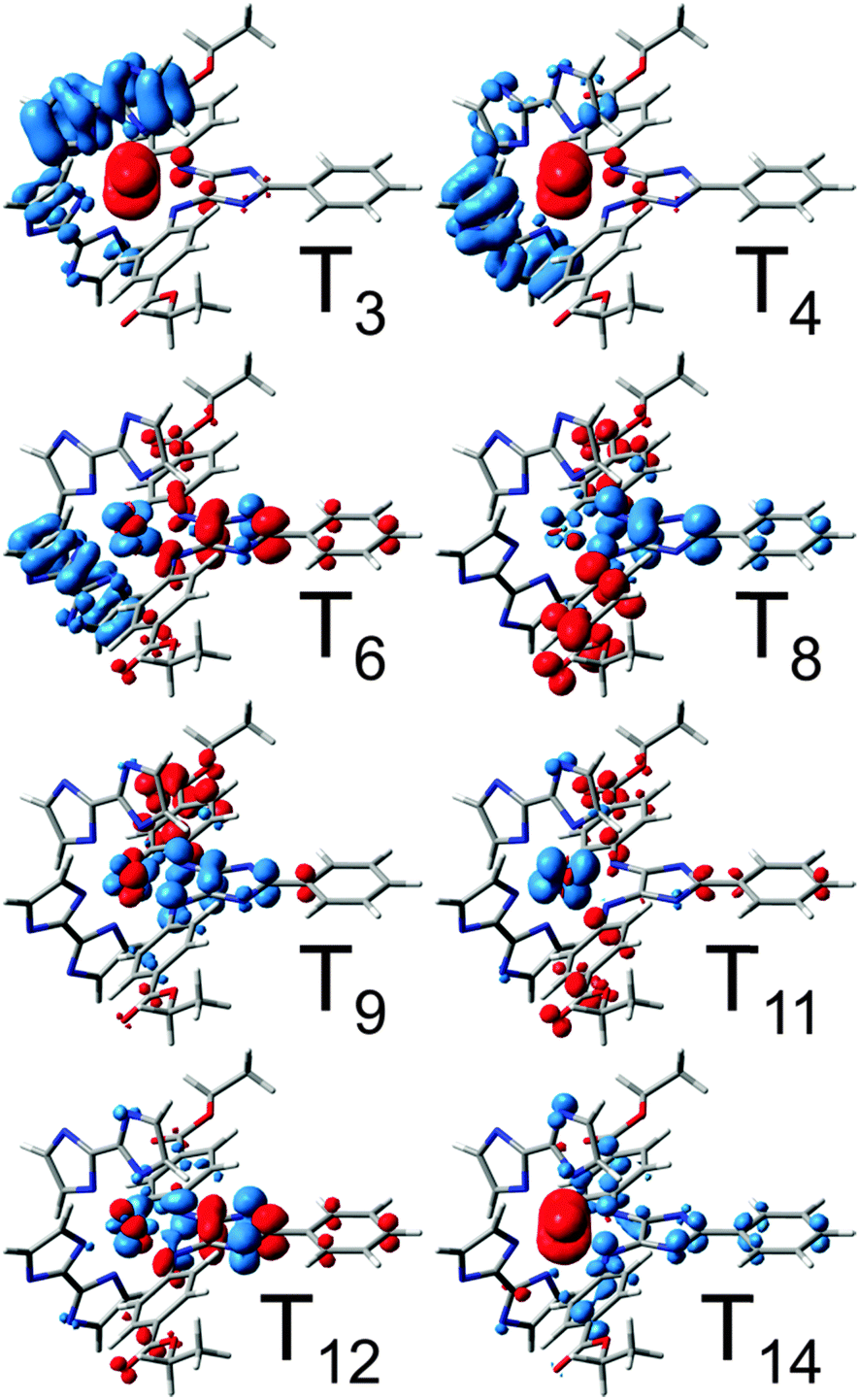 | ||
| Fig. 5 CDDs for bright triplet excitations contributing to the secondary UV-vis absorption spectrum of Ru4. CT takes place from blue to red. | ||
Thus, Ru4 renders a highly promising candidate for two-electron storage in the scope of supramolecular photocatalysis. SEC methods will be applied in order to further investigate the multi-photoelectron donor and storage capacity of Ru4.
3.2 Spectro-electrochemical evaluation
Experimentally, time-resolved spectroscopy is the method of choice to derive structure–dynamic relationships of photoinduced charge-separation cascades ranging from the femto- to nanosecond time scales. However, photophysical and photochemical processes in supramolecular photocatalysis involve long-lived photoexcited intermediates (microsecond time scale), e.g. the (singly) charge-separated species of Ru4. Thus, SEC methods are the ideal tool to study photoinduced intra-molecular redox couples by means of photooxidation and photoreduction. In SEC, the photooxidized/photoreduced species are modeled by the structural closely related electrochemically oxidized/reduced species. The photophysical and photochemical properties upon (electrochemical) oxidation and reduction reveal the multi-photoelectron donor and storage capacities of the system.Firstly, the multi-photoelectron donor capacity of Ru4 will be evaluated based on oxidative SEC. Oxidation of Ru4 yields the singly oxidized doublet species; while no prominent structural alterations are predicted upon single oxidation, merely a slight contraction of the RuN bonds as well as an increase of the torsion of the substituted terminal phenyl moieties with respect to the central imidazole moiety are observed (for detailed information see Table S5, ESI†). These slight structural changes are correlated to oxidation at the ruthenium center, 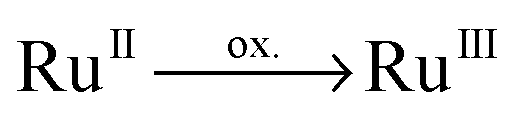 , whereas the electron hole is localized via the dxz orbital (MO dxz(200β) in Table S7, ESI†).
