Proton coupled electron transfer from the excited state of a ruthenium(II) pyridylimidazole complex†
Received
20th January 2016
, Accepted 29th March 2016
First published on 8th April 2016
1. Introduction
Hydrogen atom transfer (HAT) is important in enzymes and in synthetic organic chemistry, for example for hydrogenations of unsaturated compounds such as ketones and imines. It would be attractive to use visible light to perform HAT reactions under mild reaction conditions, and therefore we explored the (formal) HAT chemistry of photoexcited [Ru(bpy)2pyimH]2+ (bpy = 2,2′-bipyridine; pyimH = 2-(2′-pyridyl)imidazole). In pure HAT the transferred electron and proton originate from the same donor orbital, whereas in unidirectional proton coupled electron transfer (PCET) the transfer of a net hydrogen atom occurs from different donor orbitals.1–5 This is in fact the case for excited [Ru(bpy)2pyimH]2+ because the metal center acts as an electron donor, whereas the proton is released from the pyimH ligand. While early PCET studies have focused largely on reactions between molecules in their electronic ground states,6–11 photoinduced PCET is now receiving increasing attention.1,12–25 Formal HAT between a transition metal complex in its 3MLCT excited-state and various reaction partners either across a salt bridge,14,20–23 or via hydrogen bonding interactions have been explored.13,24,25 Even hydride transfer from the excited state of an iridium complex was reported recently.26 In order to predict the reactivity of an excited state, thermodynamic quantities such as its redox potential and acidity constant must be known.27,28 For HAT reactions, the determination of bond dissociation free energies (BDFEs) is useful, while for PCETs the calculation of formal BDFEs has proven meaningful.28 This is possible for reactions in the ground state as well as for reactions with excited species. Based on this concept, photochemical conversions of ketones to ketyls could be rationalized.2,16,17
The [Ru(bpy)2pyimH]2+ complex (Fig. 1) has long been known,29 in particular Haga and coworkers explored a variety of ruthenium and osmium complexes with pyimH and related (deprotonatable) ligands.30–35 Later, Gray and coworkers explored the acid–base and redox chemistry of [Ru(bpy)2pyimH]2+ and related complexes in the ground and the lowest 3MLCT excited state.29,36 However, the formal BDFE of the peripheral N–H bond of [Ru(bpy)2pyimH]2+ and related complexes has never been determined, and the excited-state PCET chemistry remained unexplored, except in the case of an Ir(III) complex with a 2,2′-biimidazole ligand.14 In structurally related complexes such as [Ru(acac)2pyimH]2+ (acac = acetylacetonato), formal BDFEs were estimated for the electronic ground state, and values around 62 kcal mol−1 were found.9,37 We anticipated that [Ru(bpy)2pyimH]2+ might exhibit an unusually low N–H BDFE in its long-lived 3MLCT excited state, making it potentially an equally potent (formal) hydrogen atom donor as previously investigated metal hydride complexes in their electronic ground states.4,38
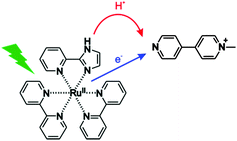 |
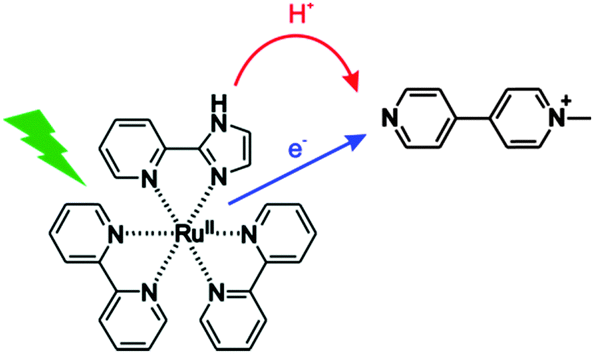
| | Fig. 1 The investigated process in this work: transfer of one electron and one proton from [Ru(bpy)2pyimH]2+ to monoquat (MQ+) upon photoexcitation, corresponding to net transfer of a hydrogen atom. | |
In the following we present the thermochemical characterization of ground and 3MLCT excited states of [Ru(bpy)2pyimH]2+ in buffered 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v:v) CH3CN/H2O. In this solvent mixture, well-defined pH values can easily be obtained by a variety of buffers and the solubility of [Ru(bpy)2pyimH]2+ as well as that of a variety of substrates is good. We find an N–H BDFE of only (43 ± 5) kcal mol−1 in the emissive 3MLCT excited state based on thermodynamic cycles and on the photoinduced PCET chemistry with monoquat (MQ+). The acceptor was chosen due to its ability to act as a combined electron–proton acceptor, the favourable spectroscopic properties of its radical form and the importance of pyridyl radicals for the reduction of CO2.39–41 The PCET reaction mechanism between photoexcited [Ru(bpy)2pyimH]2+ and MQ+ was explored in detail.
:1 (v:v) CH3CN/H2O. In this solvent mixture, well-defined pH values can easily be obtained by a variety of buffers and the solubility of [Ru(bpy)2pyimH]2+ as well as that of a variety of substrates is good. We find an N–H BDFE of only (43 ± 5) kcal mol−1 in the emissive 3MLCT excited state based on thermodynamic cycles and on the photoinduced PCET chemistry with monoquat (MQ+). The acceptor was chosen due to its ability to act as a combined electron–proton acceptor, the favourable spectroscopic properties of its radical form and the importance of pyridyl radicals for the reduction of CO2.39–41 The PCET reaction mechanism between photoexcited [Ru(bpy)2pyimH]2+ and MQ+ was explored in detail.
2 Results and discussion
Spectroscopy and thermodynamics of the ground state
pKa of [Ru(bpy)2pyimH]2+.
For determination of the pKa value of the electronic ground state, absorption spectra were recorded at different pH values between pH 3.7 and pH 10.2 (ESI,† Fig. S1a) using suitable buffers. By plotting the absorbance at the MLCT absorption maxima at 460 nm (protonated form) and 491 nm (deprotonated form) and sigmoidal fitting, pKa = 8.1 ± 0.1 was found for 1:1 (v:v) CH3CN/H2O (ESI,† Fig. S1b), in agreement with a prior study that reported pKa = 7.9 ± 0.1 in H2O containing 5% methanol.29
Table 1 Acidity constants of [Ru(bpy)2pyimH]2+ in 1:1 (v:v) CH3CN/H2O in the electronic ground state (pKa), in the long-lived 3MLCT excited state (pKa*) and in the one-electron oxidized form (pKoxa)
| pKa |
8.1 ± 0.1 |
| pKa* |
5.6 ± 0.3 |
| pKoxa |
3.6 ± 0.1 |
pKa of [Ru(bpy)2pyimH]3+.
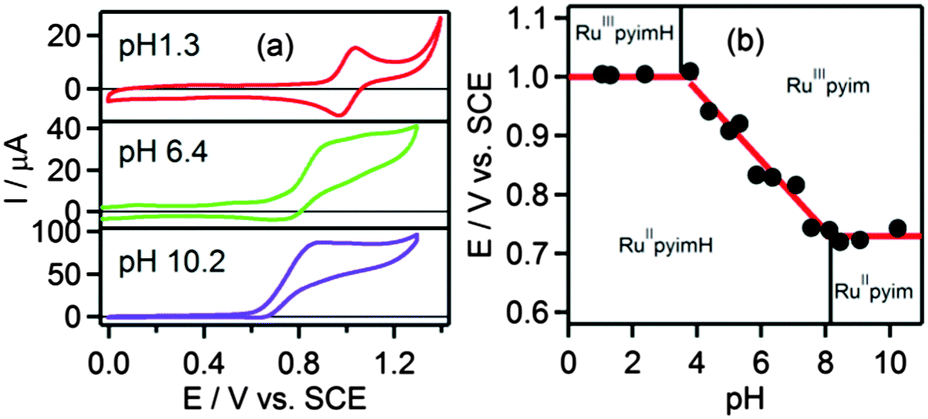
Oxidative cyclic voltammetry sweeps probing metal oxidation were performed in the pH range between pH 1 and pH 11. Characteristic voltammograms for three pH regions of interest are shown in Fig. 2a. In the acidic pH range where [Ru(bpy)2pyimH]2+ remains protonated after oxidation of RuII to RuIII, the voltammograms show one reversible oxidation wave at Eprotox = (1.00 ± 0.05) V vs. SCE with peak separations between 67 and 80 mV. This behaviour is observed up to pH = 3.6 ± 0.1, corresponding to the acidity constant of [Ru(bpy)2pyimH]3+ (pKoxa). At higher pH values oxidation is irreversible at a sweep rate of 100 mV s−1 (Fig. 2a and Fig. S5 in the ESI†) due to deprotonation of the pyimH ligand of the oxidized complex. Oxidation potentials of the deprotonated complex were estimated by determining the relevant inflection points of the oxidation waves. Plotting the oxidation potentials in volt vs. pH gives the data points for the Pourbaix diagram in Fig. 2b. In the range between pKoxa = 3.6 and pKa = 8.1 the RuII/III oxidation wave shifts cathodically with a slope of −(60 ± 4) mV per pH unit, as expected for a 1-electron-1-proton-process. Oxidation of the deprotonated complex, [Ru(bpy)2pyim]+, occurs at Edepox = (0.73 ± 0.05) V vs. SCE (Table 2). Thus, the total cathodic shift between oxidation potentials of [Ru(bpy)2pyimH]2+ and [Ru(bpy)2pyim]+ is 270 mV, which is smaller than the previously reported shift of 380 mV in neat acetonitrile.29
Table 2 Ground and excited state redox potentials (Eox, *Eox), 3MLCT energy (E0–0), emission maxima at 25 °C (λmax) and luminescence lifetimes at 25 °C under aerated and deaerated conditions of [Ru(bpy)2pyimH]2+ in 1:1 (v:v) CH3CN/H2O with 0.05 M buffer
| |
[Ru(bpy)2pyimH]2+ |
[Ru(bpy)2pyim]+ |
|
Taken from references.29,42
Determined from emission in ethanol/methanol 4:1 (v:v) at 77 K.
