
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi on the CeO2(110) and (100) surfaces: adsorption vs. substitution effects on the electronic and geometric structures and oxygen vacancies†
W. Q.
Li
ab,
S.
Goverapet Srinivasan
a,
D. R.
Salahub
*a and
T.
Heine
bc
aDepartment of Chemistry, Centre for Molecular Simulation and Institute for Quantum Science and Technology, University of Calgary, AB T2N 1N4, Canada. E-mail: dsalahub@ucalgary.ca
bDepartment of Physics and Earth Science, Jacobs University Bremen, Campus Ring 1, 28759 Bremen, Germany
cWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, Linnéstr. 2, 04103 Leipzig, Germany
First published on 30th March 2016
Abstract
We report density functional theory (DFT) calculations of the interactions of both Ni adsorbate and substitutional dopant with the ceria (110) and (100) surfaces to explain the origin of the activity of Ni/ceria catalysts. Our results indicate that the Ni adatom on the (110) surface prefers to adsorb on a two-fold bridge site over a hollow site up to 0.25 ML coverage, and the most stable position of a Ni adsorbate on the (100) surface was found to be the bridge site where the Ni atom is coordinated to two surface O atoms. The Ni+ oxidation state for the Ni adatom on the (110) surface was found to be more favorable than the Ni2+ state on the two-fold bridge site while on the (100) surface, a Ni adatom prefers its Ni2+ oxidation state over the Ni+ oxidation state. With increasing coverage, the binding energy of a Ni adatom on the (110) surface was found to decrease from −0.45 eV at 0.083 ML coverage to −0.32 eV at 0.25 ML coverage. Oxidation of the Ni adatom to Ni+ reduces one Ce4+ ion on the ceria surface to Ce3+ which preferred to be located next to the Ni+ ion in the nearest neighbor location. The Ce3+ ions on the (100) surface also prefer to stay in the vicinity of the adsorbed Ni atom, while they prefer to be located away from the Ni adatom on the (111) surface. No reduction of Ce4+ ions was observed upon substitution of Ce atoms by Ni atoms. Two Ni substituents preferred to be distributed on adjacent metal ion sites on the (110) surface. Ni adsorbate and substituent on the (110) surface were both found to induce significant structural distortions. In comparison to the pure ceria (110) and (100) surfaces, we show that a Ni adsorbate increases the energy required to create an oxygen vacancy while a Ni dopant reduces it. While multiple dopants on the (110) surface do reduce the vacancy formation energy, the degree of reduction is smaller compared to a single dopant indicating the presence of an optimum level of doping to obtain enhanced catalytic activity.
1. Introduction
Owing to their ability to cycle oxygen through redox processes and high oxygen ion mobility, ceria based compounds have been used in a variety of applications ranging from catalysts for the treatment of automobile exhausts and industrial pollutants1–3 to the water gas shift (WGS) reaction3–5 and as solid electrolytes in solid oxide fuel cells.6 The ability of ceria to cycle oxygen efficiently originates from the relative ease of the reversible removal of an oxygen ion from the surface. The formation of an oxygen vacancy results in the partial reduction of lattice cerium ions from the Ce4+ to the Ce3+ oxidation state. Numerous studies, both theoretical and experimental, have been carried out in the past to characterize the role of ceria in such applications and develop novel catalysts with higher activity. Often ceria has been used in combination with other transition metals (e.g. Pd, Pt, Au etc.) or transition metal oxides (e.g. VOx) to achieve enhanced catalytic performance. For instance, a higher activity was shown by ceria-supported VOx catalysts for the oxidative dehydrogenation of methanol,7,8 ceria-supported Au9 and Pt10,11 catalysts for CO oxidation, ceria-supported Cu, Au, Pt, Pd, Rh4,5,12–15 for the WGS reaction etc. More recently, it has been shown that the Ni/CeO2 system has an exceptional activity and selectivity for both WGS and ethanol steam reforming reactions.16–20 A detailed review of the theoretical approaches used to study ceria and heteroatom doped ceria surface energies, catalysis by ceria and their comparison to experiments was presented in a recent article by Sauer et al.21The geometric and electronic structure changes induced by a hetero metal atom on a metal oxide support have been described traditionally via ‘strong metal support interaction’.22–24 To explain the enhanced catalytic activity of transition metal/ceria catalysts, firstly it is necessary to gain a fundamental understanding of the interaction of the metal particle with the ceria support. Towards this end, a number of studies in the past have been carried out. Most of these studies have, however, focused on the adsorption of metal ions on the CeO2(111) surface, which is the most stable surface25 amongst the low-index ceria surfaces (i.e., (100), (110) and (111) surfaces). Metal-atom adsorbates that were studied include Cu,26–29 Ag,26,29 Au,26,29,30 Pt,31–33 Pd,33,34 Rh,33 and Ni.19,28 All these metal atoms adsorbed strongly on the (111) surface and were found to be oxidized. The oxidation of the metal adsorbates resulted in the partial reduction of one or more lattice Ce4+ ions to Ce3+, with the unpaired electrons localizing on the Ce 4f orbitals. An Au adsorbate was found to have a number of competing states with Au+ and Au0 oxidation states of the metal lying close in energy.35 While the (111) surface is the most stable of the low index ceria surfaces, ceria nanorods often expose the more reactive and less stable (110) and (100) surfaces.36,37 As such, an understanding of the interaction of metal adsorbates with (110) and (100) surfaces is essential to explain the origin of their activity. Very few studies in the past have investigated the interaction of transition-metal adsorbates with the (110) and (100) surfaces. Adsorption of Cu, Ag and Au on ceria (110) was reported by Cui et al.38 while Nolan39 reported the adsorption of a series of 3rd row transition metals (V, Cr, Fe), d10 metals (Cu, Ag and Au) and trivalent cations (Al, Ga, In, La). It was found that all the transition metals were oxidized (Cu, Ag and Au to their 1+ oxidation states, V and Cr to their 3+ oxidation states, Fe to its 2+ oxidation state) with the partial reduction of an equivalent number of lattice Ce4+ ions to Ce3+. It was found that amongst the studied transition metals, Cu had the largest adsorption energy. Also, the d10 metals adsorbed more strongly on the (110) surface compared to other transition metals. Amongst the trivalent cations, Al and La were oxidized to their 3+ oxidation states while Ga and In were only partially oxidized to their 2+ oxidation states. On the other hand, to the best of our knowledge, a detailed investigation of the adsorption of Ni on ceria (110) and (100) is yet to be undertaken. Experimentally it is known that Ni particles anchor more strongly on the (110) and (100) surfaces as compared to the (111) surface.40 In addition, recent experiments20 have shown that the oxidation state of Ni particles on ceria (111) could vary from Ni2+ to Ni0 depending on their surface coverage. Thus, any theoretical investigation of a transition-metal adsorbate on ceria surfaces must take into account the possibility of the presence of many competing close-lying oxidation states of the metal adsorbate. Finally, doping of ceria surfaces has also been considered as a way to enhance the oxidative capacity of the ceria surface.41 Thus, a number of studies in the past have attempted to understand the modifications to the surface properties induced by transition-metal substituents. Doping of ceria surfaces with Mn,42 Ti, Zr and Hf,43 La,44 Ni and Pd45 resulted in a strong distortion of the surface induced by the substituents along with a reduction in the energy required for the creation of an oxygen vacancy.
