
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAromaticity of the doubly charged [8]circulenes†
Gleb V.
Baryshnikov
*ab,
Rashid R.
Valiev
cd,
Nataliya N.
Karaush
b,
Dage
Sundholm
e and
Boris F.
Minaev
bf
aDivision of Theoretical Chemistry and Biology, School of Biotechnology, KTH Royal Institute of Technology, 10691 Stockholm, Sweden. E-mail: glebchem@rambler.ru; Fax: +38 0472 354463; Tel: +38 0472 376576
bDepartment of Organic Chemistry, Bohdan Khmelnitsky National University, blvd. Shevchenko 81, 18031, Cherkasy, Ukraine
cTomsk Polytechnic University, 30 Lenin Avenue, 634050, Tomsk, Russia
dTomsk State University, 36 Lenin Avenue, 634050, Tomsk, Russia
eDepartment of Chemistry, University of Helsinki, P.O. Box 55 (A.I. Virtanens plats 1), Helsinki Fin-00014, Finland
fKey Laboratory of Engineering Plastics and Beijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Science, 100190 Beijing, China
First published on 26th February 2016
Abstract
Magnetically induced current densities and current pathways have been calculated for a series of fully annelated dicationic and dianionic tetraphenylenes, which are also named [8]circulenes. The gauge including magnetically induced current (GIMIC) method has been employed for calculating the current density susceptibilities. The aromatic character and current pathways are deduced from the calculated current density susceptibilities showing that the neutral [8]circulenes have two concentric pathways with aromatic and antiaromatic character, respectively. The inner octatetraene core (the hub) is found to sustain a paratropic (antiaromatic) ring current, whereas the ring current along the outer part of the macrocycle (the rim) is diatropic (aromatic). The neutral [8]circulenes can be considered nonaromatic, because the sum of the ring-current strengths of the hub and the rim almost vanishes. The aromatic character of the doubly charged [8]circulenes is completely different: the dianionic [8]circulenes and the OC-, CH-, CH2-, SiH-, GeH-, SiH2-, and GeH2-containing dicationic species sustain net diatropic ring currents i.e., they are aromatic, whereas the O-, S-, Se-, NH-, PH- and AsH-containing dicationic [8]circulenes are strongly antiaromatic. The present study also shows that GIMIC calculations on the [8]circulenes provide more accurate information about the aromatic character than that obtained using local indices such as nuclear-independent chemical shifts (NICSs) and 1H NMR chemical shifts.
1. Introduction
Aromaticity is one of the most important and controversial concepts in chemistry characterizing the physical properties and chemical reactivity of chemical compounds.1,2 Aromaticity is a complicated property due to its multidimensional character i.e., different means to assess the degree of aromaticity might lead to various results. In addition, aromaticity cannot be obtained directly from experiment, even though it is commonly used for understanding chemical properties.2–5 Many different approaches have been proposed for assessing and quantifying the degree of aromaticity of entire molecules or of parts of molecules.1,2,6–8 The most popular ways to determine the degree of aromaticity are structural, energetic and magnetic criteria. Nucleus-independent chemical shift (NICS)8 calculations represent the most widely used magnetic criterion for evaluating aromaticity. NICS values are the magnetic shieldings with an opposite sign calculated in selected points near the molecule under consideration. NICS values are local indicators of the strength of the ring current circling around the studied molecular ring; the NICS provides qualitative information about the aromaticity, antiaromaticity or nonaromaticity of monocyclic systems like benzene, cyclobutadiene, cyclooctatetraene, furan, pyrrole, etc.8 However, the NICS approach cannot be employed for assessing the aromatic character of more complicated molecules,9,10 which has previously been discussed in a number of studies of the aromatic character of polycyclic aromatic hydrocarbons,11,12 porphyrins,13 all-metal aromatic compounds14–18 and hydrogen-bonded molecular clusters.19A more direct approach to evaluate the aromatic or antiaromatic nature of molecules as a whole is to use the gauge-including magnetically induced current (GIMIC) method.20–26 The GIMIC approach employs gauge-including atomic orbitals (GIAOs) in explicit calculations of the magnetically induced current density susceptibilities, current strengths and current pathways in molecules providing detailed information about electronic delocalization properties. Some interesting examples of GIMIC applications are current density studies on fullerene C60 and its multicharged C6010+ ion,27 [n]cycloparaphenylenes,28 nano-sized hydrocarbons,29etc.
The aromaticity concept has recently been applied to a wide variety of conjugated polyheterocyclic compounds in order to understand their chemical properties.30–34 Among them, hetero[8]circulenes possess a prominent place due to their peculiar electronic structure,35–42 symmetry,40,43,44 self-assembling possibilities45–49 and specific aromatic properties.50–53 Moreover, they also show potential as nanophotonic and microelectronic platforms for the fabrication of organic light-emitting diodes (OLEDs).54–57 The aromatic nature of this class of compounds has been extensively studied using the structural, energetic, electronic and magnetic criteria.40–44,51–53 Recently,53 we reported computational studies of the aromatic properties for a series of neutral58 hetero[8]circulenes 1–7 and their hydrocarbon analogues 8 and 9 shown in Fig. 1.
 | ||
| Fig. 1 The molecular structures of the [8]circulenes 1–13. | ||
In our previous study, we found that the [8]circulenes 1–9 are almost nonaromatic molecules (except compound 8), because the inner paratropic ring current is only slightly larger than the diatropic ring current flowing mainly along the outer part of the molecule leading to a practically vanishing total ring-current strength.53 The aromaticity of the novel Si- and Ge-containing [8]circulenes 10–13 (Fig. 1) is investigated for the first time in this work.
In the present work, GIMIC calculations are employed for characterizing the aromaticity of the doubly charged [8]circulenes and the obtained results are compared with the degree of aromaticity deduced from NICS and proton nuclear magnetic resonance (1H NMR) calculations.
2. Computational methods
The ground-state geometries of the examined species were optimized at the density functional theory (DFT) level by employing the B3LYP59,60 functional and the 6-311++G(d,p) basis set.61 The GAUSSIAN 09 package was used.62 Hessian calculations indicate that the obtained geometries of the studied species correspond to genuine minima on the potential energy surfaces at the employed level of theory.NICS indices8,63–66 were calculated at the same level of theory by employing gauge-including atomic orbitals (GIAOs).67,68 The NICS values were calculated at ghost atoms located at the center of each ring (denoted as NICS(0)) and at a distance of 1 Å above the ring center (NICS(1)). The NICS(1) values were not calculated for the non-planar species. NICS(1) indices are commonly used for better accounting of the magnetic π-electron effects65,66 while NICS(0) indices are considered for evaluating the sum of the σ- and π-electron delocalization effects.
Negative NICS values indicate that the molecule sustains a diatropic ring current, i.e., the ring is aromatic; positive NICS values correspond to paratropic ring currents, i.e., the ring is antiaromatic.69 When the NICS value is close to zero, the ring is nonaromatic.
Magnetically induced current density susceptibilities were calculated using the GIMIC method.20–24,70,71 Nuclear magnetic resonance (NMR) shielding calculations that are needed for the GIMIC calculations of the current-density susceptibilities and the ring-current susceptibilities, which are also called current densities and ring-current strengths (in nA T−1), were performed using the Turbomole software package.72 The calculated ring-current strengths can be used as an aromaticity index. For example, the current strength of the aromatic benzene molecule is 11.8 nA T−1 at the B3LYP/TZVP level as compared with −19.9 nA T−1 for the antiaromatic cyclobutadiene and 0.2 nA T−1 for the nonaromatic cyclohexane.22 The current density plots have been generated using the JMOL package.73
The calculations were performed at the PDC supercomputer of the Royal Institute of Technology (Stockholm), at the CSC supercomputer of the Finnish IT Center for Science (Finland), and at the Tomsk State University (SKIF, Russia).