, whereas the electron hole is localized via the dxz orbital (MO dxz(200β) in Table S7, ESI†).
In order to investigate the photophysics and the photochemistry of the oxidized form, i.e. charge recombination vs. multi-photoelectron donor capacity, TDDFT was applied to simulate the absorption spectrum of the singly oxidized Ru4. The calculated spectrum, depicted in Fig. 4a, is dominated by a double band structure around 500 nm, given by the bright doublet excited states D14 and D15 at 2.35 (527 nm) and 2.51 eV (494 nm), and by an UV band at approximately 370 nm, which is mainly correlated to the bright D46 at 3.35 eV (370 nm). The states D14 and D15 are of mixed character, while D14 features a LMCT transition leading to charge recombination (π*im → dxz) as well as MLCT character to the 4H-imidazole ligand. The leading transitions of D15 are mainly of ILCT and MLCT character to the 4H-imidazole, while also LLCT contribution from the bisbiimidazole sphere to 4H-imidazole is obtained. Further LLCT character (πbim → π*im) is observed for the bright D46 in the UV. Additionally, a third band in the visible region is predicted at approximately 450 nm, however, the corresponding excited states are considerably spin-contaminated. Furthermore, a weak absorbing band at ∼700 nm and a red-sided shoulder at approximately 560 nm are calculated. The electronic character of the underlying transitions for the D11 at 1.78 eV (698 nm) are found to be of MLCT nature from ruthenium towards the 4H-imidazole, with slight contributions from the π-system of the biimidazole ligands. The red-sided shoulder at 2.19 eV (565 nm) is correlated to the D13 state and leads partially to charge recombination and to MLCT towards 4H-imidazole.
To evaluate the multi-photoelectron storage capacity of Ru4, we investigate the excited state properties upon electrochemical reduction. Pronounced structural alterations upon single reduction have been determined between the equilibrium geometries of the non-reduced singlet and the singly reduced doublet. In particular, planarization of the terminal substituted phenyl moieties with respect to the central imidazole fragment from 43.5° and −30.0° in the non-reduced species to 0.5° and 18.2° in the singly reduced species takes place accompanied by partial pyramidalization (dihedral angles δ1 and δ2 and pyramidalizational angles α1 and α2 in Table S5, ESI†). Furthermore, a slight increase in the ruthenium–nitrogen bond lengths is observed. These structural changes are rationalized by means of the introduced excess charge localized in the lowest anti-bonding π*-orbital of 4H-imidazole (π*im, LUMO of the non-reduced singlet). This additional charge is stabilized by planarization of the entire 4H-imidazole ligand frame; a similar behavior was observed by means of transient absorption experiments, RR-SEC and quantum chemical calculations for structurally related polypyridyl–ruthenium–4H-imidazole complexes, however, in SEC studies such planarization was not determined until double reduction.42,46–48 The absorption spectrum of the singly reduced doublet, shown in Fig. 4c, features broad absorption bands covering the entire visible region and originates from excitations into the states D6, D7, D8, D9, D10, D11, D18 and D26. The low-lying bright and medium bright states D6, D7 and D8 at 1.77 (701 nm), 1.85 (671 nm) and 2.13 eV (582 nm) are mainly 4H-imidazole-centered ππ*-excitations, while TDDFT also predicts partial MLCT character to the 4H-imidazole. The three bright MLCT states D9, D10 and D11, between 2.26 and 2.46 eV (548–505 nm), populate low-lying π*im-orbitals. However, the physical relevance of these states is questionable due to the high amount of spin-contamination. Further MLCT excitation towards 4H-imidazole is observed by means of the medium bright excitation into the D18 state at 2.76 eV (449 nm). At 2.93 eV (426 nm) a further 4H-imidazole ILCT state (D26) is computed that features also LLCT contributions (πbim → π*im). Detailed information with respect to the excited states properties as well as the MOs involved in the leading transitions can be found in the ESI† (Tables S7 and S8).
Hence, all excitations of the singly reduced doublet species are exclusively directed towards the 4H-imidazole ligand, indicating the ligand’s capability to collect two electrons, the first one introduced electrochemically and the second one via photoexcitation. In order to further confirm this finding, calculations on the doubly reduced species have been performed within singlet and triplet multiplicity. The fully optimized equilibrium geometries of both doubly reduced spin states are similar to the structure of the singly reduced doublet. However, for the doubly reduced singlet species further planarization of the substituted terminal phenyl moieties occurs, while for the triplet species a loss of planarity is observed for one substituent (dihedral angles δ1 and δ2 in Table S5, ESI†). Both doubly reduced species of Ru4 (singlet and triplet) accumulate two additional charges in the low-lying anti-bonding orbitals of the 4H-imidazole ligand, which clearly demonstrates the ligand’s ability to collect multiple charges. In fact, the absorption spectra, shown in Fig. S1 (ESI†), of the doubly reduced forms show exclusively MLCT and ILCT excitations towards 4H-imidazole (Tables S7 and S8, ESI†), indicating even the possibility of accumulating three charges on this ligand.