|
|
E
ox [V vs. SCE] |
1.00 ± 0.05 |
0.73 ± 0.05 |
| *Eox [V vs. SCE] |
−1.1 ± 0.1 |
−1.2 ± 0.1 |
|
E
0–0 [eV] |
2.1 ± 0.1a,b |
1.9 ± 0.1b |
|
λ
max [nm] 25 °C |
625 ± 5 |
675 ± 5 |
|
τ [ns] aerated |
110 ± 10 |
50 ± 5 |
|
τ [ns] deaerated |
210 ± 20 |
70 ± 7 |
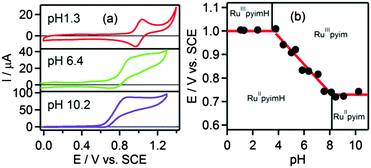 |
| | Fig. 2 (a) Voltammograms of [Ru(bpy)2pyimH]2+ in 1:1 (v:v) CH3CN/H2O with 0.05 M buffer at different pH values, (b) Pourbaix diagram of [Ru(bpy)2pyimH]2+. The slope between pH 3.6 and pH 8.1 is −(60 ± 4) mV per pH. A comprehensive set of voltammograms is shown in Fig. S5 of the ESI.† | |
Excited state properties
Excited state pKa* of [Ru(bpy)2pyimH]2+.
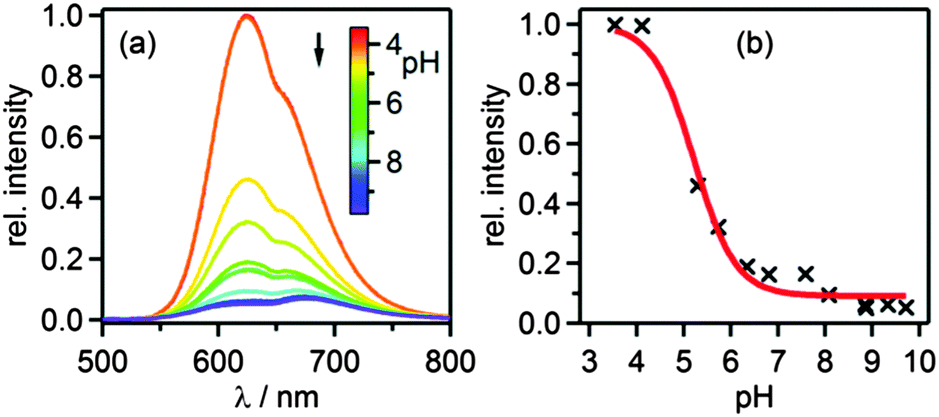
Luminescence spectra of [Ru(bpy)2pyimH]2+ were measured between pH 3 and pH 10 in 1:1 (v:v) CH3CN/H2O containing 0.05 M buffer to control the pH (Fig. 3a). All spectra were recorded at identical complex concentration and their intensity was normalized to the intensity of the most acidic sample; excitation occurred into the isosbestic point at 474 nm (Fig. S1a, ESI†). The decrease in intensity is due to different luminescence quantum yields of the protonated and deprotonated complex and due to deprotonation in the excited state. Emission decays were measured at 630 nm to determine the lifetimes of the protonated (τprot) and deprotonated 3MLCT state (τdep) in aerated and deaerated solution (Table 2). For these measurements the complex was excited at 532 nm with laser pulses of ca. 10 ns duration. The acidity constant in the long-lived 3MLCT-state (pKa*) was then determined from the inflection point of the steady-state emission titration curve (pHi = 5.2 ± 0.2) in Fig. 3b and the excited-state lifetimes of protonated (τprot) and deprotonated complex (τdep).| | | pKa* = pHi + log[τprot/τdep] | (1) |
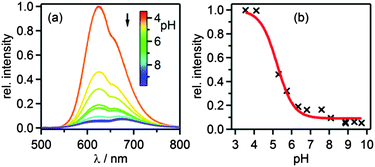 |
| | Fig. 3 (a) Luminescence of 40 μM [Ru(bpy)2pyimH]2+ in aerated 1:1 (v:v) CH3CN/H2O with 0.05 M buffer at different pH values following excitation into the isosbestic point at 474 nm. (b) Relative luminescence intensity at 625 nm vs. pH. | |
With eqn (1) one obtains pKa* = 5.6 ± 0.3.27 Based on the emission maxima of [Ru(bpy)2(pyimH)]2+ and [Ru(bpy)2(pyim)]+ at 25 °C, the Förster equation yields pKa* = 5.3 ± 0.6 (ESI,† page S6). This value is in good agreement with that determined from the luminescence titration, and also with the previously reported pKa* in water with 5% methanol.29 The increase of acidity in the excited state compared to the ground state indicates that the 3MLCT state is localized on the bpy spectator ligands, as noted earlier.36
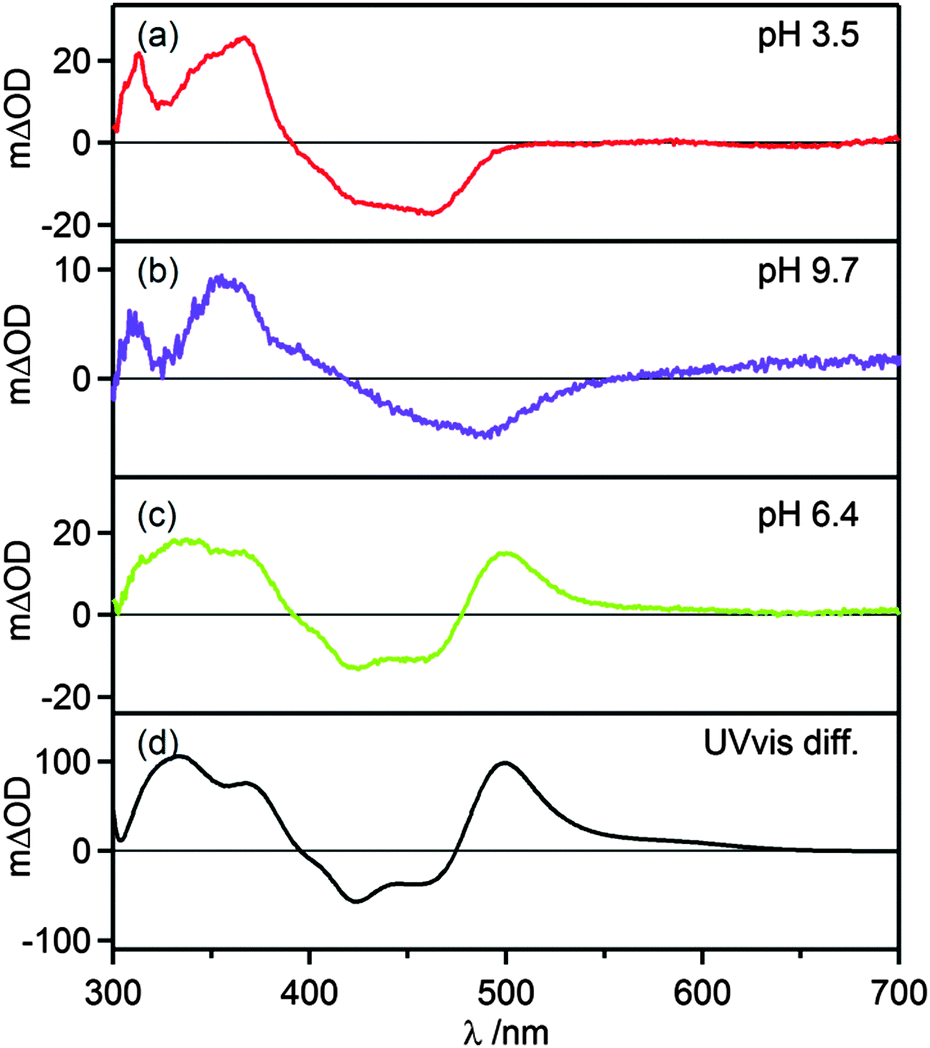
Transient absorption spectra were measured using pulsed laser excitation at 532 nm. Under acidic conditions (Fig. 4a), [Ru(bpy)3]2+-like transient spectra were observed, exhibiting a bleach around 450 nm and increased intensity around 370 nm.43 At basic pH the MLCT band is red-shifted (ESI,† Fig. S1), and therefore the MLCT-bleach in transient absorption is red-shifted as well (Fig. 4b). Transients that were recorded in the pH range between pH 5.0 and 8.1 exhibit a prominent feature at 500 nm (Fig. 4c) which can be explained by deprotonation in the excited state and the accumulation of deprotonated complex in the ground state. This interpretation is confirmed by a subtraction of the ground-state UV-vis spectra of the protonated and deprotonated complex (Fig. 4d) which also exhibits the prominent positive feature at 500 nm. The temporal evolution of this signal at pH 6.4 in presence of 0.05 M acetic acid/sodium acetate buffer and ca. 10−5 M complex concentration is shown below. In the absence of any reaction partner the formation of [Ru(bpy)2pyim]+, the deprotonated complex in its ground state, occurs with a time constant of τ = (60 ± 10) ns. Re-protonation then occurs with a time constant of τ = (106 ± 10) ns as discussed later. The kinetics of these deprotonation and protonation events are dictated by the buffer concentration. At pH 6.3 the quenching constant kq is (8.3 ± 0.5) × 108 L mol−1 s−1 based on a Stern–Volmer luminescence quenching experiment (ESI,† Fig. S7).