3. Results and discussion
3.1. Molecular structures
Bond lengths of the optimized structures for the doubly charged [8]circulenes 1–13 and for the novel neutral species 10–13 are given in Table 1. The studied O-, N-, CO- and CH-containing neutral [8]circulenes are planar with a strong bond-length alternation in the inner cyclooctatetraene (COT) core. It should be noted that the O-, N-and CO-containing circulenes 1, 2, and 5 possess the highest possible D4h symmetry point group, while the CH-containing compound 8 was designed with the D2h symmetry in accordance with the experimental X-ray data for this species.74,75 The more symmetrical form of compound 8 (within D4h symmetry) also has a minimum on the potential energy surface. In the framework of the DFT/B3LYP/6-311++G(d,p) approach both D2h and D4h species are degenerate by the total energy values, whereas the spin–flip (SF) DFT approach indicated that the D4h form has a higher energy (34.7 kJ mol−1 at the SF-TD-BHHLYP/6-311G(d,p) level of approximation) than the D2h model.74 Moreover, Tobe et al. found that the D2h structure possesses the almost closed-shell electronic structure (with a very small diradical admixture) while the D4h structure has the tetraradical character. On this basis, Tobe et al. suggested that the lower symmetry D2h structure may equilibrate by valence tautomerization via the D4h isomer as a transition state.74,75| Circulene | Symmetry |
r
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (CClong) (CClong)
|
r
2(CCshort)
|
R
3(CCradial)
|
r
4(CX)
|
r
5(CC side)
|
r
6(CC top)
|
r
7(CC side)* |
|---|---|---|---|---|---|---|---|---|
| X denotes heteroatoms. The bond lengths of the outer side of the five-membered hydrocarbon ring in molecules 2, 8, and 9 are indicated with an asterisk (*). | ||||||||
| 12+ | D 4h | 1.435 | 1.384 | 1.416 | 1.369 | 1.384 | 1.425 | — |
| 12− | D 4h | 1.421 | 1.406 | 1.418 | 1.399 | 1.379 | 1.447 | — |
| 22+ | D 4h | 1.444 | 1.444 | 1.436 | 1.200 | 1.369 | 1.421 | 1.490 |
| 22− | D 4h | 1.489 | 1.387 | 1.435 | 1.247 | 1.399 | 1.385 | 1.461 |
| 32+ | D 2d | 1.485 | 1.409 | 1.445 | 1.729 | 1.384 | 1.392 | — |
| 32− | D 2d | 1.467 | 1.440 | 1.448 | 1.762 | 1.374 | 1.410 | — |
| 42+ | D 2d | 1.484 | 1.407 | 1.443 | 1.879 | 1.382 | 1.399 | — |
| 42− | D 2d | 1.469 | 1.437 | 1.446 | 1.909 | 1.372 | 1.413 | — |
| 52+ | D 4h | 1.473 | 1.368 | 1.429 | 1.375 | 1.405 | 1.385 | — |
| 52− | C s | 1.430 | 1.421 | 1.426 | 1.401 | 1.392 | 1.415 | — |
| 62+ | D 2d | 1.466 | 1.418 | 1.441 | 1.823 | 1.377 | 1.412 | — |
| 62− | D 2d | 1.461 | 1.444 | 1.448 | 1.823 | 1.373 | 1.417 | — |
| 72+ | D 2d | 1.467 | 1.421 | 1.438 | 1.959 | 1.373 | 1.417 | — |
| 72− | D 2d | 1.464 | 1.443 | 1.446 | 1.947 | 1.371 | 1.420 | — |
| 8 | D 2h | 1.483 | 1.384 | 1.435 | — | 1.389 | 1.400 | 1.450 |
| 82+ | C 2 | 1.479 | 1.360 | 1.455 | — | 1.409 | 1.384 | 1.409 |
| 82− | C 2h | 1.491 | 1.388 | 1.457 | — | 1.418 | 1.378 | 1.407 |
| 9 | D 2d | 1.488 | 1.412 | 1.418 | — | 1.386 | 1.389 | 1.498 |
| 92+ | D 2d | 1.462 | 1.432 | 1.436 | — | 1.373 | 1.409 | 1.490 |
| 92− | D 2d | 1.448 | 1.448 | 1.441 | — | 1.372 | 1.427 | 1.506 |
| 10 | C 2 |
1.499
1.517 |
1.391
1.403 |
1.442
1.459 1.466 |
1.801, 1.760
1.854, 1.812 |
1.396, 1.394
1.416, 1.419 |
1.394
1.365 |
— |
| 102+ | D 2d | 1.513 | 1.390 | 1.454 | 1.701, 1.791 | 1.401 | 1.390 | |
| 102− | C 2 | 1.516 |
1.410
1.408 |
1.441
1.444 |
1.855, 1.859 |
1.396
1.401 |
1.383
1.390 |
— |
| 11 | C 2 |
1.499
1.520 |
1.401
1.400 |
1.440
1.438 1.448 1.439 |
1.859, 1.867
1.964, 1.959 |
1.400, 1.396
1.402, 1.405 |
1.381
1.378 |
— |
| 112+ | D 2d | 1.513 | 1.394 | 1.447 | 1.873 | 1.398 | 1.382 | — |
| 112− | C 2 | 1.515 |
1.409
1.411 |
1.435
1.436 |
1.957
1.963 |
1.392
1.396 |
1.387
1.386 |
— |
| 12 | D 2d | 1.514 | 1.413 | 1.429 | 1.863 | 1.388 | 1.389 | — |
| 122+ | D 2d | 1.457 | 1.434 | 1.444 | 1.887 | 1.373 | 1.422 | — |
| 122− | D 2d | 1.507 | 1.407 | 1.446 | 1.837 | 1.405 | 1.380 | — |
| 13 | D 2d | 1.513 | 1.415 | 1.425 | 1.945 | 1.387 | 1.391 | — |
| 132+ | D 2d | 1.460 | 1.434 | 1.440 | 1.967 | 1.372 | 1.423 | — |
| 132− | D 2d | 1.463 | 1.447 | 1.451 | 1.935 | 1.374 | 1.421 | — |
The other [8]circulenes (except 9) possess nonplanar saddle-shaped structures due to the sectorial excess of the macrocycle in accordance with the Wynberg–Dopper sectorial model.76 The Wynberg–Dopper sectorial model is described in detail in paragraph II.1 of ref. 40. The pure hydrocarbon compound 9 has a planar tetraphenylene skeleton except for the outer nonplanar CH2 moieties, whose hydrogens break the ideal flat structure.
The doubly charged [8]circulenes retain the molecular shape of the neutral molecules except the imidogen-containing compound 5, whose N–H moieties deviate out-of-plane for the dianion. The doubly charged tetra-hetero[8]circulenes 3, 4, 6, and 7 are very nonplanar due to the increase in the C–X distances as compared with the same bond lengths of the neutral molecules. The dicationic circulene 22+ and dianionic hydrocarbon-type [8]circulene 92− lack bond-length alternation in the inner eight-membered ring, whose C–C bond lengths of 1.444 Å and 1.448 Å are practically equal suggesting that the aromatic nature of the COT core is similar to that of octathia[8]circulene and its dianion.52
The geometry optimizations at the DFT level yielded nonplanar molecular structures for the novel hetero[8]circulenes 10–13 (Fig. 1). The molecular structures belong to the C2 point group for circulenes 10 and 11 and to D2d for 12 and 13. The reason for the low symmetry of the neutral circulenes 10 and 11 and for their dianions is the two sp3-hybridized silicon and germanium atoms. Similar distortions of the molecular structure upon reduction have previously been reported for tetraza[8]circulene 5.52 The doubly oxidized circulene dications 102+ and 112+ have high symmetry, belonging to the D2d point group.