These findings are generally in accordance with the results obtained for related terpyridine–ruthenium–4H-imidazole complexes by Zedler et al. in ref. 47 and 48. However, low-lying terpyridine π*terpy-orbitals in terpyridine–ruthenium–4H-imidazole complexes (e.g.RuTerpy) allow merely two-electron storage on 4H-imidazole for the doubly reduced singlet species. While in triplet multiplicity, one of the two excess charges resides on 4H-imidazole and the other one on the terpyridine ligand. Thus, only the 4H-imidazole ligands bearing EWGs, e.g. ester groups, are able to stabilize the doubly reduced singlet state, while EDGs, e.g. methyl and dimethylamino groups, favor the population of the unsuitable doubly reduced triplet state. In order to verify if multi-charge accumulation in bisbiimidazole–ruthenium–4H-imidazole complexes is also restricted to substitution with EWGs, we rationalized the primary (singlet) and secondary photoexcitation (triplet) as well as the electronic ground states of the doubly reduced singlet and triplet species of Ru4Me, where the terminal ester groups are replaced by methyl groups (molecular structure in Fig. S2a and structural data in Table S9, ESI†). The calculated primary and secondary absorption spectra of Ru4Me (Fig. S2b, detailed excited state properties and MOs shown in Tables S10 and S11, ESI†) reveal no contributions of excited states populating π*bim-orbitals. Furthermore, SEC studies on the doubly reduced singlet and triplet species show that in both multiplicities the two excess charges are localized on the 4H-imidazole ligand (the singlet and triplet frontier orbitals π*im(171), π*im(171α) and π*im(172α) for the doubly reduced Ru4Me are illustrated in Table S12, ESI†). Thus, in contrast to terpyridine–ruthenium–4H-imidazole complexes the spectroscopic properties in bisbiimidazole–ruthenium–4H-imidazole complexes can be easily tuned by functionalization with EWGs as well as with EDGs without affecting the multi-electron storage capacity of the 4H-imidazole ligand.
The SEC approach assumes that long-lived photoexcited intermediates can be modeled by structurally, closely related electrochemically-generated species and thus neglects the structural and electronic impact of photooxidation (of the ruthenium core, 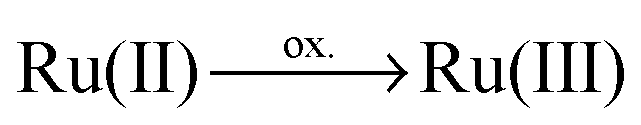 ) and photoreduction (of the ligand) in reductive and oxidative SEC measurements, respectively. In order to evaluate such an approximation we compare the absorption spectra of the singly oxidized and the singly reduced doublet (Fig. 4a and c) with the spectrum of the non-reduced triplet species (secondary photoexcitation, Fig. 4b). Therefore, specific electronic excited states, namely one ILCT state of πim → π*im nature present in all three species, three π*im → π*im ILCT states (not present for the singly oxidized species) and three LMCT charge recombination states of the non-reduced triplet and the singly oxidized species (not present in the singly reduced complex), have been selected. For states of equivalent character similar excitation energies and oscillator strengths have been obtained for all three species, while the excitation energies of the oxidized Ru4 are found to be mostly 0.1–0.2 eV higher than those for the non-reduced triplet. Likewise, slightly smaller energies (approximately 0.0–0.14 eV) have been calculated for the singly reduced complex with respect to the triplet species. The similar excitation energies and oscillator strengths for the corresponding states of the three species demonstrate that photophysical properties of photoexcited intermediates can be modeled using the SEC approach. Detailed information with respect to the electronic states, energies and oscillator strengths are collected in Table S13 of the ESI.†
) and photoreduction (of the ligand) in reductive and oxidative SEC measurements, respectively. In order to evaluate such an approximation we compare the absorption spectra of the singly oxidized and the singly reduced doublet (Fig. 4a and c) with the spectrum of the non-reduced triplet species (secondary photoexcitation, Fig. 4b). Therefore, specific electronic excited states, namely one ILCT state of πim → π*im nature present in all three species, three π*im → π*im ILCT states (not present for the singly oxidized species) and three LMCT charge recombination states of the non-reduced triplet and the singly oxidized species (not present in the singly reduced complex), have been selected. For states of