In this article, we have carried out a detailed analysis of the interaction of a Ni adsorbate and a substituent with the ceria (110) and (100) surfaces. Considering the points mentioned above, in this article we have attempted to answer the following questions:
(1) What are the preferred adsorption site and oxidation state for a Ni adsorbate?
(2) What is the adsorption energy of a Ni adsorbate and how does it compare with the adsorption energies of the other transition metal atoms reported in the literature?
(3) What is the nature of the distortions induced in the geometric and electronic structure of the surface by the Ni adsorbate?
(4) How do the above quantities vary as a function of the surface coverage of the Ni adsorbate?
(5) How do multiple Ni substituents prefer to distribute themselves and how do they compare with a mono-Ni substituent?
(6) How is the oxygen vacancy formation energy affected by Ni adsorption and substitution?
Section 2 describes the details of the computational methods used in this work while Section 3 discusses our results. Conclusions from this work are presented in Section 4.
2. Computational methods and details
DFT calculations have been performed to understand the interaction of Ni adsorbate/substituents with the ceria surface. All the calculations were performed using the VASP software.46–49 The valence electronic states were expanded in a basis of plane waves. The core–valence interaction was described through the Projector Augmented Wave (PAW) approach.50,51 A plane wave kinetic energy cutoff of 400 eV was used. The Perdew–Burke–Ernzerhof (PBE) GGA was used to describe the exchange–correlation interactions. The DFT+U52 approach was used to describe the localized 4f and 3d electronic states in Ce and Ni respectively. In line with earlier works,53,54 we choose a Hubbard-U value of 4.5 eV and 4.6 eV for the Ce 4f and Ni 3d states respectively. The Hubbard-U value results in a lattice constant of 5.44 Å for bulk CeO2 (fluorite structure, Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group), 3.83 Å and 6.01 Å (a and c lattice parameter values) for bulk Ce2O3 (Pm1 space group), 4.17 Å for bulk NiO (Fmm space group) in close agreement with the experimental values of 5.41 Å for CeO2, 3.89 Å and 6.06 Å for Ce2O3, and 4.17 Å for NiO. Our computed lattice parameters agree well with earlier reported values for CeO2 (5.48 Å),54 Ce2O3 (3.87 and 5.93 Å)55 and NiO (4.19 Å).56 All our calculations utilized ‘accurate’ precision settings in VASP. Complete cell optimizations and a re-relaxation for the bulk systems were performed to avoid any artifacts due to Pulay stress. Spin–orbit coupling (SOC) was not considered since our calculations indicated that the inclusion of SOC has only a minor effect on the electronic structure, for instance the band gap of CeO2 changes only by 70 meV (see Fig. S1 in ESI†). We have also ignored the effect of variations in energy due to different occupations of the m projections of the 4f orbitals on Ce and 3d orbitals on Ni.57 Table T1 in the ESI† gives a comparison of the results obtained with DFT/PBE, DFT/PBE+U and experiment. Clearly, our method of choice (DFT/PBE+U) is in much better agreement with experiments as compared to DFT/PBE.
m space group), 3.83 Å and 6.01 Å (a and c lattice parameter values) for bulk Ce2O3 (Pm1 space group), 4.17 Å for bulk NiO (Fmm space group) in close agreement with the experimental values of 5.41 Å for CeO2, 3.89 Å and 6.06 Å for Ce2O3, and 4.17 Å for NiO. Our computed lattice parameters agree well with earlier reported values for CeO2 (5.48 Å),54 Ce2O3 (3.87 and 5.93 Å)55 and NiO (4.19 Å).56 All our calculations utilized ‘accurate’ precision settings in VASP. Complete cell optimizations and a re-relaxation for the bulk systems were performed to avoid any artifacts due to Pulay stress. Spin–orbit coupling (SOC) was not considered since our calculations indicated that the inclusion of SOC has only a minor effect on the electronic structure, for instance the band gap of CeO2 changes only by 70 meV (see Fig. S1 in ESI†). We have also ignored the effect of variations in energy due to different occupations of the m projections of the 4f orbitals on Ce and 3d orbitals on Ni.57 Table T1 in the ESI† gives a comparison of the results obtained with DFT/PBE, DFT/PBE+U and experiment. Clearly, our method of choice (DFT/PBE+U) is in much better agreement with experiments as compared to DFT/PBE.
In order to study the effect of Ni adsorbate coverage on Ni/ceria interactions, 3D boundary conditions were used throughout and, hence, the surfaces were modelled using the repeated slab method in which a finite number of crystal layers are used to generate two identical surfaces via the introduction of a vacuum gap perpendicular to the surface. In the case of the (110) surface, two different CeO2(110) surface models, a (2 × 1) surface and a (3 × 2) surface, were considered. Both the surfaces were generated from a conventional CeO2 unit cell consisting of 12 atoms (4 Ce atoms and 8 O atoms in a fluorite structure with Fmm space group). The (2 × 1) surface model consisted of 60 atoms (20 Ce atoms and 40 O atoms) with simulation box sizes of 10.89 Å × 7.7 Å, while the (3 × 2) surface model consisted of 180 atoms (60 Ce atoms and 120 O atoms) with box sizes of 16.33 Å × 15.4 Å. Both the slabs were 5 atomic layers thick and a vacuum gap of 15 Å was used in the direction perpendicular to the surface. Thus, the surface layer of the (2 × 1) slab contained 4 Ce atoms while that of the (3 × 2) slab contained 12 Ce atoms. Adsorption of one Ni atom then corresponds to a coverage of 0.25 ML on the (2 × 1) surface and 0.083 ML on the (3 × 2) surface. Interactions of a Ni substituent with the CeO2(110) surface were studied for the larger (3 × 2) surface only. Mono-substitution and di-substitution of surface Ce atoms with Ni atoms were considered to investigate the effect of the Ni substituent concentration. Thus, mono-substitution corresponds to 8.3% Ni concentration, while di-substitution corresponds to 16.6% Ni concentration at the surface. The (100) surface was modelled as an oxygen-terminated surface with half of the oxygen atoms in a chessboard-like arrangement. It is stoichiometric and has been found to be the most stable polar-compensated (100) plane in the past.58 A (3 × 3) expansion of the (100) surface was used for our calculations, and includes 11 atomic layers and 135 atoms (45 CeO2 units) with simulation box sizes of 11.54 Å × 11.54 Å. Adsorption of one Ni atom then corresponds to a coverage of 0.111 ML. Mono-substitution corresponds to 11.1% Ni concentration at the surface. These models were further used to compute the formation energies of charge-compensating and active O vacancies.