The novel [8]circulenes 10–13 have a strong bond-length alternation of the C–C bonds in the inner COT core with variations of 0.098–0.120 Å in the bond lengths, which can be compared with 0.136 Å for the strongly antiaromatic free COT that also possesses D2d symmetry.40 The bond lengths are given in Table 1. The bond-length alternation of the COT bonds in the inner COT core of dianionic and dicationic molecules does not exceed 0.026 Å, except for 122−, whose bond-length alternation is 0.1 Å. The small bond-length alternation indicates that the doubly charged molecules are more aromatic than the neutral ones. However, it is well known that equal bond lengths cannot be used as the only criterion for aromaticity, because some molecules with almost equal bond lengths are not aromatic, as discussed in ref. 65. Other criteria characterizing the aromaticity such as chemical reactivity and energetic criteria also have some limitations, whereas magnetic properties such as 1H NMR chemical shifts, magnetic susceptibility exaltation, and NICS and GIMIC indices are perhaps more reliable than the classical criteria. They are therefore the most often used means for characterizing aromatic and antiaromatic compounds. In the next section, we discuss aromatic characters obtained using GIMIC, NICS and 1H NMR calculations.
3.2. GIMIC studies of the neutral [8]circulenes 8–13
The signed modulus plots of the calculated magnetically induced current densities, current pathways and current strengths for the neutral [8]circulenes 8–13 are shown in Fig. 2. Current density data are given for the neutral and the corresponding charged molecules. The aromatic character of the neutral [8]circulenes 8 and 9 has been previously discussed in ref. 52 and 53. The calculated current strengths circling around the macrocycle of the neutral and doubly charged tetraphenylenes 1–13 are summarized in Table 2, which also reports how the net diatropic (aromatic) and paratropic (antiaromatic) ring currents of the studied molecules depend on the molecular charge.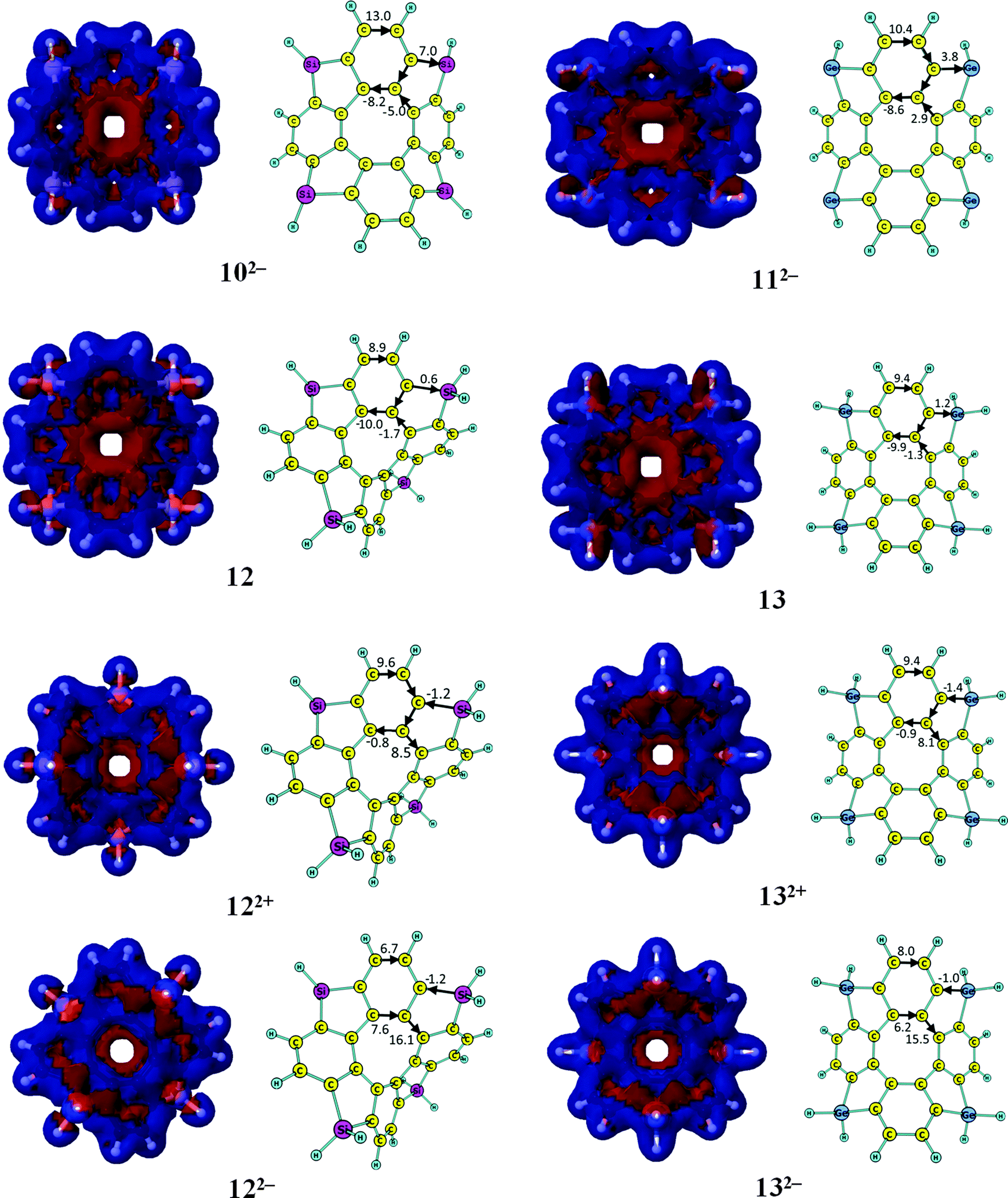 | ||
| Fig. 2 The signed modulus of the magnetically induced current densities, current strengths and current pathways for the neutral and doubly charged [8]circulenes 8–13. Paratropic current densities are shown in red and the diatropic ones in blue. | ||
| Species | Current | Total current strength, nA T−1 | Conclusion | Source | ||
|---|---|---|---|---|---|---|
| rim-system | hub-system | Balance | ||||
| 1 | d | p | p > d | −2.1 | Weakly antiaromatic | Ref. 52 |
| 12+ | p | d | p > d | −55 | Antiaromatic | This work |
| 12− | d | d | 2d | 22 | Aromatic | This work |
| 2 | d | p | p > d | −4 | Weakly antiaromatic | Ref. 51 |
| 22+ | d | d | 2d | 12 | Aromatic | This work |
| 22− | d | p | d > p | 7.8 | Aromatic | This work |
| 3 | d | p | d ≈ p | −1 | Almost nonaromatic | Ref. 51 |
| 32+ | p | d | p > d | −84 | Strongly antiaromatic | This work |
| 32− | d | d | 2d | 19.5 | Aromatic | This work |
| 4 | d | p | d ≈ p | −1 | Almost nonaromatic | Ref. 51 |
| 42+ | p | d | p > d | −74 | Strongly antiaromatic | This work |
| 42− | d | d | 2d | 17.3 | Aromatic | This work |
| 5 | d | p | d ≈ p | −0.5 | Almost nonaromatic | Ref. 52 (ESI) |
| 52+ | d | p | p > d | −3 | Weakly antiaromatic | This work |
| 52− | d | d | 2d | 21 | Aromatic | This work |
| 6 | d | p | p > d | −2.5 | Weakly antiaromatic | Ref. 51 |
| 62+ | p | d | p > d | −66 | Strongly antiaromatic | This work |
| 62− | d | d | 2d | 17 | Aromatic | This work |
| 7 | d | p | p > d | −4 | Weakly antiaromatic | Ref. 51 |
| 72+ | p | d | p > d | −8 | Antiaromatic | This work |
| 72− | d | d | 2d | 15.4 | Aromatic | This work |
| 8 | p | p | 2p | −40 | Strongly antiaromatic | Ref. 51 |
| 82+ | d | p | d > p | 19.6 | Aromatic | This work |
| 82− | d | p | d > p | 11.8 | Aromatic | This work |
| 9 | d | p | p > d | −2.7 | Weakly antiaromatic | Ref. 51 |
| 92+ | d | p | d > p | 105 | Strongly aromatic | This work |
| 92− | d | d | 2d | 21.9 | Aromatic | This work |
| 10 | p | p | 2p | −22.4 | Antiaromatic | This work |
| 102+ | d | p | d > p | 10.5 | Aromatic | This work |
| 102− | d | p | d > p | 4.0 | Weakly aromatic | This work |
| 11 | p | p | 2p | −8 | Antiaromatic | This work |
| 112+ | d | p | d > p | 11.0 | Aromatic | This work |
| 112− | d | p | d > p | 1.8 | Weakly aromatic | This work |
| 12 | d | p | d ≈ p | −0.9 | Almost nonaromatic | This work |
| 122+ | d | d | 2d | 8.5 | Aromatic | This work |
| 122− | d | d | 2d | 14.5 | Aromatic | This work |
| 13 | d | p | d ≈ p | −0.5 | Almost nonaromatic | This work |
| 132+ | d | d | 2d | 9.0 | Aromatic | This work |
| 132− | d | d | 2d | 14.0 | Aromatic | This work |
In Table 2, one sees that most of the neutral [8]circulenes are practically nonaromatic, since they do not sustain any strong net ring currents around the macroring. The net ring currents vanish for 1–7, and 9, because the diatropic ring current in the rim and the paratropic contribution to the ring current in the hub are almost equal. In 6, 7 and 9, the benzoic rings also sustain local ring currents. The [8]circulenes with CH, SiH and GeH moieties (8, 10 and 11) are antiaromatic sustaining net paratropic (i.e., negative) ring currents, whose strength decreases with increasing atomic number. The same trend is also obtained for the corresponding sp3-hybridized CH2-, SiH2- and GeH2-containing compounds (9, 12 and 13). However, they sustain very weak net ring currents of −2.7 to −0.5 nA T−1, implying that they are globally almost nonaromatic. In neutral 12 and 13, the four benzoic rings are locally aromatic, whereas very weak currents pass between the aromatic benzoic rings via the five-membered rings (Fig. 2).