equivalent character similar excitation energies and oscillator strengths have been obtained for all three species, while the excitation energies of the oxidized Ru4 are found to be mostly 0.1–0.2 eV higher than those for the non-reduced triplet. Likewise, slightly smaller energies (approximately 0.0–0.14 eV) have been calculated for the singly reduced complex with respect to the triplet species. The similar excitation energies and oscillator strengths for the corresponding states of the three species demonstrate that photophysical properties of photoexcited intermediates can be modeled using the SEC approach. Detailed information with respect to the electronic states, energies and oscillator strengths are collected in Table S13 of the ESI.†
3.3 Protonation effects
Excited state properties and relaxation pathways in ruthenium–biimidazole complexes are known to be highly pH-dependent.15,56,58,60,61 Thus, the impact of single protonation on the UV-vis absorption of Ru4, i.e. the unidirectional CT towards 4H-imidazole, as well as on the multi-(photo)electron storage capacity of 4H-imidazole needs to be verified. Three possible singly protonated species have been selected based on a natural bond orbital (NBO) analysis (Fig. S3, ESI†), yielding partial charges of −0.592 and −0.594 for the non-bonding nitrogen atoms of the biimidazole ligands, −0.634 and −0.640 for the non-bonding nitrogen atoms of the 4H-imidazole moiety and −0.651 and 0.655 for the carbonyl groups of the ester substituents. Comparison of the optimized equilibrium structures of the three singly protonated singlet species (biimidazole, 4H-imidazole and carbonyl) with Ru4 (deprotonated) showed no pronounced structural alterations, merely protonation at the carbonyl group leads to planarization of the associated phenyl–4H-imidazole fragment. Detailed structural data on the singly protonated species is collected in Table S14 (ESI†).Despite the partial charge of merely −0.594 (lowest charge of the three possible positions) at the non-bonding biimidazole nitrogen atoms, protonation at the biimidazole yields the most stable singly protonated structure, whereas protonation at the 4H-imidazole (∼−0.640) and the carbonyl group (highest charge ∼−0.655) is 0.49 and 2.01 eV less stable. The optimized triplet equilibrium geometries are very similar to the respective singlet structures, while for protonation at biimidazole and 4H-imidazole a slight increase of planarity is observed (δ1 and δ2 in Table S14, ESI†), and an opposite behavior is obtained for the carbonyl protonated species. However, the computed singlet–triplet gaps, obtained between the respective optimized equilibrium structures, highly depend on the protonation site. While for the deprotonated and at the biimidazole protonated Ru4, gaps of 0.39 eV have been calculated; gaps of merely 0.11 and 0.05 eV are predicted for single protonation at the 4H-imidazole and the carbonyl moieties.
The pH-influence and the protonation site on the excited states properties are investigated by means of UV-vis absorption. Single protonation at the biimidazole ligand does not affect the primary absorption spectrum considerably, with respect to the deprotonated Ru4 (Fig. S4a and b, ESI†). The medium bright and bright MLCT states S3, S4, S13 and S14 are predicted at 1.42 (870 nm), 1.91 (650 nm), 2.92 (425 nm) and 3.11 eV (398 nm). Additionally, two bright ILCT states, S10 and S22, are calculated with excitation energies of 2.72 (456 nm) and 3.45 eV (359 nm), respectively. Generally, all excited states feature a hypsochromic shift of approximately 0.06 to 0.22 eV upon protonation. The electronic transitions involved in these MLCT and ILCT states are exclusively directed towards low-lying anti-bonding orbitals of 4H-imidazole. The triplet excitations underlying the simulated UV-vis spectrum upon secondary photoexcitation (Fig. S4b, ESI†) feature similar hypsochromic shifts upon protonation at the biimidazole. Weakly absorbing LMCT states (T3, T4, T5 and T17) are calculated at 1.14 (1088 nm), 1.64 (755 nm), 1.80 (687 nm) and 2.85 eV (434 nm), whereas merely T4 is associated to charge recombination. A further medium bright charge recombination state (LMCT, T12) is predicted at 2.57 eV (483 nm). Besides, bright 4H-imidazole-centered ILCT states (T7 and T15) with excitation energies of 1.92 (645 nm) and 2.75 eV (451 nm), and one bright MLCT state (T10, 2.27 eV, 545 nm) dominate the visible region of the secondary absorption spectrum.