For the (100) surface, the atomic coordinates of all the atoms were allowed to relax fully, but for the (110) surface, atoms in the bottom two layers were frozen at their bulk positions due to the large size of the system (180 atoms). For both the slabs, the lattice parameters were fixed at the above-mentioned values during optimization. Geometry optimizations were deemed to have converged when the forces dropped below 0.03 eV Å−1. For the electronic structure, the SCF energy convergence was set to a threshold of 10−5 eV. A Pulay mixing scheme59 as implemented in VASP was used for charge-density mixing in the SCF solution. For the (3 × 3) slab of the (100) surface, during geometry optimization, the Brillouin zone was sampled using a 2 × 2 × 1 Monkhorst–Pack (MP) grid. For the (2 × 1) slab of the (110) surface, the Brillouin zone was sampled using a (1 × 2 × 1) MP k-point mesh. Later, a finer (4 × 6 × 1) MP k-point mesh was used to compute the energies at the optimized structure. For the (3 × 2) slab of the (110) surface, the Brillouin zone was sampled using the Γ-point approximation.
CeO2(s), Ni(s), Ce(s) and O2(g) were used as the reference to compute the formation energies in eqn (1)–(8). Calculations on the face-centered cubic (fcc) Ni(s) and γ-Ce(s) were performed on a one-atom unit cell using a 16 × 16 × 16 Monkhorst–Pack k-point mesh, resulting in an optimized lattice parameter of 3.474 Å for Ni(s) and 5.208 Å for Ce(s), in good agreement with the experimental value of 3.52 Å and 5.16 Å respectively. The local magnetic moment on the Ni atom in Ni(s) was found to be ∼0.78 μB while it was ∼1 μB on the Ce atom in Ce(s). Calculations on the O2(g) molecule were performed in a 10 Å × 10 Å × 10 Å box in the triplet state using the Γ-point approximation to sample the Brillouin zone. The optimized O2 bond length was found to be 1.234 Å.
3. Results and discussion
In bulk CeO2 (fluorite structure, Fmm space group), Ce4+ and O2− are characterized by eight- and four-fold coordination, respectively. The relaxed O–Ce bond length of 2.355 Å is close to the experimental value of 2.343 Å. The CeO2(110) surface consists of six-coordinated Ce4+ ions and three-coordinated O2− ions. On the (100) surface, Ce4+ and O2− are six- and two-coordinated, respectively. The relaxed surface was found to have a Ce–O bond length of 2.326 Å on the (110) surface and 2.191 Å on the (100) surface. We have also investigated the CeO2(111) surface, in which Ce4+ and O2− are seven- and three-coordinated, respectively. The O–Ce bond length 2.360 Å is close to the bulk O–Ce bond length 2.355 Å. The surfaces were diamagnetic with all the Ce ions in their Ce4+ oxidation state and O ions in their O2− oxidation state. The surface energies of CeO2(110), (100) and (111) were found to be 1.06, 1.44 and 0.60 J m−2, respectively. They are in very good agreement with earlier reported values (1.06 J m−2 for (110), 1.44 J m−2 for (100) and 0.68 J m−2 for the (111) surface) in the literature.25,60–62 It is known that the order of stability of CeO2 low index surfaces is CeO2(111) > CeO2(110) > CeO2(100).25
3.1 Ni adsorption on CeO2 surfaces
 | ||
| Fig. 1 Initial structures of Ni adsorption on the ceria (110) surface. (a) Bridge site (b) Hollow site. Insets show the front view of the structures. Ce atoms are yellow, O atoms are red and blue, Ni atom is grey. In order to distinguish the surface O atoms from other O atoms in the slab, they are shown in blue colour. | ||
Thus, in addition to the adsorption site, any detailed analysis of the interaction of a transition-metal adsorbate on CeO2 surfaces must consider the oxidation state of the adsorbed transition metal atom. As such, we have studied configurations where the Ni atom is in its Ni+ and Ni2+ oxidation states on both the bridge and hollow sites. In its Ni+ oxidation state, the Ni atom donates an electron from its 4s orbital to the 4f orbital of a surface Ce, thereby reducing it from Ce4+ to Ce3+. Similarly, when Ni is in its Ni2+ oxidation state, two surface Ce are reduced from Ce4+ to Ce3+. It is now well known that the reduced Ce3+ could be located in one or more of the many surface metal ion sites and various configurations must be investigated to determine the most favorable distribution of Ce3+ on the surface. In order to do so, we have followed the procedure outlined by Kullgren et al.64 First we perform a geometry optimization using a different PAW potential (where the Ce 4f electron is included in the core) on those Ce atoms that we wish to be reduced to Ce3+, while the rest of the Ce atoms use the full valence pseudopotential. In the second step we continue the optimization from the converged structure in the previous step with all the Ce atoms described using the full valence pseudopotential. At the end of each calculation, the magnetizations on the Ce ions were checked to ensure that the reduced Ce3+ ion was located at the same position as chosen initially. This was done by looking at the local magnetization on the Ce3+ ion which should be ca. 1 μB due to an electron localized in its f orbital while the remaining Ce4+ ions have a magnetic moment of 0 μB. The reaction energy for the adsorption of a Ni atom on CeO2(110) was computed using eqn (1):
| Ni(s) + CeO2(surf) → Ni–CeO2(surf) | (1) |
Fig. 2 and 3 show the optimized structures and reaction energies for the adsorption of Ni on hollow and bridge sites respectively. From Fig. 2 we see that on a hollow site, the Ni ion prefers a Ni2+ oxidation state over a Ni+ oxidation state, with the two reduced Ce3+ ions located at sites (1,2). The reaction energy for the adsorption of a Ni2+ ion on a hollow site at 0.25 ML coverage was found to be −0.248 eV while Ni+ adsorption was disfavored. The local structure around the Ni2+ ion was nearly planar with the four Ni–O bond lengths equal to 1.936 Å. Comparing this with the Ni–O bond length of 2.085 Å in solid NiO, we see that the Ni–O bond lengths are shortened upon adsorption of a Ni2+ ion on a hollow site. The four O2− ions that coordinate with the adsorbed Ni2+ are pulled away from their normal positions on the surface, with the Ce3+–O2− distance increased to 2.835 Å from 2.326 Å. On a bridge site, however (see Fig. 3), we see that both Ni+ and Ni2+ oxidation states have stable configurations, with Ni+ being the preferred oxidation state. As mentioned earlier, for a given oxidation state of the adsorbate, various distributions of the reduced Ce3+ ions are possible. Here we have investigated three different Ce3+ distributions for Ni2+ adsorbate and two different distributions for Ni+ adsorbate, as shown in Fig. 3. For the Ni2+ adsorbate, two Ce3+ ions located diagonally across the Ni2+ ion at the (1,3) site