3.3. GIMIC studies of the doubly charged [8]circulene anions
The aromatic nature of the doubly charged circulenes 1–13 differs completely from that of the neutral ones. The current densities, current pathways and strengths are given in Fig. 2 and 3. The ring-current strengths in Table 2 show that the dianionic species generally possess aromatic character as they sustain net diatropic ring currents. The net ring currents for the dianions are positive mainly because the ring current in the inner COT core changes direction when adding two electrons. However, the current pathways of the doubly charged anions are more complicated than that. In 12−, 32−, 42−, and 52−, the diatropic ring current passes the benzoic ring along the rim and the hub, whereas at the five-membered ring the rim current splits into an outer and inner branch of almost equal strengths. In 22−, a diatropic current flows along the rim passing the formal single bonds of the carbonyl group, whereas a weak paratropic current circles around the hub. In 62−, 72−, 92−, 122− and 132−, the outer moieties of the five-membered rings block the ring current, whereas the hub sustains a strong diatropic ring current that splits into an outer and inner route of about equal strengths at the benzoic rings. 82− sustains a diatropic ring current along the rim and a weaker paratropic one in the hub. In addition the five-membered rings sustain local ring currents. The net current for 82− is diatropic (i.e.82− is aromatic) in good agreement with the suggestions of Tobe et al. done on the basis of NICS(1) calculations.75102− and 112− sustain a diatropic ring current along the rim and a weak paratropic current in the hub. The benzoic rings also sustain local ring currents. For the studied molecules, the COT core gains aromatic character when adding two electrons leading to hub-aromaticity or to reduced hub-antiaromaticity and the ring current along the outer perimeter is in most cases diatropic, which we call rim-aromaticity. However, for some of the doubly charged anions, the outer moieties of the five-membered rings practically block the current forcing the main ring current to take the inner route. The sum of the strengths of hub- and rim-currents yields the net ring current that determines the global aromatic character.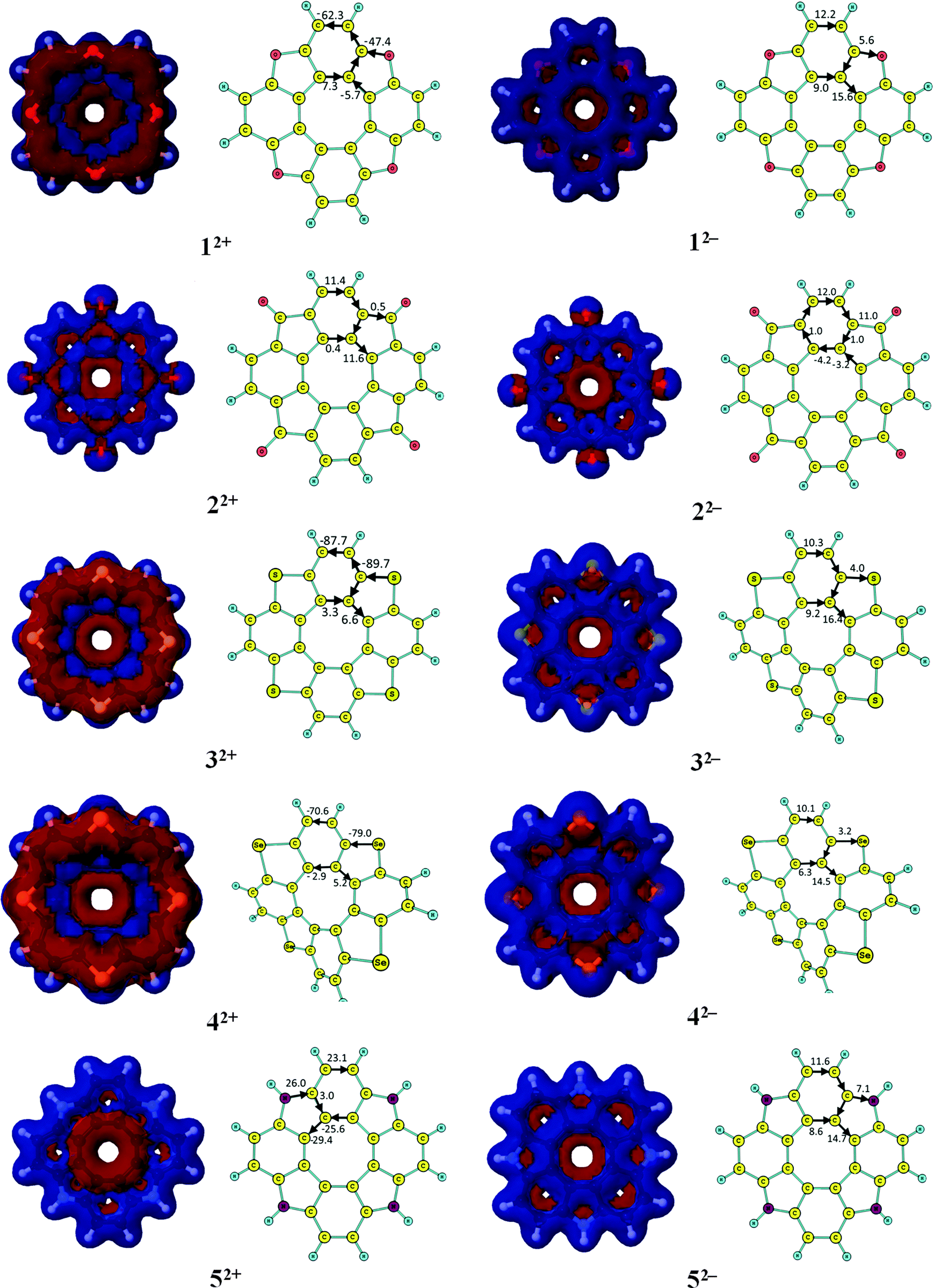
 | ||
| Fig. 3 The signed modulus of the magnetically induced current densities, current strengths and current pathways for doubly charged [8]circulenes 1–7. Paratropic current densities are shown in red and the diatropic ones in blue. | ||
3.4. GIMIC studies of the doubly charged [8]circulene cations
The current pathways of the dications are even more irregular than the ones for the dianions. The current densities, current pathways and strengths are shown in Fig. 2 and 3. The 12+ ion sustains a strong paratropic rim current. The benzoic rings sustain a local ring current and a weak paratropic current passes on the inside of the five-membered ring leading to strong antiaromaticity. The ring current in 22+ takes the outer route at the benzoic ring and the inner route at the five-membered rings. Almost no current passes along the hub at the benzoic rings and along the rim of the five-membered rings. 32+ sustains a strong paratropic rim current and a weak diatropic hub current. The five-membered rings sustain weak local paratropic ring currents. 