Protonation at the energetically less favored protonation sites, namely at the 4H-imidazole (+0.49 eV) and the carbonyl group (+2.01 eV), leads to (partially) considerable alterations of the absorption spectrum upon primary and secondary photoexcitation with respect to the deprotonated Ru4 (Fig. S4c and d, ESI†). The performed TDDFT calculations on the primary absorption of the investigated singly protonated forms of Ru4 revealed a preservation of the unidirectional CT towards the 4H-imidazole ligand, independent of the protonation site. However, the excitation energies of the electronic states are found to be site-dependent; in general, a blue-shift of the absorption bands and the underlying electronic excitations increase with respect to the deprotonated Ru4 from protonation at the biimidazole, to the 4H-imidazole and to the carbonyl group. Tables S15–S17 (ESI†) collect excited state properties such as excitation energies, oscillator strengths and electronic character for all three singly protonated species upon primary and secondary photoexcitation, while the respective MOs involved in the main electronic transitions are summarized in Tables S18–S20 of the ESI.† The secondary absorption spectra also do not feature CT towards the biimidazole (π*bim), however, the contribution of charge recombination states (π(*)im → dxz) to the absorption profile is highly site specific. This phenomenon is exemplarily investigated by means of the dark LMCT charge recombination state T7 of the deprotonated Ru4 upon secondary photoexcitation. The MOs in β-spin involved in the leading transition, that is πim → dxz, the HOMO (πbim), and the lowest π*im-orbital are shown in Fig. 6. The analogous MOs of the singly protonated species (biimidazole, 4H-imidazole and carbonyl) are illustrated accordingly, while all orbital energies are given with respect to the HOMO (β-spin) of the deprotonated complex. As can be seen in Fig. 6, single protonation at biimidazole leads to a similar stabilization of all four MOs. However, protonation at 4H-imidazole substantially lowers the energy of the πim/π*im-orbitals, while πbim and dxz are less affected. Protonation at the carbonyl group, associated with planarization of the respective phenyl–4H-imidazole fragment, stabilizes π*im even more, while the orbital energy of dxz increases, thus the order of the π*im and the dxz orbital is inverted. Consequently, the excitation energies and oscillator strengths of the triplet charge recombination states (πim → dxz), that is T7, T4, T11 and T16 of the deprotonated and the three singly protonated forms (biimidazole, 4H-imidazole and carbonyl), feature distinct alterations (for details see Fig. S5, ESI†). While the πim → dxz charge recombination state (T7) in the deprotonated Ru4 is found at 1.87 eV (664 nm) with an oscillator strength of 0.009, and upon single protonation at the biimidazole (T4) at 1.64 eV (755 nm) with an oscillator strength of 0.014, the excitation energy and oscillator strengths are significantly enhanced for the other two protonation sites. The respective bright (f = 0.225) triplet state of the 4H-imidazole protonated species (T11) is obtained at 2.27 eV (546 nm), and the excitation energy and oscillator strength of the respective state (T16) for protonation at the carbonyl are further increased to 2.40 eV (517 nm) and f = 0.256. This way, the influence of undesired charge recombination states in the secondary absorption process increases tremendously upon protonation at the 4H-imidazole and the carbonyl group. Therefore, protonation at these sites is unsuitable to achieve multi-photoinduced charge separation. However, these structures are found to be 0.49 and 2.01 eV less stable than the appropriate biimidazole protonated species, but admittedly, kinetic effects have not been addressed in this study.
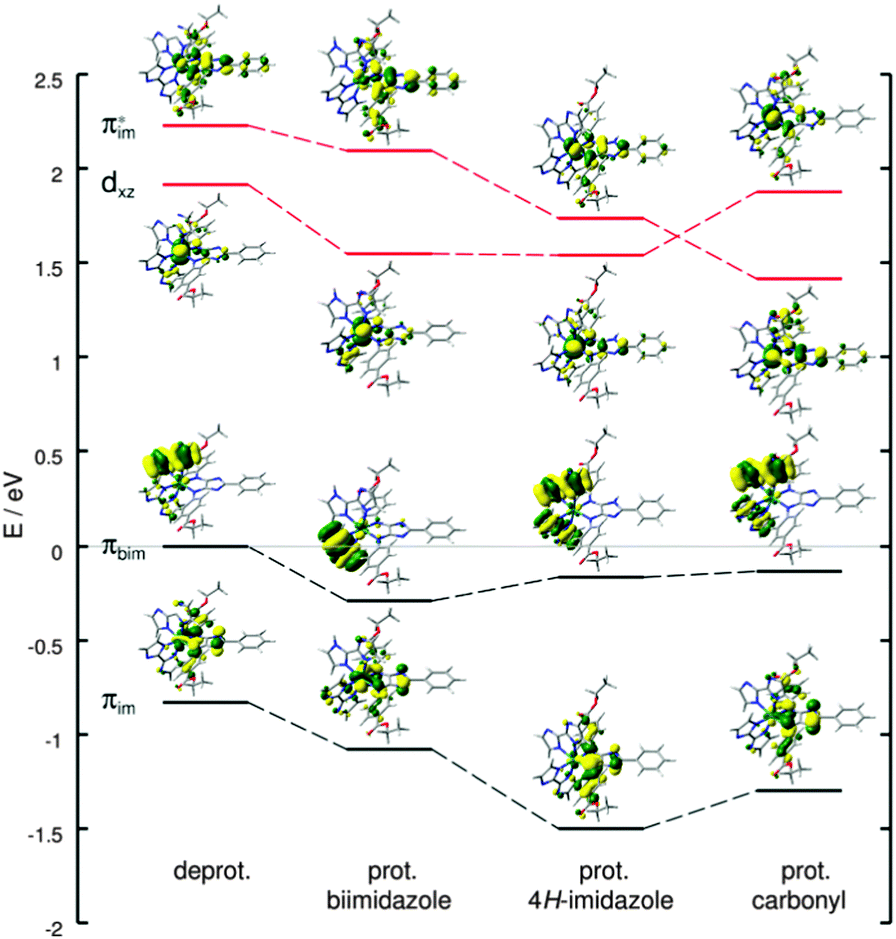 | ||
| Fig. 6 β-Spin orbitals πim and dxz involved in the leading transition of the charge recombination states T7 (deprot.), T4 (prot. biimidazole), T11 (prot. 4H-imidazole) and T16 (prot. carbonyl), and β-spin orbitals πbim and π*im involved in charge separation. All orbital energies are given accordingly to the HOMO(β) of the deprotonated triplet form of Ru4. | ||
4. Conclusions