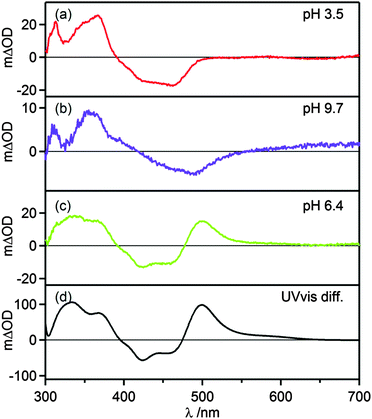 |
| | Fig. 4 (a–c) Transient absorption spectra of 20 μM [Ru(bpy)2pyimH]2+ in 1:1 (v:v) CH3CN/H2O with 0.05 M buffer at different pH values. Excitation occurred at 532 nm with laser pulses of ca. 10 ns duration, the spectra were recorded without time delay over a period of 200 ns. (d) Difference of ground state UV-vis spectra of protonated and deprotonated complex. | |
At any given pH and buffer concentration, the amount of accumulated [Ru(bpy)2pyim]+ in the ground state correlates with the acid–base equilibration in the 3MLCT excited state, and this reflects directly in the intensity of the transient band at 500 nm. A plot of the change in optical density at 500 nm vs. pH yields an inflection point at pH 5.6 ± 0.2 (ESI,† Fig. S4), in line with the pKa* value determined by luminescence titration and the Förster equation.
Thus, at sufficiently high pH, [Ru(bpy)2pyimH]2+ exhibits ordinary photoacid behavior similar to hydroxyarenes such as naphthols and hydroxypyrenetrisulfonate (“pyranine”).44
Excited state redox chemistry of [Ru(bpy)2pyimH]2+.
The excited state oxidation potentials (*Eox) were estimated based on the ground state redox potentials (Eox) and the 3MLCT energy (E0–0) using eqn (2).42E0–0 was determined from low-temperature luminescence spectroscopy (Fig. S2, ESI†). For [Ru(bpy)2pyimH]2+ we determined Eprot0–0 = (2.1 ± 0.1) eV29,42 and for [Ru(bpy)2pyim]+ we found Edep0–0 = (1.9 ± 0.1) eV. Based on these E0–0 values (see also Table 2), excited state redox potentials of *Eprot = −(1.1 ± 0.1) V vs. SCE and *Edep = −(1.2 ± 0.1) V vs. SCE were estimated for the protonated and deprotonated complex, respectively. Expectedly, oxidation is far easier in the 3MLCT excited state than in the electronic ground state.
BDFEs and “cube” scheme
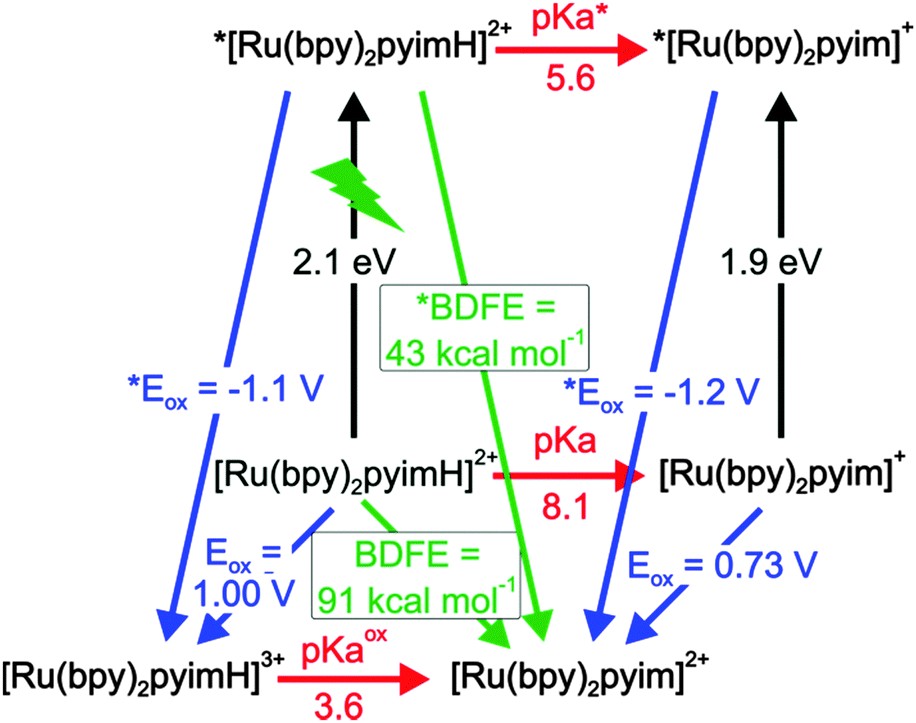
A graphical summary of all relevant thermodynamic parameters for [Ru(bpy)2pyimH]2+ is provided in Scheme 1. This so-called “cube” scheme is a three dimensional illustration displaying ground state redox potentials (blue) and acidity constants (red) on the bottom and excited state potentials (blue) and pKa values (red) on the top. 3MLCT excitation energies are represented by vertical black arrows. N–H bond dissociation free energies (BDFEs) can be estimated using eqn (3) and the experimentally determined acidity constants and oxidation potentials.2,45,46| | | BDFE (N–H) = 1.37 pKa + 23.06 E° + 57.6 kcal mol−1 | (3) |
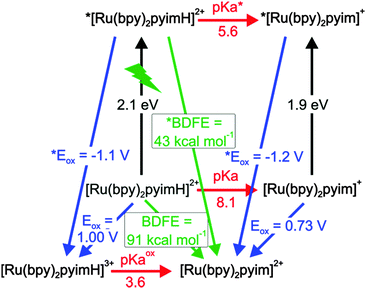 |
| | Scheme 1 Thermodynamic “cube” scheme for [Ru(bpy)2pyimH]2+ in 1:1 (v:v) CH3CN/H2O based on the data in Tables 1 and 2. Horizontal/red: pKa values, orthogonal in black: triplet energy E0–0, pointing towards the reader in blue: oxidation potentials in V vs. SCE, diagonal in green: BDFEs. | |
In eqn (3), E° must be entered in units of V vs. NHE; the last summand is a solvent-characteristic parameter describing solvation of hydrogen atoms. The resulting BDFEs for [Ru(bpy)2pyimH]2+ in the electronic ground and excited states are BDFE = (91 ± 1) kcal mol−1 and *BDFE = (43 ± 5) kcal mol−1 (green arrows in Scheme 1). The ground-state BDFE is comparable to primary and secondary amines2,47,48 but somewhat higher than related ruthenium pyridylimidazole complexes which were characterized in acetonitrile.9,37 Upon photoexcitation, the BDFE drops by 48 kcal mol−1, which is essentially equal to the energy of the absorbed visible photon. Interestingly, the resulting excited-state BDFE is comparable to metal hydride catalysts for hydrogenation reactions for which M–H BDFEs ranging from 50 to 55 kcal mol−1 for M = vanadium, 58 kcal mol−1 for M = chromium, and 68 kcal mol−1 for M = tungsten have been reported.4,38 In principle this drop in BDFE is expected to occur for other related metal complexes in the course of photoexcitation, but prior studies have not explicitly reported on this effect. Presumably this is due to the fact that in many cases the necessary redox potentials and acidity constants were not always determined in the same solvent, which complicates the application of eqn (3). Estimations based on prior work yields for the first N–H BDFE of [Ru(bpy)2(2,2′-biimidazole)]2+ a decrease from 86 to 40 kcal mol−1 between the electronic ground state and the long-lived 3MLCT state.29,49
In order to test whether the N–H BDFE is really that low, we set out to react photoexcited [Ru(bpy)2pyimH]2+ in PCET chemistry with a suitable formal H atom acceptor. We identified N-methyl-4,4′-bipyridinium (monoquat, MQ+) as a promising candidate. Meyer and coworkers already proposed the use of MQ+ for detecting PCET photoproducts because its one-electron reduced and protonated congener (MQH˙+) exhibits absorption features that can be identified unambiguously.50
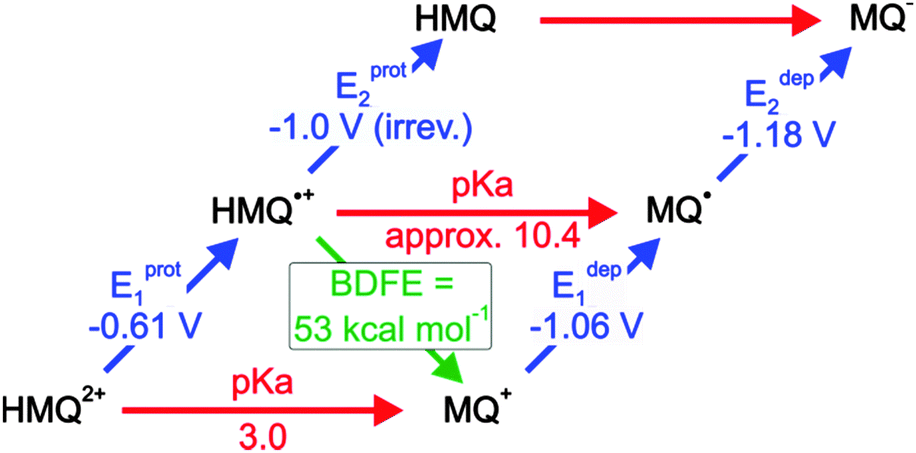
Before performing actual photochemical experiments between [Ru(bpy)2pyimH]2+ and MQ+, the thermodynamic properties of MQ+ in 1:1 (v:v) CH3CN/H2O were determined. The results from acid–base titration and electrochemical experiments are in the ESI† (Fig. S8 and S9). Here we merely report the final results in a thermodynamic “square” scheme (Scheme 2).