42+ sustains a strong paratropic rim current that splits into a weak inner branch at the benzoic rings, whereas the five-membered rings sustain local paratropic ring currents. 52+ sustains a diatropic rim current and a paratropic hub current of about the same size leading to a practically nonaromatic molecule. The five-membered rings sustain a weak local diatropic ring current. 62+ is strongly antiaromatic with a strong paratropic rim current. Paratropic currents also pass along the inner route of the five-membered rings and the benzoic rings sustain local paratropic ring currents. 72+ sustains a paratropic rim current that splits at the five-membered rings into an outer and inner branch. The benzoic rings sustain weak local paratropic ring currents. 82+ is globally aromatic sustaining a strong diatropic rim current and a very weak paratropic hub current. It agrees well with the NICS(1) calculations from ref. 75. The five-membered rings are aromatic sustaining local diatropic ring currents. 92+ is strongly aromatic sustaining a net ring current of 105 nA T−1. The benzoic rings are also very aromatic with a local ring current of 76 nA T−1. At the five-membered rings the diatropic rim current is split into an outer and inner branch. Two third of the global diatropic current passes the outer sp3-hybridized CH2 moiety of the five-membered rings. 102+ and 112+ sustain global diatropic ring currents that split into a strong outer branch and a weak inner one at the five-membered rings. The benzoic rings sustain weak local diatropic ring currents. 122+ and 132+ sustain global diatropic ring currents that take the outer route at the benzoic rings and the inner pathway at the five-membered rings. Almost no net current passes the other bonds.3.5. NICS characterization
In this section, we show that the NICS criterion cannot be employed in aromaticity studies of the [8]circulenes due to the local properties of the NICS indices. It is almost impossible to deduce a correct analysis of the complex aromatic character of the [8]circulenes by performing NICS calculations. The main differences and similarities between the NICS- and GIMIC-based results are discussed below.The NICS values in Fig. 4 and 5 show that the dianionic [8]circulenes contain a local aromatic centre as the NICS values are negative. However, it is not apparent from the NICS calculations whether the dianionic [8]circulenes are globally aromatic or not. Only for 12−–52− and 82− the NICS indices predict that molecules are globally aromatic i.e., all the five-, six- and eight-membered rings possess local aromatic character as indicated by the negative NICS indices for each of them. NICS calculations suggest that the rest of the dianions consist of local antiaromatic five- or eight-membered cycles. However, when one accepts the idea that the paramagnetic contributions from these rings are significantly smaller than the diatropic ones, the NICS- and GIMIC-based results can be considered to agree with each other.
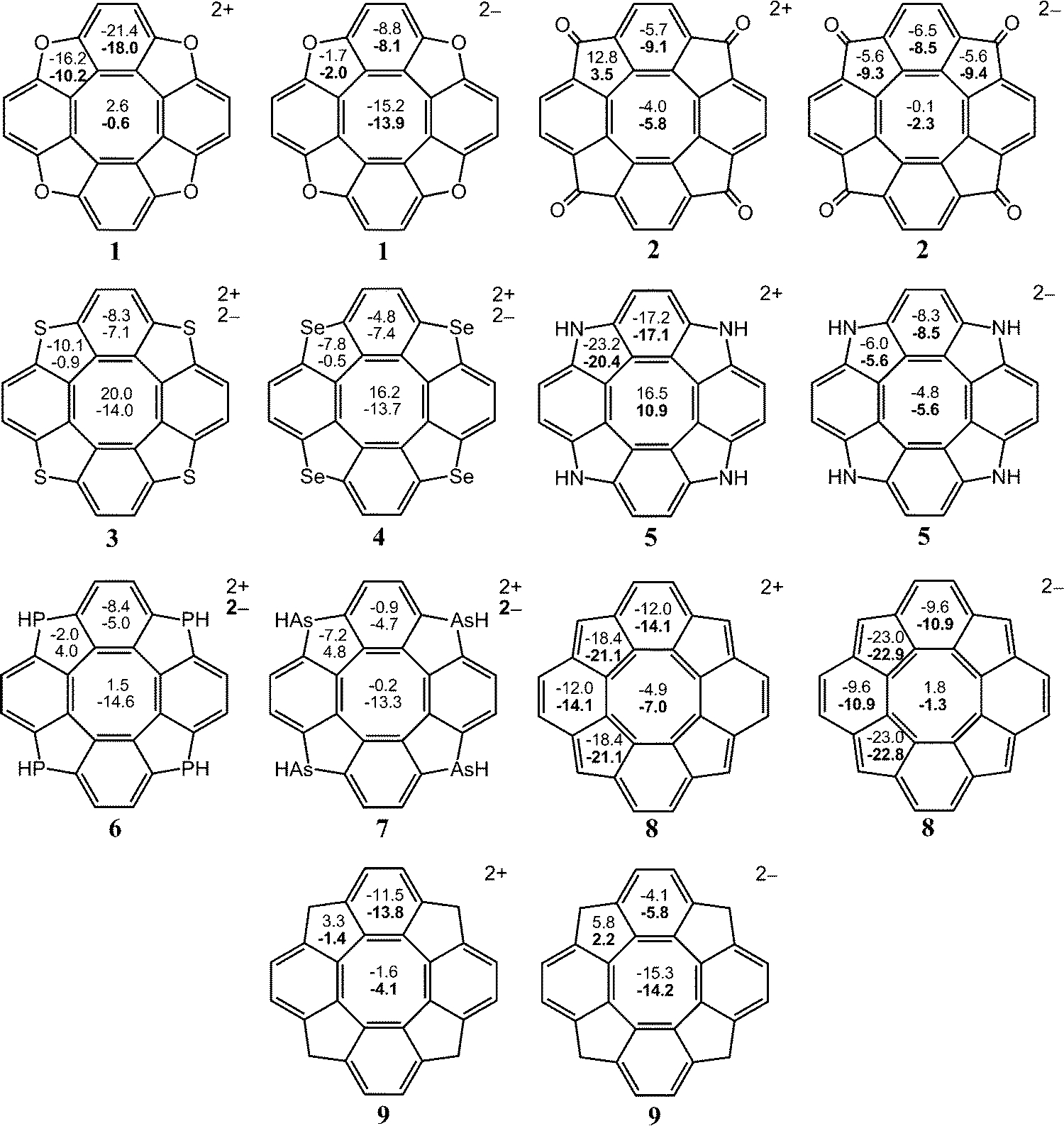 | ||
| Fig. 4 The NICS(0) and NICS(1) (in bold when available) indices for the doubly charged circulenes 1–9. | ||
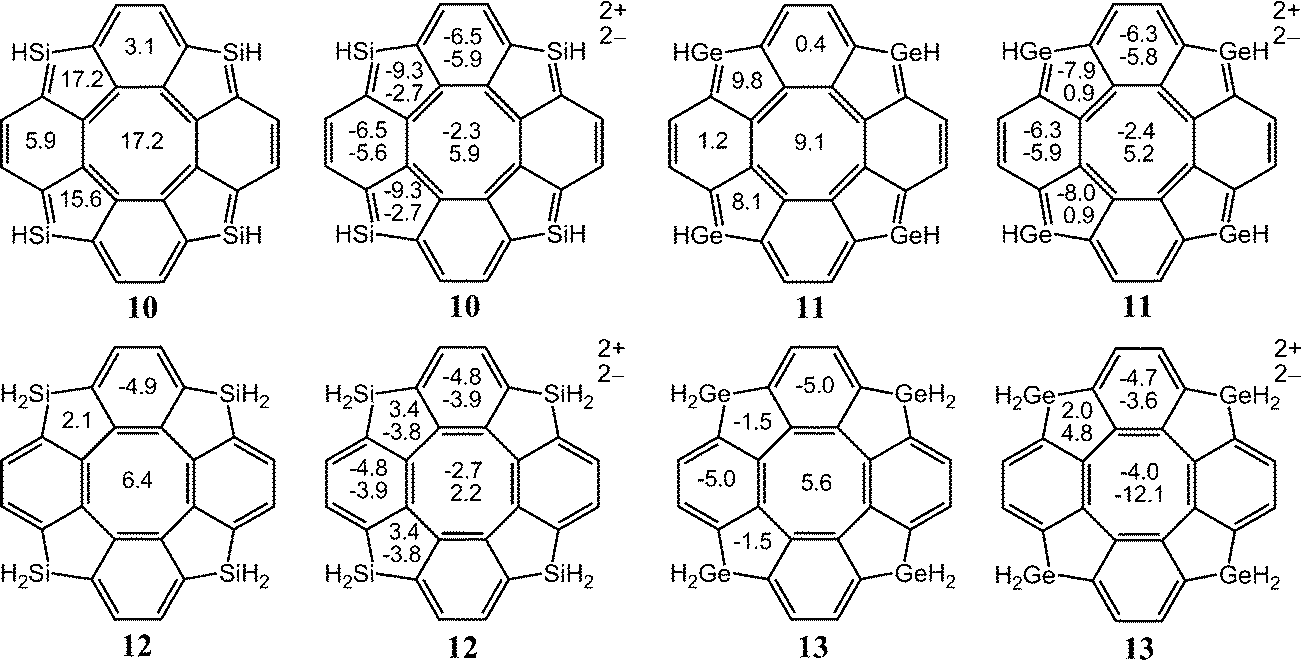 | ||
| Fig. 5 The NICS(0) indices for the neutral and doubly charged [8]circulenes 10–13. | ||