A theoretical design concept to achieve unidirectional CT and multi-photoinduced charge separation in 4H-imidazole–ruthenium-based dyes is reported. The class of 4H-imidazole–ruthenium–polypyridyl complexes combines the ability to accumulate multiple (photoexcited) charges on 4H-imidazole with a pronounced light-harvesting capacity. Therefore, such dyes render highly promising candidates for applications in solar energy conversion, i.e., in the scope of DSSCs and supramolecular photocatalysis. However, 4H-imidazole–ruthenium–polypyridyl dyes feature two major drawbacks: firstly, multi-charge separation is limited to 4H-imidazoles bearing EWGs to stabilize the excess charges, which confines the tuning of optical properties. Secondly, photoexcitation of these complexes is always directed to both the 4H-imidazole and the polypyridine ligand spheres. To overcome these limitations, we introduce non-electron-accepting biimidazole-based ligands. Four complexes utilizing such biimidazole ligands replacing the polypyridyl sphere have been studied using TDDFT methods. One complex featuring a bisbiimidazole ligand sphere, Ru4, shows broad absorption bands from the NIR towards the UV region, covering the entire visible region based on the unidirectional MLCT and ILCT states towards the 4H-imidazole. Simulations on the respective triplet species, modeling a secondary photoexcitation, point to the dye’s potential to transfer even a second photoexcited electron towards the 4H-imidazole ligand, which is a prerequisite for supramolecular catalytic hydrogen evolution. In addition, merely minor contributions of charge recombination states, regaining the initial ruthenium(II) species, to the secondary absorption have been determined. SEC studies on the electrochemically singly and doubly reduced complex demonstrated the system’s ability to stabilize even three charges on the 4H-imidazole ligand. In contrast to the respective polypyridyl dyes, 4H-imidazole–ruthenium–bisbiimidazole complexes are capable of multi-photoinduced charge separation with EWGs as well as with EDGs on 4H-imidazole. Thus, the optical properties can be easily modified without affecting the electron storage capacity. The photophysical and photochemical properties of biimidazole-based ruthenium complexes are known to be highly pH-dependent. Therefore, the influence of site-specific protonation (at the biimidazole, the 4H-imidazole and the carbonyl group) was investigated upon primary and secondary photoexcitation. Depending on the protonation site, hypsochromic shifts between 0.06 and 0.33 eV have been predicted upon site-specific single protonation by the quantum chemical calculations, while the unidirectional nature of the CT is preserved. The dual charge separation properties are confirmed upon secondary photoexcitation, however, an increased influence of undesired charge recombination states is predicted for energetically unfavored protonated sites (4H-imidazole and carbonyl). This novel class of 4H-imidazole–ruthenium–bisbiimidazole black absorbers combines outstanding multi-photon light-harvesting properties with the ability of multi-photoinduced charge accumulation and pH stability. Synthesis and subsequent spectroscopic investigations will be performed on Ru4, i.e. time-resolved spectroscopy to further evaluate the dye’s capability to accumulate multiple photoinduced charges, and related 4H-imidazole–ruthenium–bisbiimidazole dyes, thus these systems will be subject of further joint experimental–theoretical studies.Acknowledgements
Support by the COST Action CM1202 Perspect-H2O is gratefully acknowledged. The author thanks Prof. Dr Sven Rau and Dr Julien Guthmuller for fruitful discussions. All calculations have been performed at the Universitätsrechenzentrum of the Friedrich Schiller University Jena and at the HP computers of the Theoretical Chemistry group in Jena.References
- A. H. Velders, H. Kooijman, A. L. Spek, J. G. Haasnoot, D. de Vos and J. Reedijk, Inorg. Chem., 2000, 39(14), 2966–2967 CrossRef CAS PubMed.
- M. J. Clarke, Coord. Chem. Rev., 2002, 232(1–2), 69–93 CrossRef CAS.
- K. Szaciłowski, W. Macyk, A. Drzewiecka-Matuszek, M. Brindell and G. Stochel, Chem. Rev., 2005, 105(6), 2647–2694 CrossRef PubMed.
- A. D. Ostrowski and P. C. Ford, Dalton Trans., 2009,(48), 10660–10669 RSC.
- D. Crespy, K. Landfester, U. S. Schubert and A. Schiller, Chem. Commun., 2010, 46(36), 6651–6662 RSC.
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48(77), 9649–9651 RSC.
- T. Becker, S. Kupfer, M. Wolfram, H. Görls, U. S. Schubert, E. V. Anslyn, B. Dietzek, S. Gräfe and A. Schiller, Chem. – Eur. J., 2015, 21(44), 15554–15563 CrossRef CAS PubMed.
- R. Siebert, A. Winter, B. Dietzek, U. S. Schubert and J. Popp, Macromol. Rapid Commun., 2010, 31(9–10), 883–888 CrossRef CAS PubMed.
- R. Siebert, A. Winter, U. S. Schubert, B. Dietzek and J. Popp, Phys. Chem. Chem. Phys., 2011, 13(4), 1606–1617 RSC.