as shown in Fig. 3(a) on the surface was found to be the most favorable distribution, with a reaction energy of −0.301 eV. Upon adsorption, the two O atoms bridged by the Ni2+ ion are pulled outward from their location on the surface. This results in an elongation of the two O2−–Ce4+ bonds to 2.464 Å from 2.326 Å. The two O2−–Ce3+ distances were 2.584 Å while the Ni2+–O2− bond distance was 1.69 Å. In comparison to Ni2+ adsorption on the hollow site, we notice that the distortions in the Ce–O bond lengths are smaller, resulting in a larger binding energy of the Ni2+ adsorbate on the bridge site. The least favorable distribution of Ce3+ ions for Ni2+ adsorbate was found to be at nearest neighbor locations (site (1,4) as shown in Fig. 3c), which was ca. 263 meV less stable compared to Ce3+ located at the (1,3) site. For the Ni+ adsorbate on a bridge site, we have investigated two different locations of the Ce3+ ion, a nearest-neighbor surface site (site 2, Fig. 3e) and a subsurface site (site 5, Fig. 3d). We find that the subsurface site is unfavorable while the nearest neighbor location for the Ce3+ ion results in adsorption energy of −0.322 eV. Adsorption of Ni+ on a bridge site causes the O2− coordinated to the Ce3+ ion to be pulled away from the surface significantly (by 1.23 Å with respect to CeO2(110)), resulting in a locally asymmetric structure around the Ni+ adsorbate. Fig. S2 in the ESI† shows the bond lengths surrounding the Ni+ adsorbate. Thus, we find that a Ni adsorbate prefers the bridge site over the hollow site. Further, on the bridge site, the Ni adsorbate prefers the Ni+ oxidation state, with the reduced Ce3+ located on the nearest neighbour surface site, over the Ni2+ oxidation state. The stabilization energy of the Ni+ oxidation state over the Ni2+ oxidation state is ca. 21 meV. In addition, the Ni+ adsorbate on the bridge site is ca. 74 meV more stable than the Ni2+ adsorbate on a hollow site. Thus, the preferred adsorption pattern for a Ni adsorbate on the CeO2(110) surface is:
| Nibridge+ > Nibridge2+ > Nihollow2+ |
 | ||
| Fig. 2 Adsorption of Ni on a hollow site on the CeO2(110) surface. (a) Ni is in its Ni2+ oxidation state with the two Ce3+ ions located at sites 1 and 2. (b) Ni is in Ni+ oxidation state with the reduced Ce3+ ion located at site 1. Ce atoms are yellow, O atoms are red and blue, and Ni atom is grey. | ||
 | ||
| Fig. 3 Adsorption of Ni on a bridge site on the CeO2(110) surface. (a–c) Ni is in its Ni2+ oxidation state with the two Ce3+ ions located at sites (1,3), (1,2) and (1,4) respectively. (d–e) Ni is in its Ni+ oxidation state with the reduced Ce3+ ion located at site 5 and 2 respectively. Inset in (d) shows the front view of the structure. Here, site 5 is the Ce ion beneath the Ni adatom. Ce atoms are yellow, O atoms are red and blue, Ni atom is grey. | ||
 | ||
| Fig. 4 Adsorption of Ni on a bridge site on the CeO2(110) (3 × 2) surface. (a–d) Ni is in its Ni+ oxidation state with the Ce3+ ion located at sites 1, 6, 5 and 7 respectively. (e–h) Ni is in its Ni2+ oxidation state with the two reduced Ce3+ ions located at sites (1,3), (1,5), (3,4) and (1,4) respectively. The insets in panels (a) and (e) show the front view of the structure. Ce ion is yellow, O atoms are red and blue, Ni atom is gray. | ||
From Fig. 4, it is clear that on a larger (3 × 2) surface (corresponding to 0.083 ML Ni coverage) as well, a Ni adsorbate prefers the Ni+ oxidation state over the Ni2+ oxidation state. This is in contrast to Ni adsorption on the CeO2(111)19 and CeO2(100) surfaces, where the Ni adsorbate oxidizes to its Ni2+ state. The most favourable location of the reduced Ce3+ ion was found to be next to the adsorbed Ni+ (see Fig. 4a) while locations far away from the adsorbed Ni+ (see Fig. 4d) or the subsurface location were found to be unfavourable. For the Ni adsorbate in the Ni2+ oxidation state, similar to the structure found for the (2 × 1) slab above, two Ce3+ ions located diagonally across the Ni2+ adsorbate was found to the most stable configuration. However, amongst the configurations studied here, while the energy difference between the least stable and the most stable Ce3+ distributions for Ni+ adsorbate was ca. 795 meV, we find this value to be reduced to only 97 meV for the Ni2+ adsorbate. Furthermore, comparing the above results with those on the smaller (2 × 1) slab (corresponding to 0.25 ML Ni coverage) presented in the previous section, the following trends emerge:
(1) As the coverage of Ni on the CeO2(110) surface increases from 0.083 ML to 0.25 ML, the adsorption energy for the most favourable configuration, i.e., Ni+ on a bridge site, reduces from −0.449 eV to −0.322 eV.
(2) With increase in coverage of Ni from 0.083 ML to 0.25 ML, the stabilization of the Ni+ oxidation state over the Ni2+ oxidation state on a bridge site decreases from ca. 49 meV to 21 meV for the most preferred Ce3+ ion distribution in the respective oxidation states of the Ni adsorbate.
(3) Amongst the configurations investigated here, with increase in coverage from 0.083 ML to 0.25 ML, the energy difference between the least and most stable Ce3+ distribution for the Ni+ adsorbate decreases from ca. 795 meV to 383 meV, while this value increases from ca. 97 meV to 263 meV for the Ni2+ adsorbate.
In comparison to the adsorption energies of other transition-metal atoms on the CeO2(110) surface,39 namely Au (−2.19 eV), Ag (−1.45 eV), Cu (−3.25 eV), V (−0.97 eV), Cr (−0.64 eV) and Fe (−0.75 eV), we find that Ni has the lowest adsorption energy amongst the transition metals reported so far. On the other hand, comparing the adsorption energy of a Ni atom on the CeO2(100) (−1.490 eV) and CeO2(111) (−0.185 eV) surfaces (see Table T2 in ESI†), we find that the order of stability of a Ni adsorbate on ceria surfaces is CeO2(100) > CeO2(110) > CeO2(111). This is in agreement with the experimental finding that the CeO2(110) and CeO2(100) surfaces bind Ni stronger than the CeO2(111) surface.40
Fig. 5 shows the total and projected density of states for a Ni adsorbate on a bridge site at 0.083 ML coverage in both Ni+ (Fig. 5a and b) and Ni2+ (Fig. 5c and d) oxidation states. The Fermi energy is set to 0 eV in all panels of Fig. 5. The common feature in both cases is the appearance of split-off states above the valence band of CeO2(110) and right below the Fermi level. While for the Ni2+ adsorbate, this state arises almost exclusively from the two electrons localized on the 4f orbitals of the two reduced Ce3+ ions, for the Ni+ adsorbate this state arises from the combination of the 3d orbital of Ni+, 4f orbital on the reduced Ce3+ ion and the 2p orbitals on the two O2− ions coordinating the Ni+ adsorbate.