 |
| | Scheme 2 Thermodynamic “square” scheme of monoquat (MQ+) in 1:1 (v:v) CH3CN/H2O. Horizontal with red arrows: pKa values, upward right with blue arrows: redox potentials in V vs. SCE, downward left with green arrow: BDFEs. The pKa of HMQ˙+ was taken from the literature.46 | |
The key finding is that the HMQ˙+ radical has an N–H BDFE of (53 ± 1) kcal mol−1. Consequently, photoexcitation of [Ru(bpy)2pyimH]2+ in presence of MQ+ is expected to lead to formal HAT, resulting in [Ru(bpy)2pyim]+ and HMQ˙+. Based on an N–H *BDFE of (43 ± 5) kcal mol−1 for the photoexcited complex (Scheme 1), the driving-force for this reaction should be −(10 ± 6) kcal mol−1, which corresponds to −(0.4 ± 0.3) eV. In the following we report on the photochemistry between [Ru(bpy)2pyimH]2+ and MQ+ as a function of pH.
From simple photoinduced ET to formal HAT
In the electronic ground state, electron transfer (ET) and proton transfer reactions (PT) between [Ru(bpy)2pyimH]2+ and MQ+ are strongly endergonic (ΔGET = +(2.1 ± 0.1) eV, ΔGPT = +(0.30 ± 0.02) eV) and therefore no ground-state chemistry occurs. Regarding excited-state chemistry, there are in fact three different pH domains which are discussed individually in the following 3 sub-sections.
Acidic pH – photoinduced electron transfer.
In the acidic range, both the complex and the acceptor are protonated in the ground and excited state. Under these conditions, the expected reaction is photoinduced electron transfer (eqn (4)).
| *[Ru(bpy)2pyimH]2+ + HMQ2+ → [Ru(bpy)2pyimH]3+ + HMQ˙+ |
| | | ΔGET = −(0.5 ± 0.2) eV | (4) |
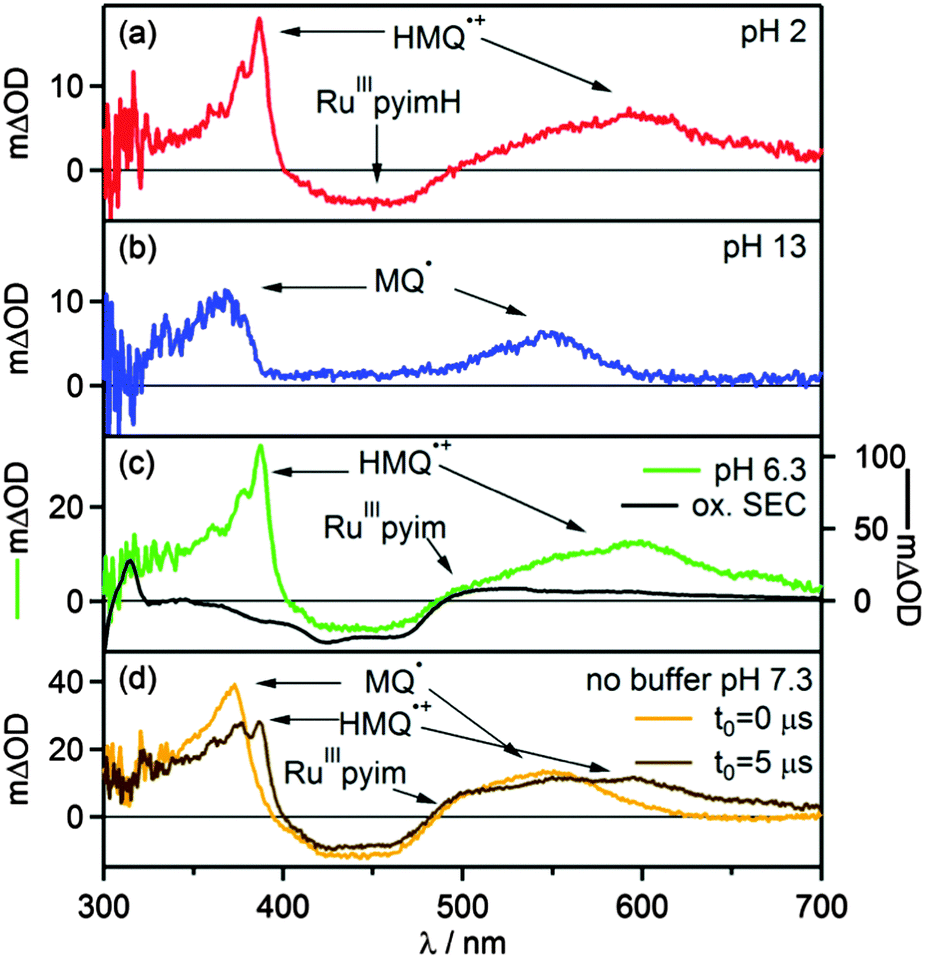
The transient absorption spectrum recorded at pH = 2 (Fig. 5a) confirms this expectation. Directly after the laser pulse one observes a bleach around 450 nm which is compatible with metal oxidation, and the signatures of the HMQ˙+ cation radical appear at 387 nm and 610 nm.46 The latter closely resemble the well-known radical of methyl viologen (MV2+).51 The photoinduced ET reaction from eqn (4) is associated with ΔGET = −(0.5 ± 0.2) eV. A Stern–Volmer experiment under acidic conditions reveals a quenching constant of kq = (6.8 ± 0.1) × 108 L mol−1 s−1 (ESI,† Fig. S14), which is comparable to what was found for the reaction of *[Ru(bpy)3]2+ with MV2+ in water (kq = 5.9 × 108 L mol−1 s−1).52,53 The thermal reverse ET from HMQ˙+ to [Ru(bpy)2pyimH]3+ in the electronic ground state then occurs on a time scale of approximately 100 μs, as determined by monitoring the HMQ˙+ signal at 610 nm (ESI,† Fig. S10).
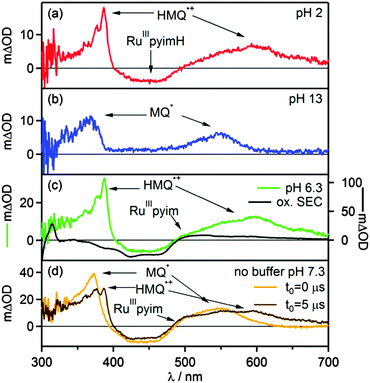 |
| | Fig. 5 Transient absorption spectra of [Ru(bpy)2pyimH]2+ in 1:1 (v:v) CH3CN/H2O recorded in presence of 60 mM MQ+ following excitation at 532 nm with laser pulses of ca. 10 ns duration. Detection occurred by integration over 200 ns. (a) 30 μM [Ru(bpy)2pyimH]2+ at pH 2 with 0.05 M buffer, data recorded with a delay time (t0) of 5 μs, (b) 30 μM [Ru(bpy)2pyimH]2+ at pH 13, data recorded with a delay time (t0) of 5 μs, (c) 50 μM [Ru(bpy)2pyimH]2+ at pH 6.3 with 0.05 M acetate buffer, data recorded with a delay time (t0) of 1 μs. (d) 50 μM [Ru(bpy)2pyimH]2+ in unbuffered in 1:1 (v:v) CH3CN/H2O at pH 7.3 recorded without delay time (orange trace) and with a time delay of 5 μs (brown trace). | |
Basic pH – ET.