- P. Coppo, M. Duati, V. N. Kozhevnikov, J. W. Hofstraat and L. De Cola, Angew. Chem., Int. Ed., 2005, 44(12), 1806–1810 CrossRef CAS PubMed.
- S. B. Meier, D. Tordera, A. Pertegás, C. Roldán-Carmona, E. Ortí and H. J. Bolink, Mater. Today, 2014, 17(5), 217–223 CrossRef CAS.
- M. E. Lippitsch, J. Pusterhofer, M. J. P. Leiner and O. S. Wolfbeis, Anal. Chim. Acta, 1988, 205, 1–6 CrossRef CAS.
- J. N. Demas and B. A. DeGraff, Anal. Chem., 1991, 63(17), 829A–837A CrossRef CAS.
- J. N. Demas and B. A. DeGraff, J. Chem. Educ., 1997, 74(6), 690–695 CrossRef CAS.
- S. Mardanya, S. Karmakar, M. Bar and S. Baitalik, Dalton Trans., 2015, 44(48), 21053–21072 RSC.
- S. Mardanya, S. Karmakar, D. Maity and S. Baitalik, Inorg. Chem., 2015, 54(2), 513–526 CrossRef CAS PubMed.
- F. Aiga and T. Tada, J. Mol. Struct., 2003, 658(1–2), 25–32 CrossRef CAS.
- Y. Pellegrin, L. L. Pleux, E. Blart, A. Renaud, B. Chavillon, N. Szuwarski, M. Boujtita, L. Cario, S. Jobic, D. Jacquemin and F. Odobel, J. Photochem. Photobiol., A, 2011, 219(2–3), 235–242 CrossRef CAS.
- B. O’Regan and M. Gratzel, Nature, 1991, 353(6346), 737–740 CrossRef.
- M. Grätzel, Inorg. Chem., 2005, 44(20), 6841–6851 CrossRef PubMed.
- M. Grätzel, Acc. Chem. Res., 2009, 42(11), 1788–1798 CrossRef PubMed.
- M. Grätzel, Pure Appl. Chem., 2001, 73(3), 459–467 CrossRef.
- S. Rau, B. Schäfer, D. Gleich, E. Anders, M. Rudolph, M. Friedrich, H. Görls, W. Henry and J. G. Vos, Angew. Chem., Int. Ed., 2006, 45(37), 6215–6218 CrossRef CAS PubMed.
- S. Tschierlei, M. Presselt, C. Kuhnt, A. Yartsev, T. Pascher, V. Sundström, M. Karnahl, M. Schwalbe, B. Schäfer, S. Rau, M. Schmitt, B. Dietzek and J. Popp, Chem. – Eur. J., 2009, 15(31), 7678–7688 CrossRef CAS PubMed.
- S. Tschierlei, M. Karnahl, M. Presselt, B. Dietzek, J. Guthmuller, L. González, M. Schmitt, S. Rau and J. Popp, Angew. Chem., Int. Ed., 2010, 49(23), 3981–3984 CrossRef CAS PubMed.
- P. Lei, M. Hedlund, R. Lomoth, H. Rensmo, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2008, 130(1), 26–27 CrossRef CAS PubMed.
- M. G. Pfeffer, B. Schaefer, G. Smolentsev, J. Uhlig, E. Nazarenko, J. Guthmuller, C. Kuhnt, M. Wächtler, B. Dietzek, V. Sundstroem and S. Rau, Angew. Chem., Int. Ed., 2015, 54(17), 5044–5048 CrossRef CAS PubMed.
- M. G. Pfeffer, T. Kowacs, M. Waechtler, J. Guthmuller, B. Dietzek, J. G. Vos and S. Rau, Angew. Chem., Int. Ed., 2015, 54(22), 6627–6631 CrossRef CAS PubMed.
- E. S. Andreiadis, M. Chavarot-Kerlidou, M. Fontecave and V. Artero, Photochem. Photobiol., 2011, 87(5), 946–964 CrossRef CAS PubMed.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114(24), 11863–12001 CrossRef PubMed.
- L. Sun, L. Hammarstrom, B. Akermark and S. Styring, Chem. Soc. Rev., 2001, 30(1), 36–49 RSC.
- F. Odobel and H. Zabri, Inorg. Chem., 2005, 44(16), 5600–5611 CrossRef CAS PubMed.
- M. K. Nazeeruddin, P. Péchy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123(8), 1613–1624 CrossRef CAS PubMed.
- H. B. Gray and A. W. Maverick, Science, 1981, 214(4526), 1201–1205 CAS.
- A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107(10), 4022–4047 CrossRef CAS PubMed.
- M. Elvington, J. Brown, S. M. Arachchige and K. J. Brewer, J. Am. Chem. Soc., 2007, 129(35), 10644–10645 CrossRef CAS PubMed.
- V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96(2), 759–834 CrossRef CAS PubMed.
- K. Zeitler, Angew. Chem., Int. Ed., 2009, 48(52), 9785–9789 CrossRef CAS PubMed.
- M. Wächtler, J. Kübel, K. Barthelmes, A. Winter, A. Schmiedel, T. Pascher, C. Lambert, U. S. Schubert and B. Dietzek, Phys. Chem. Chem. Phys., 2016, 18, 2350–2360 RSC.