 | ||
| Fig. 5 Total and projected density of states for the adsorption of Ni on CeO2(110) bridge site at 0.083 ML coverage. (a and b) Total and projected density of states for Ni adsorbate in Ni+ oxidation state. (c and d) Total and projected density of states for Ni adsorbate in Ni2+ oxidation state. The Fermi level is set to 0 in all the plots. | ||
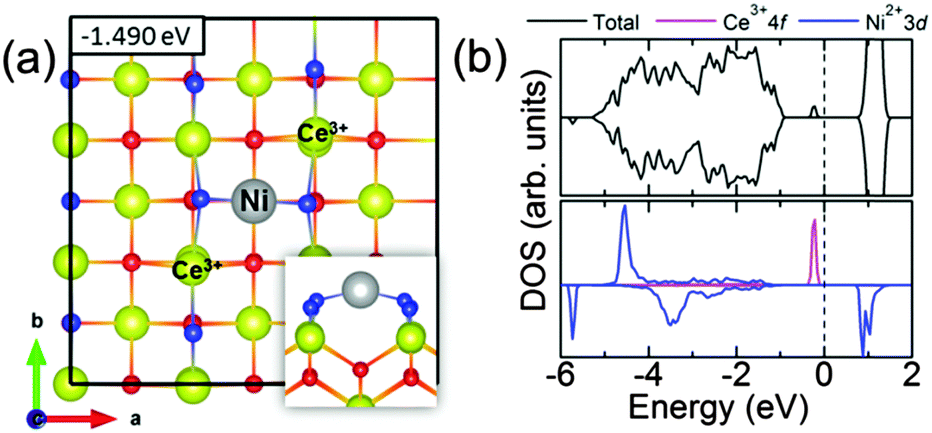 | ||
| Fig. 6 (a) The optimized atomic structure of Ni adsorbed on the CeO2(100) surface with the two Ce3+ ions located in the nearest neighbour locations. Inset shows the side view of the outermost four atomic layers. Ce atoms are yellow, O atoms are red and blue, Ni atoms is grey. (b) The total density of states (DOS) in the upper panel and projected DOS of Ce3+ 4f and Ni2+ 3d orbitals in the lower panel. The curves above (below) the horizontal axis correspond to spin-up (down) states. The Fermi level (the vertical dashed line) is set to zero. | ||
The Ce3+ ion prefers to be distributed around the Ni2+ adsorbate on the ceria (110) and (100) surfaces. This trend is in contrast to Ni2+ adsorbed on the ceria (111) surface,19 where the Ce3+ ions strongly prefer to maximize their separation from the adsorbed Ni2+ ion (see Fig. S14 and S15 in ESI†).
3.2 Ni substituent on CeO2 surfaces
| Ni(s) + CexO2x(surf) + O2(g) → NiCex−1O2x(surf) + CeO2(s) | (2) |
| 2Ni(s) + CexO2x(surf) + 2O2(g) → Ni2Cex−2O2x(surf) + 2CeO2(s) | (3) |
A number of different configurations for Ni substituents on the surface were examined to determine the most favourable location. Fig. 7 shows the most favourable configuration of a mono- and di-Ni substituted surface while Fig. S3 in the ESI† shows the geometries of various other metastable structures along with their energies relative to the most favourable configuration shown in Fig. 7.
 | ||
| Fig. 7 Ni substituent on CeO2(110) surface. (a) Optimum configuration for a mono substituent with four-coordinated Ni. (b) Optimum configuration for di Ni substituted surface. Ce atoms are yellow, O atoms are red and blue, Ni substituent is gray. ‘S’, ‘M’, ‘A’ and ‘B’ in (a) correspond to a surface Ce4+ ion, magnetic oxygen atoms, surface and sub-surface oxygen atoms respectively. 1, 2 in (b) correspond to the two Ni dopants. | ||
Doping the surface with a Ni atom results in a strong distortion of the surface with the Ni substituent moving from the surface metal ion site into the ceria lattice by about 0.69 Å, consistent with the observations of Nolan.45 The local geometry around the Ni substituent was found to be nearly planar with the Ni substituent coordinating with two surface- (designated by ‘A’ in Fig. 7a) and two subsurface O ions (designated by ‘B’ in Fig. 7a). The Ni–OA distance was found to be 1.806 Å while the Ni–OB distance was found to be 1.870 Å. For di-Ni substitution, the preferred distribution of the two Ni substituents was found to be at the nearest neighbour positions as shown in Fig. 7b. This configuration resulted in the spontaneous formation of an O2 molecule (see Fig. 7b) that remained adsorbed at around 1.9 Å above the surface. The O2 molecule was found to be in a triplet spin state with a bond length of 1.238 Å, very close to the VASP-optimized bond length of 1.234 Å. Further we note that once a dopant is introduced, the energy cost associated with the introduction of a second dopant is reduced, compared to the first dopant (−1.591 eV vs. −5.352 eV). In CeO2, Ce ions are in an oxidation state of Ce4+ while O ions are in an oxidation state of O2−. Substituting a Ce4+ ion in CeO2(110) with a divalent cation results in a charge imbalance which is compensated by spin polarization of the oxygen ion (in the case of Ni substituent, mainly on those O ions marked with the label M in Fig. 7a), leading to partially unoccupied ‘p’ orbitals on these ions. The magnetic moment on the two OM ions was found to be 0.37 μB while the magnetic moment on the Ni substituent was 0.92 μB, indicating that the Ni substituent does not get fully oxidized to the Ni2+ state. The total and projected density of states for the mono-Ni substituted surface is shown in Fig. S4 of the ESI.†
Another means to achieve charge neutrality is through the loss of one or more surface O atoms to form ‘charge compensating’ oxygen vacancies (one and two vacancies for mono- and di-Ni substituted CeO2(110) respectively). We have studied various distributions of charge compensating O vacancies on both the surfaces and computed their formation energy according to eqn (4)
 | (4) |
 | ||
| Fig. 8 (a) The optimized atomic structures of the Ni-substituted CeO2(100) surface. The lower-left inset shows the side view of the outermost four atomic layers and the lower-right inset shows the local structure of the Ni2+ ion. M1 and M2 represent the magnetic O ions surrounding the Ni substituent. Ce atoms are yellow, O atoms are red and blue and Ni atom is grey. (b) The Ni-substituted CeO2(100) surface with a charge compensating oxygen vacancy represented by a black sphere. | ||
As indicated, on the Ni-substituted (110) surface, substituting a Ce4+ ion in CeO2(110) with a lower-valent cation results in a charge imbalance which is compensated by spin polarization of the oxygen ion. The Ni substituent on the (100) surface also leads to spin-polarized O atoms, resulting in unoccupied gap states ∼0.5 eV above the top of the valence band (see Fig. S18 in ESI†). These states have mostly O 2p character and are spatially localized on the four nearest-neighbor O atoms of the Ni substituent (see the atoms marked by M1 and M2 in Fig. 8a). The two O atoms marked by M1 and M2 in Fig. 8a have a local magnetic moment of ∼0.61 and 0.23 ΩB per O atom, respectively. Here, the magnetic moment on the Ni substituent was 1.78 μB, indicating that the Ni substituent gets fully oxidized to the Ni2+ state. For reference, the calculated magnetic moment of Ni2+ in NiO was 1.65 μB. The formation energy of the Ni-substituted (100) surface was found to be −1.735 eV, while this value was found to be −0.572 eV for the (111) surface (see Table T2 in ESI†), indicating that it is easier to dope the (100) surface than the (110) and (111) surfaces, in agreement with their order of stability. The formation energy of a Ni substituent in bulk CeO2 with a 96-atom supercell (see Fig. S20 in ESI†) was calculated using eqn (5).