For the dicationic [8]circulenes, the NICS results agree with GIMIC data only for 82+, 102+, and 112+, which are predicted to be aromatic. The NICS indices for the other doubly oxidized species do not unambiguously provide the aromatic character as obtained in the GIMIC calculations. The NICS values have the opposite signs for the five-, six- and eight-membered rings, as shown in Fig. 4 and 5. Moreover, the analysis of the NICS indices for the dications 12+, 52+–72+ and the neutral 9, 12, 13 species suggests that they are aromatic rather than antiaromatic, which disagrees with the GIMIC results.
In summary, we conclude that the NICS criterion is rather unreliable for obtaining the correct description of the complex aromatic nature of the neutral and doubly charged hetero[8]circulenes due to the local character of NICS indices. However, using the GIMIC approach one can explicitly calculate the current strength and current pathways providing an accurate and detailed picture of the ring current pathways rendering it possible to determine the aromatic character of molecules with complex annelated rings.
3.6. 1H NMR chemical shift characterization
Spectroscopic measurements of proton NMR chemical shifts (1H NMR) are one of the most popular experimental methods to assess the aromaticity of polycyclic compounds. Diatropic (aromatic) ring currents deshield the protons at the outer edge of the molecule leading downfield shift of the 1H NMR signal relative to the tetramethylsilane (TMS) reference. The general aromatic chemical shift region for the protons directly connected to the aromatic framework varies in the wide range of 6.5–9 ppm and the chemical shift strongly depends on the type and position of heteroatom in the carbon skeleton.Comparisons of the calculated isotropic magnetic shielding constants with the 1H NMR shielding constant for TMS calculated at the same level of theory yield the calculated values for the 1H NMR chemical shifts in Table 3. The calculated 1H NMR chemical shifts of the hydrogens of the benzoic rings of the neutral [8]circulenes fall in the aromatic range. The exceptions are compounds 8, 10 and 11, which possess the rim-antiaromaticity implying that the corresponding NMR signals are upfield shifted, which is in good agreement with the experimental data for compound 8. The 1H NMR chemical shifts describe correctly the rim aromaticity of the neutral species as obtained in the GIMIC calculation. For the dianionic [8]circulenes, the hydrogens of the benzoic rings are characterized by aromatic 1H NMR chemical shift values. The same also holds for the benzoic protons of the NH, PH, AsH, CH (compound 8), SiH and GeH containing compounds. These results are in good agreement with the GIMIC and NICS results. For the dicationic species the aromatic character deduced from the 1H NMR chemical shifts does not agree with the ones obtained in the GIMIC calculations. The chemical shifts in Table 3 show that the hydrogens of the benzoic rings and the ones at the heteroatoms are significantly deshielded, which might indicate that the molecule sustains a strong diatropic ring current along the perimeter, which is in contradiction to the GIMIC results. We conclude that the 1H NMR chemical shift criterion as well as NICS criterion for aromaticity is not able to correctly describe the aromatic character of the hetero[8]circulene dications. For the neutral and dianionic species, the 1H NMR chemical shift indices provide a more reliable description of the aromaticity. A further limitation of the 1H NMR chemical shift criterion is that the inner COT core of the [8]circulenes does not contain any directly fused protons, which means that the criterion cannot be applied for assessing the hub aromaticity.