In the basic range, the complex and the acceptor are both deprotonated in the ground and excited state, and consequently they are expected to undergo photoinduced electron transfer according to eqn (5).
| *[Ru(bpy)2pyim]+ + MQ+ → [Ru(bpy)2pyim]2+ + MQ˙ |
| | | ΔGET = −(0.14 ± 0.15) eV | (5) |
In transient absorption spectroscopy, the neutral monoquat radical (MQ˙) with characteristic absorptions at 365 nm and 545 nm is observed.51 The expected MLCT bleach of the ruthenium complex overlaps with a positive contribution of MQ˙ hence the flat region in the spectrum between 390 and 480 nm. The Stern–Volmer luminescence quenching experiment yields a quenching constant of kq = (20.7 ± 0.3) × 108 L mol−1 s−1 (ESI,† Fig. S15), which is close to the diffusion limit. The thermal reverse ET from MQ˙ to [Ru(bpy)2pyim]2+ takes place on a time scale of 10 μs (ESI,† Fig. S11).
Middle pH range – formal HAT.
In the middle pH range the most interesting photochemistry is expected. At pH 6.3 the complex is protonated in its ground state but becomes deprotonated upon excitation to the 3MLCT state as well as upon oxidation of RuII to RuIII (Scheme 1). On the other hand, MQ+ is not protonated, but it is expected to be protonated upon one-electron reduction (Scheme 2). According to the formal N–H BDFEs determined above for photoexcited [Ru(bpy)2pyimH]2+ ((43 ± 5) kcal mol−1, Scheme 1) and for HMQ˙+ ((53 ± 1) kcal mol−1, Scheme 2) the formal hydrogen atom transfer reaction in eqn (6) should be associated with a reaction free energy of −(10 ± 6) kcal mol−1.
| *[RuII(bpy)2pyimH]2+ + MQ+ → [RuIII(bpy)2pyim]2+ + HMQ˙+ |
| | | ΔGHAT = −(0.44 ± 0.16) eV | (6) |
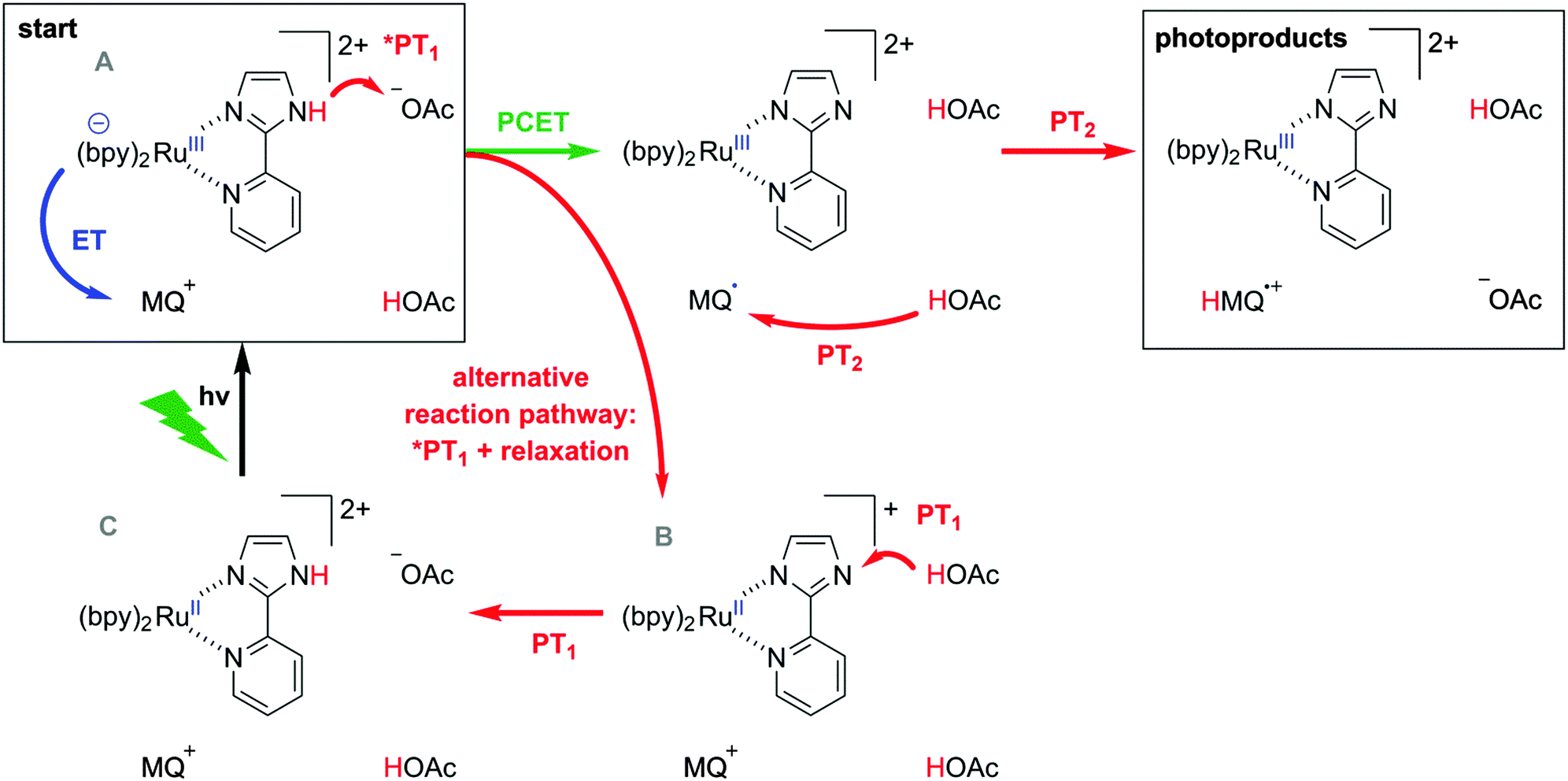
In transient absorption spectroscopy the two photoproducts from eqn (6) are indeed observed (Fig. 5c). When recording transient absorption spectra with a time delay (t0) of 1 μs there is clear evidence for HMQ˙+ (signals at 387 and 610 nm) and for [RuIII(bpy)2pyim]2+ (bleach around 450 nm), as confirmed by spectro-electrochemical (SEC) studies (black trace in Fig. 5c). However, mechanistically direct HAT between *[RuII(bpy)2pyimH]2+ and MQ+ in presence of aqueous buffer is highly improbable, particularly in view of the positive charges on both reactants. Moreover, the lowest-energetic MLCT excitation in [RuII(bpy)2pyimH]2+ involves promotion of an electron into a bpy-localized orbital rather than a pyimH orbital (see above).36 Consequently, formal HAT between photoexcited [RuII(bpy)2pyimH]2+ and MQ+ most likely involves a sequence of electron and proton transfer steps as illustrated in Scheme 3: following excitation of [RuII(bpy)2pyimH]2+, photoinduced electron transfer to MQ+ is coupled to release of the pyimH N–H proton to buffer base (acetate anion (AcO−), *PT1 in Scheme 3). This PCET process can occur either in stepwise or concerted fashion, and it results in [RuIII(bpy)2pyim]2+, MQ˙, and HOAc (PCET arrow in Scheme 3). Subsequent proton transfer (PT2 step in Scheme 3) between buffer acid (HOAc) and MQ˙ then leads to the photoproducts detected in Fig. 5c. In fact, these two reaction steps can be temporally resolved (see below).