| Ni(s) + CexO2x(s) + O2(g) → NiCex−1O2x(s) + CeO2(s) | (5) |
As concluded above, forming a ‘charge compensating’ oxygen vacancy is another means to achieve charge neutrality on a Ni-substituted surface. In order to understand better the relationship between the charge-compensating vacancy and Ni on the CeO2(100) surface, we have considered four possible configurations of the charge-compensating O vacancy on the Ni-substituted (100) surface (see Fig. S11 in ESI†) and the most stable configuration is shown in Fig. 8b. As shown in Fig. 8b, the charge-compensating O vacancy prefers to be incorporated into the host matrix close to the Ni substituent. The Ni atom relaxes outward and coordinates to four O atoms. The local structure around the Ni atom was found to be asymmetric with the Ni–O bond lengths varying between 1.981 and 2.011 Å, close to the Ni–O bond length of 2.085 Å in NiO(s). We computed the O-vacancy formation energy using eqn (4) to be −0.947 eV.
In fact, what is of interest in catalysis is the cost to remove the next so-called “active vacancy”. Hence, the formation of a second vacancy on the Ni-substituted (110) and (100) surfaces will be discussed in the next section.
| CexO2x(surf) → CexO2x−1(surf) + ½O2(g) | (6) |
| NiCexO2x(surf) → NiCexO2x−1(surf) + ½O2(g) | (7) |
| NiyCex−yO2x−y(surf) → NiyCex−yO2x−y−1(surf) + ½O2(g) | (8) |
In eqn (8), y = 1 for the mono-Ni substituted surface and y = 2 for the di-Ni substituted surface. The creation of an active O vacancy results in the partial reduction of two of the surface Ce4+ ions to Ce3+ ions. As noted earlier, a number of different distributions of Ce3+ ions on the surface are possible for a given vacancy location. The most stable configurations for the (110) and (100) surfaces are shown in Fig. 9a and 10a, respectively. The formation energy of an oxygen vacancy from the pure CeO2(110) surface computed by us (1.414 eV) compares favourably with the value reported by Kullgren et al.64 (1.56 eV, refer to Table III of their paper).
 | ||
| Fig. 9 Active oxygen vacancy on (a) pure, (b) Ni adsorbed, (c) mono-Ni substituted and (d) di-Ni substituted CeO2(110) surface. Ce4+ ions are yellow, Ce3+ ions are blue, O2− ions are red and Ni+/Ni2+ ions are grey. The position of the oxygen vacancy is shown by black spheres. Arrows in (a), (c) and (d) show the motion of an oxygen atom from its surface site to a bridging position. | ||
 | ||
| Fig. 10 The optimized atomic structure of (100) surface with active oxygen vacancies. (a) Pure (100) surface, (b) Ni substituted (100) surface, (c) Ni adsorbed (100) surface. Ce atoms are yellow, Ni atoms is grey, O atoms are red and blue. The position of the O vacancy is represented by a black sphere. The inset shows the side view corresponding to the structure in panel (c). | ||
For the Ni-adsorbed (110) surface, we showed that Ni+ and Ni2+ oxidation states for the adsorbate were close in energy. Thus we have investigated active O-vacancy formation beginning with both Ni+ and Ni2+ configurations for the adsorbate. We have investigated two different vacancy locations for the mono-Ni substituted surface and three different locations for the di-Ni substituted surface. For each of these locations various distributions of Ce3+ ions were investigated. Fig. 9 shows the most favourable configuration of an active O vacancy on all the four (110) surfaces studied here while Fig. S5–S8 in the ESI† show the structures and formation energies for various other vacancy positions.
For the Ni-adsorbed (100) surface, we have considered two possible configurations for the coexistence of a Ni adsorbate and active oxygen vacancy defect. As discussed above, two Ce3+ ions are created on the CeO2(100) surface by the formation of an oxygen vacancy or the adsorption of a Ni atom due to the transfer of electrons from the adsorbate or the vacancy defect to Ce4+. Therefore, the coexistence of a Ni adsorbate and an oxygen vacancy is expected to lead to a partially reduced surface with four Ce3+ ions. The most stable configuration is shown in Fig. 10c while the other configurations that we have studied are shown in Fig. S12 of the ESI.† The distortions around the oxygen vacancy and Ni on this surface are similar to those found around an oxygen vacancy on the pure (100) surface and Ni-adsorbed (100) surface, as shown in Fig. 6a and 10a.
From Fig. 9 and 10, we see that modifying the CeO2(110) and (100) surfaces by a Ni substitution results in a reduction in the energy cost associated with the creation of an active oxygen vacancy compared to the pristine surface, though a higher doping percentage results in a smaller change in the vacancy formation energy on the (110) surface. On the other hand, the mono-Ni adsorbate on both (110) and (100) surfaces makes it more unfavourable to create an oxygen vacancy from the surface which could lead to suppression of the activity of the catalyst. This indicates that there exists an optimal level of surface doping to achieve enhanced catalytic activity. These results are in agreement with recent experimental data which shows that increasing Ni dopant percentage beyond 7% results in a reduction in the catalytic activity of ceria nanorods.65
For mono-Ni substituted (110) and (100) surfaces, we find that the most favourable site for oxygen vacancy formation is an oxygen ion site coordinated to the Ni dopant (see Fig. 9c and 10b). On the mono-Ni substituted (110) surface, this results in significant structural rearrangement with the Ni dopant moving from a square-planar-like position to underneath a lattice metal ion site. Nolan45 on the other hand reports the most favourable site for the creation of an active oxygen vacancy on the mono-Ni substituted (110) surface to be one of the next nearest lattice sites to the Ni dopant (see Fig. S7(b) in the ESI†) with a vacancy formation energy of 1.30 eV. For the same configuration, we have computed the vacancy formation energy to be 1.39 eV (see Fig. S7(b) in the ESI†). Furthermore, the formation of an oxygen vacancy results in the migration of a neighbouring O2− ion to a bridging position on the (110) surface (Fig. 9a, c and d; arrows indicate the direction of motion of the oxygen atom). This rearrangement of an O2− ion was not observed on the (100) surface. Comparing the formation energy of an active oxygen vacancy on the pure (110) with (111) and (100) surfaces (1.983 and 1.679 eV respectively), we find that (110) is the most active amongst the three surfaces. However, upon doping the surface with a Ni substituent, the formation energy of an active O vacancy from the (100) surface reduced dramatically to only 0.355 eV, indicating that surface doping has a larger effect on the CeO2(100) surface as compared to (110).