| Circulene | Chemical shift range (δ), ppm | ||
|---|---|---|---|
| 2+ | 0 | 2− | |
| a Experimental data from Ref. 56. b Experimental data from Ref. 75. | |||
| 1 | 10.89 (8CH) | 7.80/7.68a (8CH) | 6.58 (8CH) |
| 2 | 10.18 (8CH) | 7.72 (8CH) | 8.63/8.47 (8CH) |
| 3 | 9.80 (8CH) | 8.23 (8CH) | 6.96 (8CH) |
| 4 | 6.86 (8CH) | 7.92 (8CH) | 7.05 (8CH) |
| 5 | 10.12 (8CH), 12.58 (4NH) | 7.68 (8CH), 7.95 (4NH) | 6.18 (8CH), 5.16 (4NH) |
| 6 | 9.43 (8CH), 7.73 (4PH) | 7.63 (8CH), 5.82 (4PH) | 7.07 (8CH), 6.74 (4PH) |
| 7 | 7.26 (8CH), 9.08 (4AsH) | 7.32 (8CH), 5.50 (4AsH) | 6.89 (8CH), 6.26 (4AsH) |
| 8 | 12.03 (8CH), 13.08 (4CH) | 1.24 (8CH), 0.70 (4CH)/2.86b | 8.78 (8CH), 9.42 (4CH) |
| 9 | 10.04 (8CH), 5.67 (4CH2) | 7.43 (8CH), 3.92 (4CH2) | 6.60 (8CH), 4.06 (4CH2) |
| 10 | 9.68 (8CH), 9.21 (4SiH) | 5.25–5.68 (4SiH), 4.91–5.50 (8H) | 7.18–7.19 (4SiH), 7.76–7.96 (8CH) |
| 11 | 9.44 (8CH), 9.98 (4GeH) | 6.12–7.62 (4GeH), 5.99–7.22 (8H) | 7.33 (4GeH), 7.98–7.33 (8CH) |
| 12 | 5.92–6.17 (4SiH2), 8.99 (8CH) | 5.20 (4SiH2), 7.92 (8CH) | 6.62–6.58 (4SiH2), 7.58–7.52 (8CH) |
| 13 | 6.50–6.09 (4GeH2), 8.99–9.02 (8CH) | 5.39 (4GeH2), 7.89–7.88 (8CH) | 5.33–5.35 (4GeH2), 6.96–6.89 (8CH) |
4. Conclusions
In the present work, we have presented the structural characterisation and aromaticity descriptions of the doubly charged [8]circulenes 1–9 and the novel neutral, dicationic and dianionic circulenes 10–13. We have used the three most popular magnetic criteria of aromaticity, namely the gauge including magnetically induced current (GIMIC) method, calculated nucleus-independent chemical shift (NICS) values and 1H NMR chemical shifts to solve the posed problem. We have found that the GIMIC-based results for the studies on neutral and doubly charged [8]circulenes are much more informative than the local NICS indices and 1H NMR chemical shifts, because the GIMIC approach provides very detailed information about the current pathways, which is not feasible when using the local NICS and 1H NMR chemical shifts indices.We found that most of the studied [8]circulenes (except compounds 8, 10 and 11) have a bifacial aromatic/antiaromatic character in the neutral form; the inner octatetraene core (also called hub) sustains a paratropic (antiaromatic) ring current, whereas the ring current of the outer macrocycle (the rim) is diatropic (aromatic). The aromatic character changes drastically when charging the [8]circulenes; the dianionic [8]circulenes and OC-, CH-, CH2-, SiH-, GeH-, SiH2-, and GeH2-containing dicationic species sustain net diatropic ring currents i.e., they are aromatic, whereas O-, S-, Se-, NH-, PH- and AsH-containing dicationic compounds are strongly antiaromatic, because they are dominated by the paratropic ring currents. The present computational study of the magnetically induced current densities suggests that the experimentally observed high stability of the synthesized [8]circulenes 1–5 and 9 is due to their rim-aromaticity, even though they are nonaromatic. The rim aromaticity favours substitution reactions of the [8]circulenes 1–5 and 9, analogously to benzoic compounds. The carbon atoms of the hub are protected against substitution reactions, because they are sp2 hybridized without any available hydrogen atom to replace. Addition reactions to the hub carbons would lead to a transformation from sp2 to sp3 hybridization that significantly affects most of the C–C bonds of the [8]circulene, which is expected to lead to a very high reaction barrier. Compound 8 is antiaromatic suggesting that it is unstable, which is in good agreement with experimental data.75 The same most likely also holds for the rest of the studied [8]circulenes, which have not yet been synthesized. The GIMIC approach opens up new possibilities for studies of the electronic structure of complex polycyclic aromatic compounds like [8]circulenes.
Acknowledgements
The calculations were performed with computational resources provided by the Swedish National Infrastructure for Computing (SNIC) at the Parallel Computer Center (PDC) through the project “Multiphysics Modeling of Molecular Materials”, SNIC 020/11-23 and by CSC – the Finnish IT Center for Science. We are grateful for support from the Academy of Finland projects 275845 and 266227. This research was also supported by the Ministry of Education and Science of Ukraine (project number 0115U000637). B. F. Minaev acknowledges the fellowship of Chinese Academy of Science under the CAS President's International Initiative for Visiting Scientists.References
- T. M. Krygowski and M. K. Cyrański, Chem. Rev., 2001, 101, 1385 CrossRef CAS PubMed.
- M. K. Cyrański, Chem. Rev., 2005, 105, 3773 CrossRef PubMed.
- A. R. Katritzky, P. Barczynski, G. Musumarra, D. Pisano and M. Szafran, J. Am. Chem. Soc., 1989, 111, 7 CrossRef CAS.
- M. K. Cyrański, T. M. Krygowski, A. R. Katritzky and P. v. R. Schleyer, J. Org. Chem., 2002, 67, 1333 CrossRef.
- K. Jug and A. M. Koster, J. Phys. Org. Chem., 1991, 4, 163 CrossRef CAS.
- (a) R. Gershoni-Poranne and A. Stanger, Chem. Soc. Rev., 2015, 44, 6597 RSC; (b) C. Foroutan-Nejad, S. Shahbazian and P. Rashidi-Ranjbar, Phys. Chem. Chem. Phys., 2010, 12, 12630 RSC.
- (a) S. C. A. H. Pierrefixe and F. M. Bickelhaupt, Chem. – Eur. J., 2007, 13, 6321 CrossRef CAS PubMed; (b) S. C. A. H. Pierrefixe and F. M. Bickelhaupt, Chem. – Eur. J., 2007, 13, 8371 CrossRef CAS; (c) S. C. A. H. Pierrefixe and F. M. Bickelhaupt, J. Phys. Chem. A, 2008, 112, 12816 CrossRef CAS PubMed.
- P. v. R. Schleyer, C. Maerker, H. C. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS.
- Z. Badri, S. Pathak, H. Fliegl, P. Rashidi-Ranjbar, R. Bast, R. Marek, C. Foroutan-Nejad and K. Ruud, J. Chem. Theory Comput., 2013, 9, 4789 CrossRef CAS PubMed.
- C. Foroutan-Nejad, Theor. Chem. Acc., 2015, 134, 8 CrossRef.
- J. Jusélius and D. Sundholm, Phys. Chem. Chem. Phys., 1999, 1, 3429 RSC.
- E. Steiner, P. Fowler, A. Soncini and L. W. Jenneskens, Faraday Discuss., 2007, 135, 309 RSC.
- J. Jusélius and D. Sundholm, J. Org. Chem., 2000, 65, 5233 CrossRef.
- Y.-C. Lin, J. Jusélius and D. Sundholm, J. Chem. Phys., 2005, 122, 214308 CrossRef PubMed.
- P. W. Fowler, R. W. A. Havenith and E. Steiner, Chem. Phys. Lett., 2002, 359, 530 CrossRef CAS.
- J. Jusélius, S. Michal and D. Sundholm, J. Phys. Chem. A, 2001, 105, 9939 CrossRef.
- Y.-C. Lin, D. Sundholm, J. Jusélius, L.-F. Cui, H.-J. Zhai and L.-S. Wang, J. Phys. Chem. A, 2006, 110, 4244 CrossRef CAS PubMed.
- C. A. Tsipis, Coord. Chem. Rev., 2005, 249, 2740 CrossRef CAS.
- Y.-C. Lin and D. Sundholm, J. Chem. Theory Comput., 2006, 2, 761 CrossRef CAS PubMed.
- H. Fliegl, S. Taubert, O. Lehtonen and D. Sundholm, Phys. Chem. Chem. Phys., 2011, 13, 20500 RSC.
- J. Jusélius, D. Sundholm and J. Gauss, J. Chem. Phys., 2004, 121, 3952 CrossRef PubMed.
- H. Fliegl, D. Sundholm, S. Taubert, J. Jusélius and W. Klopper, J. Phys. Chem. A, 2009, 113, 8668 CrossRef CAS PubMed.
- H. Fliegl, N. Özcan, R. Mera-Adasme, F. Pichierri, J. Jusélius and D. Sundholm, Mol. Phys., 2013, 111, 1364 CrossRef CAS.
- R. R. Valiev, H. Fliegl and D. Sundholm, J. Phys. Chem. A, 2013, 117, 9062 CrossRef CAS PubMed.
- R. R. Valiev, H. Fliegl and D. Sundholm, J. Phys. Chem. A, 2015, 119, 1201 CrossRef CAS PubMed.