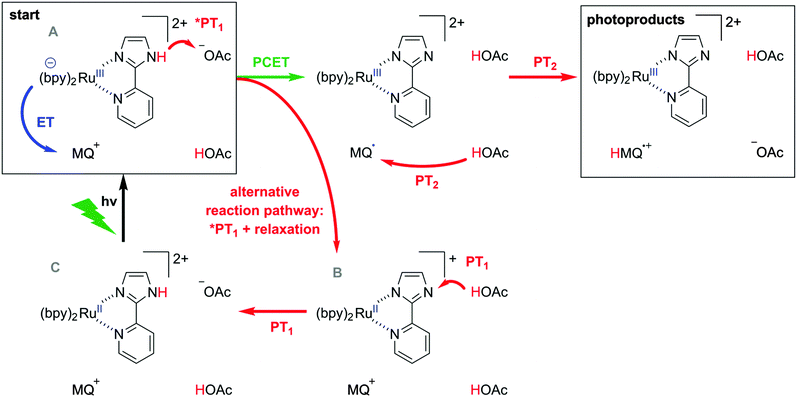 |
| | Scheme 3 Possible pathways for reaction of *[Ru(bpy)2pyimH]2+ with MQ+ in acetate-buffered solution. | |
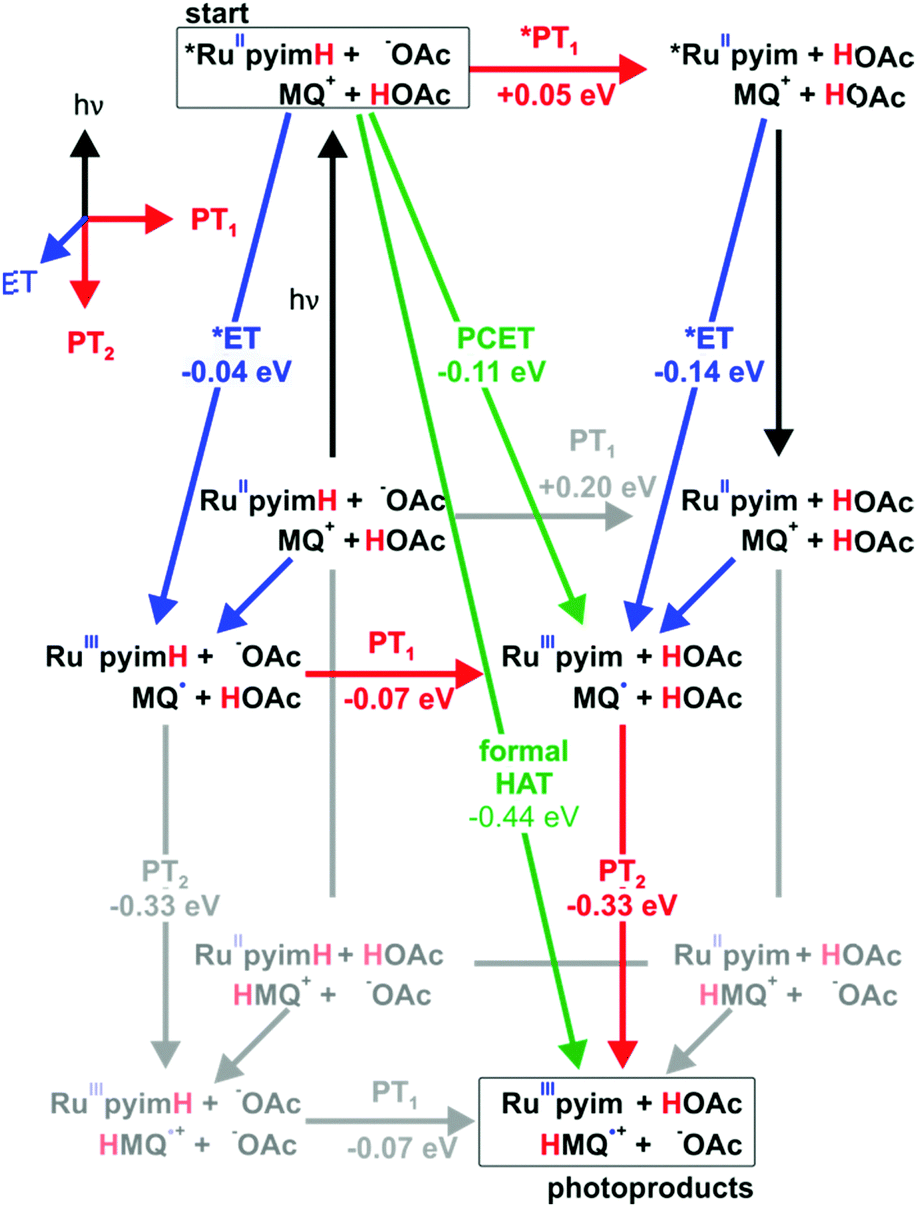
The reaction free energies for all conceivable reaction pathways are summarized in Scheme 4. The driving-forces in Scheme 4 emerge directly from the thermodynamic parameters of the two reaction partners in Schemes 1 and 2. The overall formal HAT process is dissected into photoexcitation (black arrows), electron transfer from the metal complex to monoquat (blue arrows), proton transfer from the metal complex to buffer base (PT1, red arrows), and proton transfer from buffer acid to monoquat (PT2, red arrows) as discussed above on the basis of Scheme 3.
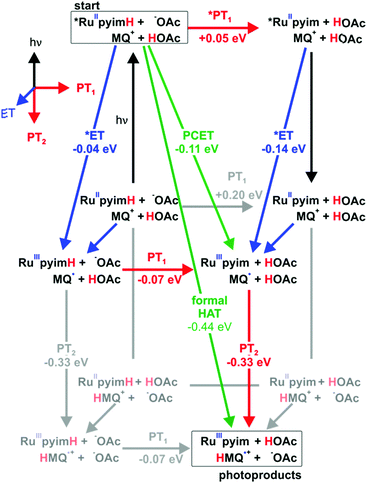 |
| | Scheme 4 Extended “cube”-scheme illustrating all reaction pathways for formal HAT between *[Ru(bpy)2pyimH]2+ and MQ+ in acetate-buffered solution (based on a combination of Schemes 1 and 2). Excited state chemistry is shown in the upper level, ET processes are pointing towards the reader, PT1 (complex to buffer base) is shown in horizontal direction and PT2 (buffer acid to monoquat) is shown in the two lower levels. Driving forces for the individual reaction steps are based on experimentally determined redox potentials and acidity constants. | |
From the starting point at the top left corner of Scheme 4, concerted proton-electron transfer (CPET) to form [RuIII(bpy)2pyim]2+, MQ˙, and protonated buffer base (HOAc) is a plausible initial reaction pathway since ΔGPCET = −(0.11 ± 0.16) eV for this process (green arrow). However, a sequence of electron and proton transfer events cannot be excluded on thermodynamic grounds and would be equally compatible with our experimental data.
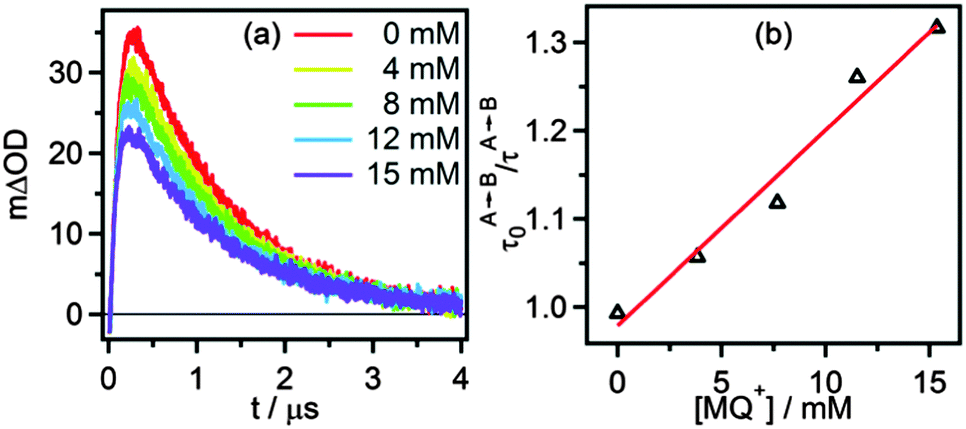
Classical Stern–Volmer luminescence quenching experiments could not be performed for determination of the kinetics of the initial PCET process (green arrow in Scheme 3) because the luminescence of [Ru(bpy)2pyimH]2+ is strongly quenched in presence of buffer molecules (kq = (8.3 ± 0.5) × 108 L mol−1 s−1 as described earlier). Therefore, we performed experiments at 5 mM buffer concentration for which we used a different spectroscopic observable to monitor the kinetics of the relevant PCET process: when measuring the transient absorption spectrum immediately after laser excitation, an additional band at 500 nm becomes observable. Based on the data in Fig. 4c and d, this band can be attributed unambiguously to [RuII(bpy)2pyim]+, i.e., to the deprotonated RuII complex in the electronic ground state. This species accumulates in a side-reaction to PCET as illustrated in the lower line, right side of Scheme 3; a subset of all excited complexes undergoes PCET chemistry to the photoproducts shown in the top right corner of Scheme 3 whereas another subsets merely acts as a photoacid (lower line, right side). Thus, the intensity of the transient absorption signal at 500 nm is a measure for the amount of ruthenium complexes that have been photoexcited but that have not undergone PCET chemistry. The temporal evolution of the transient signal at 500 nm shows classical A → B → C reaction kinetics (Fig. 6a), with species A corresponding to *[RuII(bpy)2pyimH]2+, species B being [RuII(bpy)2pyim]+, and species C corresponding to the protonated ground state as shown in Scheme 3. In absence of MQ+ the signal at 500 nm rises with τA→B = (105 ± 10) ns and decays with τB→C = (1.0 ± 0.1) μs. With increasing MQ+ concentration τA→B decreased, τB→C remained constant and the maximum intensity of the signal decreased. A plot of τA→B0/τA→Bvs. [MQ+] is shown in Fig. 6b. A linear regression fit yields a pseudo-Stern–Volmer constant (KSV′) of (22 ± 2) L mol−1 and a quenching constant (kq′) of (2.1 ± 0.4) × 108 L mol−1 s−1. This PCET-quenching constant is on the same order of magnitude as the *PT1-quenching constant and therefore both processes are competitive.