- S. Kupfer, J. Guthmuller, M. Wachtler, S. Losse, S. Rau, B. Dietzek, J. Popp and L. Gonzalez, Phys. Chem. Chem. Phys., 2011, 13(34), 15580–15588 RSC.
- M. Wächtler, S. Kupfer, J. Guthmuller, J. Popp, L. González and B. Dietzek, J. Phys. Chem. C, 2011, 115(48), 24004–24012 Search PubMed.
- M. Wächtler, S. Kupfer, J. Guthmuller, S. Rau, L. González and B. Dietzek, J. Phys. Chem. C, 2012, 116(49), 25664–25676 Search PubMed.
- J. Schindler, S. Kupfer, M. Wächtler, J. Guthmuller, S. Rau and B. Dietzek, ChemPhysChem, 2015, 16(5), 1061–1070 CrossRef CAS PubMed.
- Y. Zhang, S. Kupfer, L. Zedler, J. Schindler, T. Bocklitz, J. Guthmuller, S. Rau and B. Dietzek, Phys. Chem. Chem. Phys., 2015, 17(44), 29637–29646 RSC.
- S. Kupfer, M. Wächtler, J. Guthmuller, J. Popp, B. Dietzek and L. González, J. Phys. Chem. C, 2012, 116(37), 19968–19977 CAS.
- M. Wächtler, M. Maiuri, D. Brida, J. Popp, S. Rau, G. Cerullo and B. Dietzek, ChemPhysChem, 2013, 14(13), 2973–2983 CrossRef PubMed.
- L. Zedler, S. Kupfer, I. R. de Moraes, M. Wächtler, R. Beckert, M. Schmitt, J. Popp, S. Rau and B. Dietzek, Chem. – Eur. J., 2014, 20(13), 3793–3799 CrossRef CAS PubMed.
- L. Zedler, S. Kupfer, S. Krieck, R. Beckert, S. Rau, M. Schmitt, J. Popp and B. Dietzek, Asian J. Phys., 2016, 25(2) Search PubMed.
- M.-A. Haga, Inorg. Chim. Acta, 1980, 45, L183–L184 CrossRef CAS.
- M.-A. Haga, Inorg. Chim. Acta, 1983, 75, 29–35 CrossRef CAS.
- A. M. Bond and M. Haga, Inorg. Chem., 1986, 25(25), 4507–4514 CrossRef CAS.
- M. Haga, T. Matsumura-Inoue and S. Yamabe, Inorg. Chem., 1987, 26(25), 4148–4154 CrossRef CAS.
- T. Ohno, K. Nozaki and M. Haga, Inorg. Chem., 1992, 31(4), 548–555 CrossRef CAS.
- D. P. Rillema, R. Sahai, P. Matthews, A. K. Edwards, R. J. Shaver and L. Morgan, Inorg. Chem., 1990, 29(2), 167–175 CrossRef CAS.
- B. Dietzek, W. Kiefer, J. Blumhoff, L. Böttcher, S. Rau, D. Walther, U. Uhlemann, M. Schmitt and J. Popp, Chem. – Eur. J., 2006, 12(19), 5105–5115 CrossRef CAS PubMed.
- M. Bräutigam, M. Wächtler, S. Rau, J. Popp and B. Dietzek, J. Phys. Chem. C, 2012, 116(1), 1274–1281 Search PubMed.
- C. Herrmann, J. Neugebauer, M. Presselt, U. Uhlemann, M. Schmitt, S. Rau, J. Popp and M. Reiher, J. Phys. Chem. B, 2007, 111(21), 6078–6087 CrossRef CAS PubMed.
- H.-J. Mo, Y.-L. Niu, M. Zhang, Z.-P. Qiao and B.-H. Ye, Dalton Trans., 2011, 40(32), 8218–8225 RSC.
- J. Meyer-Ilse, S. Bauroth, M. Brautigam, M. Schmitt, J. Popp, R. Beckert, N. Rockstroh, T. D. Pilz, K. Monczak, F. W. Heinemann, S. Rau and B. Dietzek, Dalton Trans., 2014, 43(47), 17659–17665 RSC.
- S. Rau, T. Büttner, C. Temme, M. Ruben, H. Görls, D. Walther, M. Duati, S. Fanni and J. G. Vos, Inorg. Chem., 2000, 39(7), 1621–1624 CrossRef CAS PubMed.
- S. Das, D. Saha, S. Karmakar and S. Baitalik, J. Phys. Chem. A, 2012, 116(21), 5216–5226 CrossRef CAS PubMed.
- J. Pressler, R. Beckert, S. Rau, R. Menzel, E. Birckner, W. Guenther and H. Goerls, Z. Naturforsch., B: Chem. Sci., 2012, 67(4), 367–372 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, G. Z. J. Bloino, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, A. J. A. O. Yazyev, C. P. R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77(2), 123–141 CrossRef CAS.
- B. Mennucci, C. Cappelli, C. A. Guido, R. Cammi and J. Tomasi, J. Phys. Chem. A, 2009, 113, 3009 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp00911e |
| This journal is © the Owner Societies 2016 |