4. Conclusions and concluding remarks
In this work, we have undertaken a systematic study of the interaction of a Ni adsorbate and Ni substituents with the CeO2(110) and (100) surfaces using density functional theory calculations. The following conclusions can be derived from this work:(1) A Ni atom prefers to adsorb on a bridge site as compared to a hollow site on the (110) surface, and the adsorption of Ni induces strong geometric distortions locally around the adsorbate. On the (100) surface, the most stable position of a Ni adsorbate was found to be the bridge site where the Ni atom is coordinated to two surface O atoms.
(2) On a bridge site, up to 0.25 ML coverage on the (110) surface, a Ni adsorbate prefers its Ni+ oxidation state over the Ni2+ oxidation state with the reduced Ce3+ ion located next to the Ni+ adsorbate. Location of the Ce3+ ion far away from the Ni+ adsorbate or at a subsurface position was found to be unfavourable. For the Ni2+ oxidation state, the most favourable distribution of Ce3+ ions was found to be when they were located diagonally across the Ni2+ adsorbate on the neighbouring sites. On the (100) surface, a Ni adsorbate prefers its Ni2+ oxidation state over the Ni+ oxidation state. The Ce3+ ions on the (100) surface prefer to stay in the vicinity of the adsorbed Ni or the oxygen vacancy, while they prefer to separate from the Ni adatom or the oxygen vacancy on the (111) surface.
(3) As the coverage of Ni on CeO2(110) surface increases from 0.083 ML to 0.25 ML, the adsorption energy for the most favourable configuration, i.e., Ni+ on a bridge site, decreases from −0.449 eV to −0.322 eV.
(4) With increase in coverage of Ni from 0.083 ML to 0.25 ML, the stabilization of the Ni+ oxidation state over the Ni2+ oxidation state on a bridge site decreases from ca. 49 meV to 21 meV for the most preferred Ce3+ ion distribution in the respective oxidation states of the Ni adsorbate.
(5) Adsorption of Ni introduces split-off-states above the CeO2(110) and (100) valence band. In its Ni+ oxidation state on the (110) surface, these states arise from the f orbital on the reduced Ce3+ ion, the d orbital of the Ni+ adsorbate and the p orbital of the oxygen ions that are coordinated to the Ni+ adsorbate. However, in its Ni2+ oxidation state on both (110) and (100) surfaces, these states arise from the f orbitals on the two reduced Ce3+ ions only.
(6) In contrast to Ni adsorption, a Ni substituent does not induce the reduction of a Ce4+ to Ce3+. However, it induces strong structural distortion locally, with the Ni substituent moving into a four-coordinated nearly planar environment from the six-coordinated metal ion site on the (110) surface.
(7) The nearest neighbour locations were found to be the most favourable distribution of the two Ni substituents on the (110) surface. Further, this resulted in spontaneous ejection of an O2 molecule from the surface.
(8) Substitution of surface Ce atoms by Ni atoms resulted in a reduction in the energy required to create an active oxygen vacancy while a Ni adsorbate caused an increase in the formation energy of an active O vacancy. The degree of reduction in the vacancy formation energy points to the presence of an optimum level of surface doping to achieve enhanced catalytic activity.
The effect of these surface modifications on the catalytic activity of ceria catalysts for WGS and methanation reactions is a subject of an ongoing study in our group.
Notes
W.Q. Li and S. G. S are joint first authors in this article.Acknowledgements
The authors acknowledge the computational resources provided by WestGrid and Compute Canada for this work. Discovery Grant funding (project RGPIN-2014-06606) from NSERC (Canada) to D. R. S. and funding from European Research Council (ERC StGC3ENV) are gratefully acknowledged. W. L. acknowledges support from the Marie Cuire IRSES project TEMM1P.References
- A. Trovarelli, Catal. Rev., 1996, 38, 439–520 CAS.
- A. Trovarelli, C. de Leitenburg, M. Boaro and G. Dolcetti, Catal. Today, 1999, 50, 353–367 CrossRef CAS.
- R. J. Gorte, AIChE J., 2010, 56, 1126–1135 CAS.
- Q. Fu, A. Weber and M. Flytzani-Stephanopoulos, Catal. Lett., 2001, 77, 87–95 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- S. P. S. Badwal, D. Fini, F. T. Ciacchi, C. Munnings, J. A. Kimpton and J. Drennan, J. Mater. Chem. A, 2013, 1, 10768–10782 CAS.
- M. V. Ganduglia-Pirovano, C. Popa, J. Sauer, H. Abbott, A. Uhl, M. Baron, D. Stacchiola, O. Bondarchuk, S. Shaikhutdinov and H.-J. Freund, J. Am. Chem. Soc., 2010, 132, 2345–2349 CrossRef CAS PubMed.
- T. Kropp, J. Paier and J. Sauer, J. Am. Chem. Soc., 2014, 136, 14616–14625 CrossRef CAS PubMed.
- S. Carrettin, P. Concepción, A. Corma, J. M. López Nieto and V. F. Puntes, Angew. Chem., Int. Ed., 2004, 43, 2538–2540 CrossRef CAS PubMed.
- T. Bunluesin, E. S. Putna and R. J. Gorte, Catal. Lett., 1996, 41, 1–5 CrossRef CAS.
- T.-S. Nguyen, F. Morfin, M. Aouine, F. Bosselet, J.-L. Rousset and L. Piccolo, Catal. Today, 2015, 253, 106–114 CrossRef CAS.
- T. Bunluesin, R. J. Gorte and G. W. Graham, Appl. Catal., B, 1998, 15, 107–114 CrossRef CAS.
- Y. Li, Q. Fu and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2000, 27, 179–191 CrossRef CAS.
- S. Hilaire, X. Wang, T. Luo, R. J. Gorte and J. Wagner, Appl. Catal., A, 2001, 215, 271–278 CrossRef CAS.
- J. A. Rodriguez, P. Liu, J. Hrbek, J. Evans and M. Pérez, Angew. Chem., Int. Ed., 2007, 46, 1329–1332 CrossRef CAS PubMed.