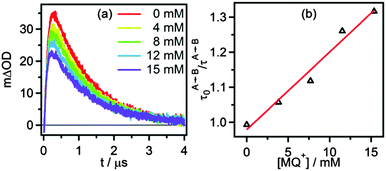 |
| | Fig. 6 (a) Change in optical density at 500 nm as a function of time after 532 nm excitation of 40 μM [Ru(bpy)2pyimH]2+ in 1:1 (v:v) CH3CN/H2O containing 5 mM acetate buffer and increasing concentrations of MQ+. (b) Pseudo Stern–Volmer plot based on the kinetics for step A → B with constant B → C lifetime, see text for details. | |
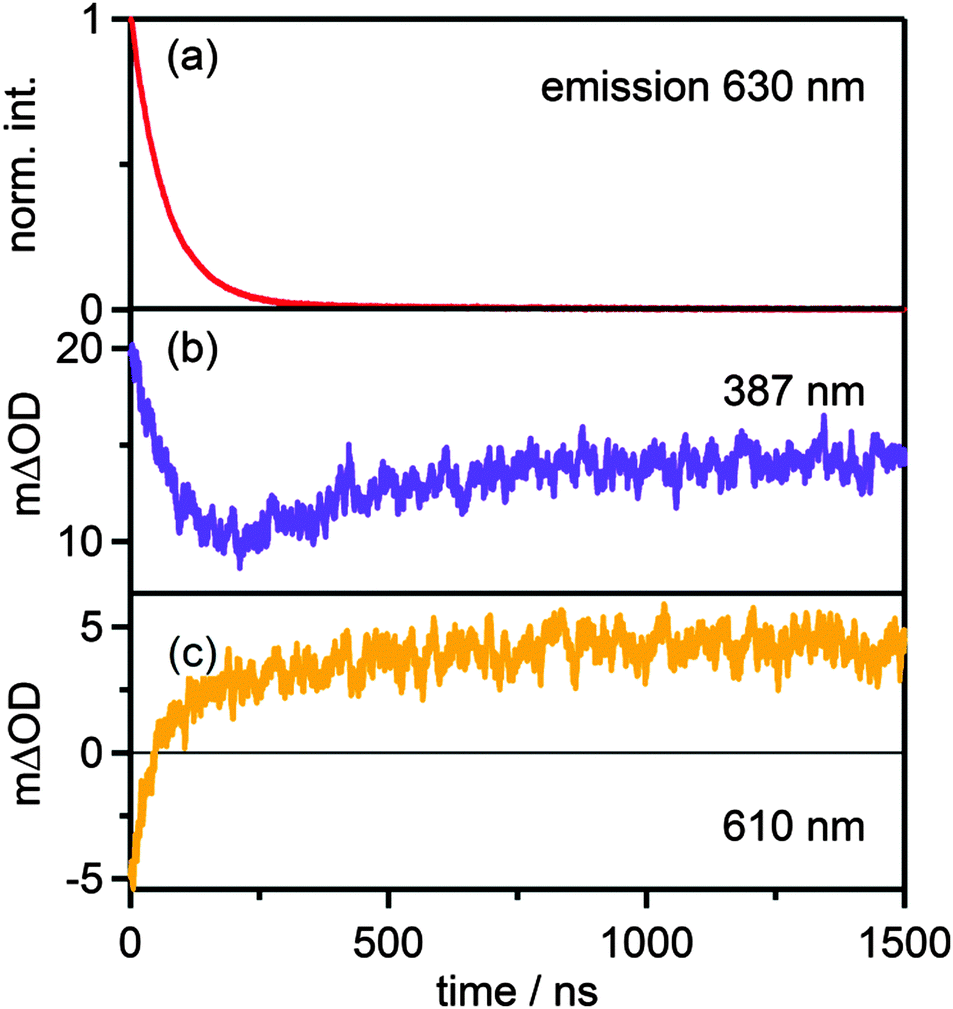
In buffered solution at pH 6.3 the final PCET photoproducts (i.e. [RuIII(bpy)2pyim]2+ and HMQ˙+) are formed within the first microsecond at buffer concentrations between 5 and 50 mM (Fig. 5c). When monitoring the transient absorption signals at 387 nm and 610 nm (Fig. 7b and c) it becomes evident that the formation of HMQ˙+ occurs more slowly than deactivation of the 3MLCT excited state of the [RuII(bpy)2pyimH]2+ complex (Fig. 7a). Based on the emission decay at 630 nm, the 3MLCT lifetime under these conditions is (70 ± 7) ns. Biexponential fits to the transients at 387 nm and 610 nm yield time constants of (70 ± 7) ns and (250 ± 25) ns with different signs of amplitude, corresponding to the 3MLCT lifetime and the time constant for formation of HMQ˙+, respectively. Thus, photoexcited [RuII(bpy)2pyimH]2+ disappears more rapidly than HMQ˙+ forms, and this can be explained by rapid PCET (green arrow in Scheme 3) followed by slow PT2 (red arrow in Scheme 3). By increasing the buffer concentration the kinetics of these processes is accelerated as described in the ESI† (Fig. S12).
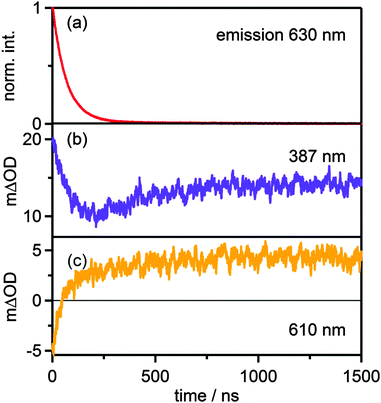 |
| | Fig. 7 (a) Decay of the emission signal at 630 nm and temporal evolution of the transient absorption signal at 387 nm (b) and at 610 nm (c) in the reaction of 40 μM [Ru(bpy)2pyimH]2+ with 15 mM MQ+ in 1:1 (v:v) CH3CN/H2O at pH 6.3 with 5 mM acetate buffer. | |
In absence of buffer the reaction kinetics are different. The absence of buffer base leaves MQ+ and water molecules as potential proton acceptors. The luminescence quenching experiment performed with acetate buffer (Fig. S7, ESI†) clearly shows that buffer base is a far better proton acceptor than water vis-à-vis photoexcited [RuII(bpy)2pyimH]2+. Transient absorption spectra recorded in 1:1 (v:v) CH3CN/H2O in absence of buffer are shown in Fig. 5d. The spectrum recorded without time delay (orange trace in Fig. 5d) is compatible with the formation of neutral MQ˙ radical as a primary photoproduct, exhibiting a characteristic broad absorption centered on 545 nm. After 5 μs the population of MQ˙ has decreased and HMQ˙+ can be detected (brown trace in Fig. 5d). Thus, the overall photochemistry is the same as in presence of buffer (PCET followed by PT2), but the kinetics are much different. The time constant for protonation of MQ˙ is (2.5 ± 0.3) μs based on the temporal evolution of the transient signal at 610 nm (ESI,† Fig. S13a) which is a factor of 10 slower than in the presence of 5 mM acetate buffer. Stern–Volmer luminescence quenching studies yielded kq = (3.4 ± 0.5) × 108 L mol−1 s−1 for the initial excited-state quenching process. When going from CH3CN/H2O to CH3CN/D2O, the protonation of MQ˙ (PT2) is slowed down by a factor of 2.6, and the initial excited state quenching process becomes a factor of (1.4 ± 0.6) slower (Table 3). In principle, an H/D kinetic isotope effect of 1.4 would be compatible with concerted electron–proton transfer (CPET), but definitive assignment of the PCET step in Scheme 3 (green arrow) either to concerted or consecutive electron and proton transfer steps is currently not possible.
Table 3 Stern–Volmer constants (KSV) and bimolecular luminescence quenching constants (kq) for *[Ru(bpy)2pyimH]2+ in presence of MQ+ in deaerated 1:1 (v:v) CH3CN/H2O at different pH values in absence and presence of buffer
| Conditions |
K
SV [L mol−1] |
k
q [108 L mol−1 s−1] |
Process |
| Buffered pH 2 |
143 ± 13 |
6.8 ± 0.1 |
ET |
| Unbuffered pH 13 |
124 ± 12 |
20.7 ± 0.3 |
ET |
| Buffered pH 6.3 |
34 ± 2 |
5.7 ± 0.3 |
PCET |
| Unbuffered H2O pH 7.3 |
70 ± 5 |
3.4 ± 0.5 |
PCET |
| Unbuffered D2O pD 7.3 |
58 ± 6 |
2.5 ± 0.8 |
PCET |
3 Summary and conclusion
The thermodynamic properties of [Ru(bpy)2pyimH]2+ in its electronic ground state and in the long-lived 3MLCT excited state in 1:1 (v:v) CH3CN/H2O were explored in detail. In the ground state the formal BDFE of the N–H bond is in the range of primary and secondary amines ((91 ± 1) kcal mol−1),2,47,48 but upon excitation with a visible photon the BDFE drops by roughly 50 kcal mol−1 to only (43 ± 5) kcal mol−1. Thus, photoexcitation leads to a formal N–H BDFE in the range of metal hydride complexes which are used as hydrogenation catalysts in their electronic ground states.4,38 Transient absorption spectroscopy demonstrates that photoexcited [Ru(bpy)2pyimH]2+ and (N-methyl-4,4′-bipyridinium, MQ+) undergo a formal HAT reaction, thereby confirming the finding of a very low formal N–H BDFE in the ruthenium complex; in HMQ˙+ the N–H BDFE is (53 ± 1) kcal mol−1. Mechanistically, formal HAT between these two reactants is found to proceed via a sequence of PCET and proton transfer reaction steps involving buffer or solvent molecules. More generally, our study demonstrates that aromatic imines can be reduced from the excited state of [Ru(bpy)2pyimH]2+.