- R. R. Valiev, H. Fliegl and D. Sundholm, Phys. Chem. Chem. Phys., 2015, 17, 14215 RSC.
- M. P. Johansson, J. Jusélius and D. Sundholm, Angew. Chem., Int. Ed., 2005, 44, 1843 CrossRef CAS PubMed.
- S. Taubert, D. Sundholm and F. Pichierri, J. Org. Chem., 2010, 75, 5867 CrossRef CAS PubMed.
- S. Taubert, J. Jusélius, D. Sundholm, W. Klopper and H. Fliegl, J. Phys. Chem. A, 2008, 112, 13584 CrossRef CAS PubMed.
- V. T. T. Huong, T. B. Tai and M. T. Nguyen, Phys. Chem. Chem. Phys., 2012, 14, 14832 RSC.
- V. T. T. Huong, H. T. Nguyen, T. B. Tai and M. T. Nguyen, J. Phys. Chem. C, 2013, 117, 10175 CAS.
- G. V. Baryshnikov, N. N. Karaush and B. F. Minaev, Chem. Heterocycl. Compd., 2014, 50, 349 CrossRef CAS.
- W. Wei, F.-Q. Bai, B.-H. Xia, H.-B. Chen and H.-X. Zhang, Chem. Res. Chin. Univ., 2013, 29, 962 CrossRef CAS.
- A. Kerim, New J. Chem., 2014, 38, 3783 RSC.
- K. Y. Chernichenko, V. V. Sumerin, R. V. Shpanchenko, E. S. Balenkova and V. G. Nenajdenko, Angew. Chem., Int. Ed., 2006, 45, 7367 CrossRef CAS PubMed.
- B. F. Minaev, G. V. Baryshnikov and V. A. Minaeva, Comput. Theor. Chem., 2011, 972, 68 CrossRef CAS.
- N. N. Karaush, B. F. Minaev, G. V. Baryshnikov and V. A. Minaeva, Opt. Spectrosc., 2014, 116, 33 CrossRef CAS.
- T. B. Tai, V. T. T. Huong and M. T. Nguyen, Chem. Commun., 2013, 49, 11548–11550 RSC.
- N. N. Karaush, R. R. Valiev, G. V. Baryshnikov, B. F. Minaev and H. Ågren, Chem. Phys., 2015, 459, 65 CrossRef CAS.
- G. V. Baryshnikov, B. F. Minaev and V. A. Minaeva, Russ. Chem. Rev., 2015, 84, 455 CrossRef CAS.
- T. B. Tai, H. T. T. Vu and M. T. Nguyen, RSC Adv., 2015, 5, 24167 RSC.
- X. Xiong, C.-L. Deng, B. F. Minaev, G. V. Baryshnikov, X.-S. Peng and H. N. C. Wong, Chem. – Asian J., 2014, 9, 1 CrossRef.
- T. N. Gribanova, N. S. Zefirov and V. I. Minkin, Pure Appl. Chem., 2010, 82, 1011 CrossRef CAS.
- G. V. Baryshnikov, V. A. Minaeva, B. F. Minaev, N. N. Karaush, X.-D. Xiong, M.-D. Li, D. L. Phillips and H. N. C. Wong, Spectrochim. Acta, Part A, 2015, 151, 247 CrossRef PubMed.
- G. V. Baryshnikov, B. F. Minaev, N. N. Karaush and V. A. Minaeva, Phys. Chem. Chem. Phys., 2014, 16, 6555 RSC.
- G. V. Baryshnikov, B. F. Minaev, N. N. Karaush and V. A. Minaeva, RSC Adv., 2014, 4, 25843 RSC.
- N. N. Karaush, G. V. Baryshnikov and B. F. Minaev, RSC Adv., 2015, 5, 24299 RSC.
- N. N. Karaush, G. V. Baryshnikov, B. A. Minaeva and B. F. Minaev, New J. Chem., 2015, 39, 7815 RSC.
- J. Yu, Q. Sun, Y. Kawazoe and P. Jena, Nanoscale, 2014, 6, 14962 RSC.
- S. Radenković, I. Gutman and P. Bultinck, J. Phys. Chem. A, 2012, 116, 9421 CrossRef PubMed.
- G. V. Baryshnikov, B. F. Minaev, M. Pittelkow, C. B. Nielsen and R. Salcedo, J. Mol. Model., 2013, 19, 847 CrossRef CAS PubMed.
- G. V. Baryshnikov, R. R. Valiev, N. N. Karaush and B. F. Minaev, Phys. Chem. Chem. Phys., 2014, 16, 15367 RSC.
- G. V. Baryshnikov, N. N. Karaush, R. R. Valiev and B. F. Minaev, J. Mol. Model., 2015, 21, 136 CrossRef PubMed.
- C. B. Nielsen, T. Brock-Nannestad, T. K. Reenberg, P. Hammershoj, J. B. Christensen, J. W. Stouwdam and M. Pittelkow, Chem. – Eur. J., 2010, 16, 13030 CrossRef CAS PubMed.
- J. Eskildsen, T. Reenberg and J. B. Christensen, Eur. J. Org. Chem., 2000, 1637 CrossRef CAS.
- T. Brock-Nannestad, C. B. Nielsen, M. Schau-Magnussen, P. Hammershoj, T. K. Reenberg, A. B. Petersen, D. Trpcevski and M. Pittelkow, Eur. J. Org. Chem., 2011, 6320 CrossRef CAS.
- A. Dadvand, F. Cicoira, K. Y. Chernichenko, E. S. Balenkova, R. M. Osuna, F. Rosei, V. G. Nenajdenko and D. F. Perepichka, Chem. Commun., 2008, 5354 RSC.
- H. Erdtman and H.-E. Högberg, Tetrahedron Lett., 1970, 11, 3389 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision C.02, Gaussian, Inc, Wallingford, CT, 2009 Search PubMed.
- T. K. Zywietz, H. Jiao, P. V. R. Schleyer and A. De Meijere, J. Org. Chem., 1998, 63, 3417 CrossRef CAS.
- P. R. Schleyer, Chem. Rev., 2001, 101, 1115 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- A. Stanger, J. Org. Chem., 2006, 71, 883 CrossRef CAS PubMed.
- K. Wolinski, J. F. Hilton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251 CrossRef CAS.
- J. R. Cheeseman, G. W. Trucks, T. A. Keith and J. M. Frisch, J. Chem. Phys., 1996, 104, 5497 CrossRef CAS.
- G. Monaco, P. W. Fowler, M. Lillington and R. Zanasi, Angew. Chem., Int. Ed., 2007, 46, 1889 CrossRef CAS PubMed.
- R. R. Valiev, H. Fliegl and D. Sundholm, Phys. Chem. Chem. Phys., 2014, 16, 11010 RSC.
- R. R. Valiev and V. N. Cherepanov, Int. J. Quantum Chem., 2013, 113, 2563 CrossRef CAS.
- F. Furche, R. Ahlrichs, C. Hättig, W. Klopper, M. Sierka and F. Weigend, Comput. Mol. Biosci., 2014, 4, 91 CrossRef CAS.
- JMOL: an open-source Java viewer for chemical structures in 3D., http://www.jmol.org.
- S. Nobusue, H. Miyoshi, A. Shimizu, I. Hisaki, K. Fukuda, M. Nakano and Y. Tobe, Angew. Chem., Int. Ed., 2015, 54, 2090 CrossRef CAS PubMed.
- H. Miyoshi, S. Nobusue, A. Shimizu and Y. Tobe, Chem. Soc. Rev., 2015, 44, 6560 RSC.
- J. H. Dopper and H. Wynberg, J. Org. Chem., 1975, 40, 1957 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates and the corresponding total energy of the singlet ground state of the studied [8]circulenes 1–13. See DOI: 10.1039/c6cp00365f |
| This journal is © the Owner Societies 2016 |