- G. Zhou, L. Barrio, S. Agnoli, S. D. Senanayake, J. Evans, A. Kubacka, M. Estrella, J. C. Hanson, A. Martínez-Arias, M. Fernández-García and J. A. Rodriguez, Angew. Chem., Int. Ed., 2010, 49, 9680–9684 CrossRef CAS PubMed.
- L. Barrio, A. Kubacka, G. Zhou, M. Estrella, A. Martínez-Arias, J. C. Hanson, M. Fernández-García and J. A. Rodriguez, J. Phys. Chem. C, 2010, 114, 12689–12697 CAS.
- S. Senanayake, J. Evans, S. Agnoli, L. Barrio, T.-L. Chen, J. Hrbek and J. Rodriguez, Top. Catal., 2011, 54, 34–41 CrossRef CAS.
- J. Carrasco, L. Barrio, P. Liu, J. A. Rodriguez and M. V. Ganduglia-Pirovano, J. Phys. Chem. C, 2013, 117, 8241–8250 CAS.
- J. Carrasco, D. López-Durán, Z. Liu, T. Duchoň, J. Evans, S. D. Senanayake, E. J. Crumlin, V. Matolín, J. A. Rodríguez and M. V. Ganduglia-Pirovano, Angew. Chem., Int. Ed., 2015, 54, 3917–3921 CrossRef CAS PubMed.
- J. Paier, C. Penschke and J. Sauer, Chem. Rev., 2013, 113, 3949–3985 CrossRef CAS PubMed.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 CrossRef CAS.
- J. A. Farmer and C. T. Campbell, Science, 2010, 329, 933–936 CrossRef CAS PubMed.
- G. L. Haller and D. E. Resasco, Adv. Catal., 1989, 36, 173–235 CAS.
- M. Nolan, S. Grigoleit, D. C. Sayle, S. C. Parker and G. W. Watson, Surf. Sci., 2005, 576, 217–229 CrossRef CAS.
- M. M. Branda, N. C. Hernández, J. F. Sanz and F. Illas, J. Phys. Chem. C, 2010, 114, 1934–1941 CAS.
- L. Szabová, M. F. Camellone, M. Huang, V. Matolín and S. Fabris, J. Chem. Phys., 2010, 133, 234705 CrossRef PubMed.
- Z. S. Lu, Z. X. Yang and K. Hermansson, Adv. Mater. Res., 2011, 213, 166–171 CrossRef CAS.
- Y. Tang, H. Zhang, L. Cui, C. Ouyang, S. Shi, W. Tang, H. Li and L. Chen, J. Power Sources, 2012, 197, 28–37 CrossRef CAS.
- Z. P. Liu, S. J. Jenkins and D. A. King, Phys. Rev. Lett., 2005, 94, 196102 CrossRef PubMed.
- Z. Yang, Z. Lu and G. Luo, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 075421 CrossRef.
- A. Bruix, K. M. Neyman and F. Illas, J. Phys. Chem. C, 2010, 114, 14202–14207 CAS.
- Z. S. Lu and Z. X. Yang, J. Phys.: Condens. Matter, 2010, 22, 475003 CrossRef PubMed.
- A. D. Mayernick and M. J. Janik, J. Chem. Phys., 2009, 131, 084701 CrossRef PubMed.
- M. M. Branda, N. J. Castellani, R. Grau-Crespo, N. H. de Leeuw, N. C. Hernandez, J. F. Sanz, K. M. Neyman and F. Illas, J. Chem. Phys., 2009, 131, 094702 CrossRef PubMed.
- H.-X. Mai, L.-D. Sun, Y.-W. Zhang, R. Si, W. Feng, H.-P. Zhang, H.-C. Liu and C.-H. Yan, J. Phys. Chem. B, 2005, 109, 24380–24385 CrossRef CAS PubMed.
- R. Si and M. Flytzani-Stephanopoulos, Angew. Chem., Int. Ed., 2008, 47, 2884–2887 CrossRef CAS PubMed.
- L. Cui, Y. Tang, H. Zhang, L. G. Hector, C. Ouyang, S. Shi, H. Li and L. Chen, Phys. Chem. Chem. Phys., 2012, 14, 1923–1933 RSC.
- M. Nolan, J. Chem. Phys., 2012, 136, 134703 CrossRef PubMed.
- X. Du, D. Zhang, L. Shi, R. Gao and J. Zhang, J. Phys. Chem. C, 2012, 116, 10009–10016 CAS.
- W. Tang, Z. Hu, M. Wang, G. D. Stucky, H. Metiu and E. W. McFarland, J. Catal., 2010, 273, 125–137 CrossRef CAS.
- D. García Pintos, A. Juan and B. Irigoyen, J. Phys. Chem. C, 2013, 117, 18063–18073 Search PubMed.
- M. Nolan, J. Phys. Chem. C, 2009, 113, 2425–2432 CAS.
- Y. Irene and N. Michael, J. Phys.: Condens. Matter, 2010, 22, 135004 CrossRef PubMed.
- M. Nolan, J. Mater. Chem., 2011, 21, 9160–9168 RSC.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- M. Cococcioni and S. de Gironcoli, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035105 CrossRef.
- X. Wang, M. Shen, J. Wang and S. Fabris, J. Phys. Chem. C, 2010, 114, 10221–10228 CAS.
- J. Graciani, A. M. Márquez, J. J. Plata, Y. Ortega, N. C. Hernández, A. Meyer, C. M. Zicovich-Wilson and J. F. Sanz, J. Chem. Theory Comput., 2011, 7, 56–65 CrossRef CAS PubMed.
- W.-B. Zhang, N. Yu, W.-Y. Yu and B.-Y. Tang, Eur. Phys. J. B, 2008, 64, 153–158 CrossRef CAS.
- J. P. Allen and G. W. Watson, Phys. Chem. Chem. Phys., 2014, 16, 21016–21031 RSC.
- N. V. Skorodumova, M. Baudin and K. Hermansson, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 075401 CrossRef.
- P. Pulay, Chem. Phys. Lett., 1980, 73, 393–398 CrossRef CAS.
- N. V. Skorodumova, M. Baudin and K. Hermansson, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 075401 CrossRef.
- M. Molinari, S. C. Parker, D. C. Sayle and M. S. Islam, J. Phys. Chem. C, 2012, 116, 7073–7082 CAS.
- T. Kropp and J. Paier, J. Phys. Chem. C, 2015, 119, 23021–23031 CAS.
- W. Song and E. J. M. Hensen, J. Phys. Chem. C, 2013, 117, 7721–7726 CAS.
- J. Kullgren, K. Hermansson and C. Castleton, J. Chem. Phys., 2012, 137, 044705 CrossRef PubMed.
- A. Romero-Nunez and G. Diaz, RSC Adv., 2015, 5, 54571–54579 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The optimal structures along with their formation energies for various Ni/CeO2(110), (100) and (111) systems are provided. See DOI: 10.1039/c6cp00738d |
| This journal is © the Owner Societies 2016 |