Acknowledgements
This work was supported by the Swiss National Science Foundation through grant number 200021_146231/1.
Notes and references
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. Granville and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- S. Hammes-Schiffer, J. Am. Chem. Soc., 2015, 137, 8860–8871 CrossRef CAS PubMed.
- M. Bourrez, R. Steinmetz, S. Ott, F. Gloaguen and L. Hammarström, Nat. Chem., 2015, 7, 140–145 CrossRef CAS PubMed.
- J. M. Mayer, D. A. Hrovat, J. L. Thomas and W. T. Borden, J. Am. Chem. Soc., 2002, 124, 11142–11147 CrossRef CAS PubMed.
- T. Irebo, S. Y. Reece, M. Sjödin, D. G. Nocera and L. Hammarström, J. Am. Chem. Soc., 2007, 129, 15462–15464 CrossRef CAS PubMed.
- T. Irebo, M.-T. Zhang, T. F. Markle, A. M. Scott and L. Hammarström, J. Am. Chem. Soc., 2012, 134, 16247–16254 CrossRef CAS PubMed.
- V. W. Manner, A. D. Lindsay, E. A. Mader, J. N. Harvey and J. M. Mayer, Chem. Sci., 2012, 3, 230 RSC.
- A. Wu, J. Masland, R. D. Swartz, W. Kaminsky and J. M. Mayer, Inorg. Chem., 2007, 46, 11190–11201 CrossRef CAS PubMed.
- J. P. Roth, J. C. Yoder, T. Won and J. M. Mayer, Science, 2001, 294, 2524–2526 CrossRef CAS PubMed.
- J. P. Roth and J. M. Mayer, Inorg. Chem., 1999, 38, 2760–2761 CrossRef CAS PubMed.
- O. S. Wenger, Coord. Chem. Rev., 2015, 46, 150–158 CrossRef.
- J. Nomrowski and O. S. Wenger, Inorg. Chem., 2015, 54, 3680–3687 CrossRef CAS PubMed.
- J. C. Freys, G. C. Bernardinelli and O. S. Wenger, Chem. Commun., 2008, 4267–4269 RSC.
- T. T. Eisenhart and J. L. Dempsey, J. Am. Chem. Soc., 2014, 136, 12221–12224 CrossRef CAS PubMed.
- H. G. Yayla and R. Knowles, Synlett, 2014, 2819–2826 CAS.
- K. T. Tarantino, P. Liu and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 10022–10025 CrossRef CAS PubMed.
- J. J. Concepcion, M. K. Brennaman, J. R. Deyton, N. V. Lebedeva, M. D. E. Forbes, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2007, 129, 6968–6969 CrossRef CAS PubMed.
- N. V. Lebedeva, R. D. Schmidt, J. J. Concepcion, M. K. Brennaman, I. N. Stanton, M. J. Therien, T. J. Meyer and M. D. E. Forbes, J. Phys. Chem. A, 2011, 115, 3346–3356 CrossRef CAS PubMed.
- Y. Deng, J. A. Roberts, S.-M. Peng, C. K. Chang and D. G. Nocera, Angew. Chem., Int. Ed., 1997, 36, 2124–2127 CrossRef CAS.
- J. P. Kirby, J. A. Roberts and D. G. Nocera, J. Am. Chem. Soc., 1997, 119, 9230–9236 CrossRef CAS.
- J. Rosenthal, J. M. Hodgkiss, E. R. Young and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 10474–10483 CrossRef CAS PubMed.
- E. R. Young, J. Rosenthal, J. M. Hodgkiss and D. G. Nocera, J. Am. Chem. Soc., 2009, 131, 7678–7684 CrossRef CAS PubMed.
- C. Bronner and O. S. Wenger, J. Phys. Chem. Lett., 2012, 3, 70–74 CrossRef CAS.
- C. Bronner and O. S. Wenger, Inorg. Chem., 2012, 51, 8275–8283 CrossRef CAS PubMed.
- S. M. Barrett, C. L. Pitman, A. G. Walden and A. J. M. Miller, J. Am. Chem. Soc., 2014, 136, 14718–14721 CrossRef CAS PubMed.
-
J. F. Ireland and P. A. H. Wyatt, Adv. Phys. Org. Chem., Elsevier, 1976, vol. 12, pp. 131–221 Search PubMed.
- C. R. Waidmann, A. J. M. Miller, C.-W. A. Ng, M. L. Scheuermann, T. R. Porter, T. A. Tronic and J. M. Mayer, Energy Environ. Sci., 2012, 5, 7771–7780 CAS.
- M.-A. Haga, Inorg. Chim. Acta, 1983, 75, 29–35 CrossRef CAS.
- M.-A. Haga and A. Tsunemitsu, Inorg. Chim. Acta, 1989, 164, 137–142 CrossRef CAS.
- A. M. Bond and M. Haga, Inorg. Chem., 1986, 25, 4507–4514 CrossRef CAS.
- M. Haga, M. M. Ali, S. Koseki, K. Fujimoto, A. Yoshimura, K. Nozaki, T. Ohno, K. Nakajima and D. J. Stufkens, Inorg. Chem., 1996, 35, 3335–3347 CrossRef CAS.
- M.-A. Haga, Inorg. Chim. Acta, 1980, 45, L183–L184 CrossRef CAS.
- M. Haga, T. Ano, T. Ishizaki, K. Kano, K. Nozaki and T. Ohno, J. Chem. Soc., Dalton Trans., 1994, 263 RSC.
- X. Xiaoming, M. Haga, T. Matsumura-Inoue, Y. Ru, A. W. Addison and K. Kano, J. Chem. Soc., Dalton Trans., 1993, 2477 RSC.
- K. M. Lancaster, J. B. Gerken, A. C. Durrell, J. H. Palmer and H. B. Gray, Coord. Chem. Rev., 2010, 254, 1803–1811 CrossRef CAS.
- A. Wu and J. M. Mayer, J. Am. Chem. Soc., 2008, 130, 14745–14754 CrossRef CAS PubMed.
- J. Choi, M. E. Pulling, D. M. Smith and J. R. Norton, J. Am. Chem. Soc., 2008, 130, 4250–4252 CrossRef CAS PubMed.
- A. J. Morris, R. T. McGibbon and A. B. Bocarsly, ChemSusChem, 2011, 4, 191–196 CrossRef CAS PubMed.
- J. Chen, K. Wu, B. Rudshteyn, Y. Jia, W. Ding, Z.-X. Xie, V. S. Batista and T. Lian, J. Am. Chem. Soc., 2016, 138, 884–892 CrossRef CAS PubMed.
- D. J. Boston, C. Xu, D. W. Armstrong and F. M. MacDonnell, J. Am. Chem. Soc., 2013, 135, 16252–16255 CrossRef CAS PubMed.
- A. A. Vlcek, E. S. Dodsworth, W. J. Pietro and A. B. P. Lever, Inorg. Chem., 1995, 34, 1906–1913 CrossRef CAS.
- A. Yoshimura, M. Z. Hoffman and H. Sun, J. Photochem. Photobiol., A, 1993, 70, 29–33 CrossRef CAS.
- L. M. Tolbert and K. M. Solntsev, Acc. Chem. Res., 2002, 35, 19–27 CrossRef CAS PubMed.
- J. W. Park, Y. Kim and J. M. Kim, Bull. Korean Chem. Soc., 1993, 174–175 CAS.
- A. Harriman, G. R. Millward, P. Neta, M. C. Richoux and J. M. Thomas, J. Phys. Chem., 1988, 92, 1286–1290 CrossRef CAS.
- F. G. Bordwell, X. Zhang and J. P. Cheng, J. Org. Chem., 1991, 56, 3216–3219 CrossRef CAS.
- F. G. Bordwell, X. M. Zhang and J. P. Cheng, J. Org. Chem., 1993, 58, 6410–6416 CrossRef CAS.
- D. P. Rillema, R. Sahai, P. Matthews, A. K. Edwards, R. J. Shaver and L. Morgan, Inorg. Chem., 1990, 29, 167 CrossRef CAS.
- C. J. Gagliardi, B. C. Westlake, C. A. Kent, J. J. Paul, J. M. Papanikolas and T. J. Meyer, Coord. Chem. Rev., 2010, 254, 2459–2471 CrossRef CAS.
- P. S. Braterman and J. I. Song, J. Org. Chem., 1991, 56, 4678–4682 CrossRef CAS.
-
D. M. Roundhill, Photochemistry and Photophysics of Metal Complexes, Springer US, Boston, MA, 1994 Search PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: additional optical spectroscopic and electro-chemical data. See DOI: 10.1039/c6cp00437g |
|
| This journal is © the Owner Societies 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence