
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Temperature effect on the build-up of exponentially growing polyelectrolyte multilayers. An exponential-to-linear transition point†
Anna S.
Vikulina
abc,
Yuri G.
Anissimov
d,
Prateek
Singh
be,
Vladimir Z.
Prokopović
b,
Katja
Uhlig
b,
Magnus S.
Jaeger
bf,
Regine
von Klitzing
g,
Claus
Duschl
b and
Dmitry
Volodkin
*ab
aSchool of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham NG11 8NS, UK. E-mail: dmitry.volodkin@ntu.ac.uk
bFraunhofer IZI-BB, Am Mühlenberg 13, 14424, Potsdam, Germany. E-mail: vladimir.prokopovic@izi-bb.fraunhofer; claus.duschl@izi-bb.fraunhofer.de; katja.uhlig@izi-bb.fraunhofer.de; Fax: +49 331 58187 399; Tel: +49 331 58187 300
cThe Faculty of Fundamental Medicine, Laboratory of Medical Biophysics, Lomonosov Moscow State University, Moscow, 119991, Russia. E-mail: vikulina.msu@gmail.com
dSchool of Natural Sciences and Micro and Nano Technology Centre, Griffith University, Australia. E-mail: y.anissimov@griffith.edu.au; Tel: +61 (07) 555 28496 ext. 28496
eLaboratory of Developmental Biology, Department of Medical Biochemistry and Molecular Biology, Institute of Biomedicine, University of Oulu, PO Box 5000, 90014 Oulu, Finland. E-mail: psingh@mail.student.oulu.fi; Fax: +358 (0)8 553 1141; Tel: +358 (0)294 481200
fFederal Institute for Risk Assessment (BfR), Max-Dohrn-Strasse 8-10, 10589 Berlin, Germany. E-mail: magnus.jaeger@bfr.bund.de; Tel: +49 30184123457
gStranski-Laboratorium für Physikalische und Theoretische Chemie, Technische Universität Berlin, Strasse des 17. Juni 124, D-10623 Berlin, Germany. E-mail: klitzing@mailbox.tu-berlin.de
First published on 10th February 2016
Abstract
In this study, the effect of temperature on the build-up of exponentially growing polyelectrolyte multilayer films was investigated. It aims at understanding the multilayer growth mechanism as crucially important for the fabrication of tailor-made multilayer films. Model poly(L-lysine)/hyaluronic acid (PLL/HA) multilayers were assembled in the temperature range of 25–85 °C by layer-by-layer deposition using a dipping method. The film growth switches from the exponential to the linear regime at the transition point as a result of limited polymer diffusion into the film. With the increase of the build-up temperature the film growth rate is enhanced in both regimes; the position of the transition point shifts to a higher number of deposition steps confirming the diffusion-mediated growth mechanism. Not only the faster polymer diffusion into the film but also more porous/permeable film structure are responsible for faster film growth at higher preparation temperature. The latter mechanism is assumed from analysis of the film growth rate upon switching of the preparation temperature during the film growth. Interestingly, the as-prepared films are equilibrated and remain intact (no swelling or shrinking) during temperature variation in the range of 25–45 °C. The average activation energy for complexation between PLL and HA in the multilayers calculated from the Arrhenius plot has been found to be about 0.3 kJ mol−1 for monomers of PLL. Finally, the following processes known to be dependent on temperature are discussed with respect to the multilayer growth: (i) polymer diffusion, (ii) polymer conformational changes, and (iii) inter-polymer interactions.
1. Introduction
Polyelectrolyte multilayer films (or multilayers) are self-assembled surface coatings with physical and chemical properties being controlled with high accuracy thanks to a multistep preparation approach – the layer-by-layer (LbL) deposition.1 Multilayers are formed by alternate adsorption of polyanions and polycations on a solid substrate. The substrate to be coated is almost non-restricted by the shape or size, allowing one to generate a variety of geometrically possible functional surfaces. A wide range of polymers that can be employed for multilayer preparation has allowed the fabrication of surfaces with diverse functions. Besides a progressive increase in a number of non-biological applications of PEMs,2,3 research over the past few years has shown a strong tendency of focusing on bio-related applications in the fields of biomimetic materials, biocompatible surfaces and controlled drug release devices.4–8 This increase in bio-related applications is attributed to a variety of biopolymers used to prepare the films aiming at a certain biological response. For example, cell adherent surfaces,9–14 non-adherent/anti-fouling surfaces15 and surfaces exhibiting specific adhesion properties16,17 for both in vitro and in vivo applications have been designed. The multilayer films and their components can be tailored to respond to variations in salt concentration,18,19 pH,20 temperature,21 and to external stimulation such as remote light activation.22–26 An exhaustive list of biological and non-biological applications can be found in recent work and reviews on these topics.3,8,27–37The interaction between the polymers employed determines the build-up regimes of the LbL formation, i.e. the exponential and the linear one.38–41 This concerns room temperature and reasonable salt concentration such as below the physiological one. Poly(styrene sulfonate)/poly(allylamine hydrochloride) (PSS/PAH) multilayers are typical examples of linearly growing films.42 For such films polymer molecules interact strongly with each other and do not diffuse into the film; polymer diffusion is limited by interdigitating of polymer chains with each other.
Poly(L-lysine)/hyaluronic acid (PLL/HA) multilayers are some of the most studied exponentially growing systems. The low charge density (compared to synthetic polymers) of HA and its rigidity may be responsible for rather weak inter-polymer interactions resulting in rather fast polymer diffusion in the film.43,44 These polymers possess biocompatible and biodegradable properties that have promoted a wide usage of PLL/HA films in the field of bio-related applications. However, the exact growth mechanism of the exponential-like growing films is still unknown. The commonly accepted model is based on intensive polymer (PLL) diffusion in and out of the film during the build-up process.44
Interestingly, when forming the film, after a certain number of bilayers at the transition point the exponential growth switches to the linear growth regime. The reason for this transition has not been fully understood up to now.40,45,46 However, the transition point is reported to be independent of the film deposition time and the polymer molecular weight,38,39 which seems to be contradictory to a diffusion-mediated mechanism of the film growth. Understanding the exponential-like growth mechanism is of high importance for fundamental research as well as for practical applications because it provides the rational for tuning the film thickness, composition, and macroscopic properties such as the elasticity and stability.
Though the build-up for two types of LbL films has been studied as a function of salt concentration47 and pH,48,49 just in a few studies film growth as a function of temperature has been addressed.41,49–51 The temperature is an easily adjustable stimulus allowing the potential tuning of the film thickness by simple heating or cooling of the polymer solutions during film preparation. In addition, most bio-related applications of multilayers occur at 37 °C while the assembly is carried out at room temperature. Hence it is important to understand how do they perform as a function of temperature.
The literature reports on various effects of the temperature on the film growth. For linearly growing mutilayers, PSS/poly(diallyl-dimethylammonium) or (PSS/PDADMA) and PSS/PAH, an increase of the temperature results in a switch from the linear to the exponential-like growth regimes indicating more “fluidity” present in the films. This effect was shown to be ion-specific.50,52 Another study on gelatin/poly(galacturonic) films49 showed a decrease in the film mass and thickness with an increase in the preparation temperature suggesting a strong impact of H-bonding on the inter-polymer interactions in the film. However, the effect that temperature has on the build-up of exponentially growing films is not much described in the literature. Studies on this matter may help to understand the mechanism of multilayer growth. In particular the details of the relation between the transition point and temperature may provide new insights into the growth mechanism.
In this work we have studied the influence of temperature on the growth of PLL/HA multilayers, which serve as a model of exponential-like growing multilayers. The growth rate has been evaluated by the direct analysis of the polymer (PLL) content in the prepared film after film decomposition, which is a robust and reliable approach for a detailed growth analysis. For understanding the mechanism of the film growth upon temperature variation, we analysed growth profiles at fixed preparation temperatures (in the range from 25 to 85 °C). The effect of preparation temperature on the film growth and the effect of the temperature change for the as-prepared films are considered. The position of the transition point has also been investigated. Finally, literature findings on temperature-mediated aspects in polymer diffusion, polymer conformational changes, and polymer complexation in multilayers are discussed in order to identify their impact on the film growth.
2. Materials and methods
2.1 Chemicals
Poly-L-lysine hydrobromide (PLL, 28 kDa), poly(ethylenimine) (PEI, 50% weight solution in water), PLL–FITC (fluorescein isothiocyanate (FITC) labelled PLL, labelling ratio ∼0.01 for fluorescent dye per polymer unit), Tris and NaCl were purchased from Sigma-Aldrich, Germany and HA (as sodium hyaluronate, 360 kDa) was purchased from Lifecore Biomedical, USA. Polymer solutions were prepared in Milli-Q water at a concentration of 0.5 mg ml−1, and were filtered through a 0.22 μm filter. The PEI solution used was of 1 mg ml−1 concentration prepared in Milli-Q water. The rinsing buffer was 10 mM Tris and 15 mM NaCl, with pH 7.4.2.2 PLL/HA film preparation
The substrates used for LbL deposition were No. 1 thick circular glasses from Marienfeld (12 mm diameter). The glasses were cleaned with 2% Hellmanex at 60 °C for 15 min, and then twice with 1 M HCl for 15 min at 60 °C followed by an adequate rinsing in Milli-Q water. The PEM films were prepared using a dip robot (Riegler and Kirstein GmbH, Germany) by sequential dipping of the glass slides into the 0.5 mg ml−1 PLL and HA solutions in Tris buffer (10 mM Tris, 15 mM NaCl buffer (pH 7.4)) as described elsewhere53 with small variations. Briefly, the slides were dipped in the polymer solution for 5 minutes with 5 iterations of 1 minute each. Between each polymer dipping, the slides were rinsed in the rinsing buffer three times, 90 seconds each. This was repeated to get a desired number of polyelectrolyte deposition steps. One bilayer (PLL/HA) corresponds to two polymer deposition steps. The terminating layer was always HA, for instance (PLL/HA)30 is referred to as the 30 bilayer film. PLL was mixed with PLL–FITC (0.5 mg ml−1) at a ratio of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v). After film preparation, the samples were stored in glass vials in TRIS buffer. The samples were stored in the dark at 4 °C, covered with an aluminium foil.
:1 (v/v). After film preparation, the samples were stored in glass vials in TRIS buffer. The samples were stored in the dark at 4 °C, covered with an aluminium foil.
2.3 Quantification of adsorbed PLL
Samples terminated with HA were rinsed in Tris-buffer (15 mM NaCl), and the polymer was removed from the substrate glass in 0.2 ml of 0.1 N NaOH. The solution was pipetted a few times to ensure complete removal of the film. 1.8 ml of 0.4 M Tris buffer was added, the solution was transferred into the cuvettes and fluorescence intensity was measured using a spectrofluorometer (Perkin Elmer UV-Vis). Excitation of PLL–FITC was set to 488 nm, and the emission was taken at 515 nm. The amount of polymers deposited in the film was quantified by taking fluorescence readings of the PLL–FITC using a standard calibration curve (Fig. S1, ESI†).2.4 Confocal microscopy
For film thickness determination a confocal laser scanning microscope (510 Meta, Zeiss, Germany) equipped with an argon laser and a 63×/1.4 oil immersion objective was employed. For image acquisition, the pinhole was set to 1 Airy unit (image slice of approximately 0.7 μm).3. Results and discussion
3.1 Film growth at various temperatures
Fig. 1 shows the increase of mass coverage of PLL during build-up of PLL/HA multilayers prepared at different temperatures ranging from 25 to 85 °C. The amount of PLL of the films formed has been measured after the film decomposition using 0.1 M NaOH. At high pH the PLL is uncharged resulting in the reduction of electrostatic interaction with HA followed by the film dissolution. The PLL content is given in absolute mass values. However these values are proportional to the film mass because the PLL to HA molar ratio is almost independent of the number of deposited layers.54 Another proof is that the polymer density in the film also does not depend on the number of bilayers because the film hydration state (water content) is the same for both low and high numbers of bilayers.55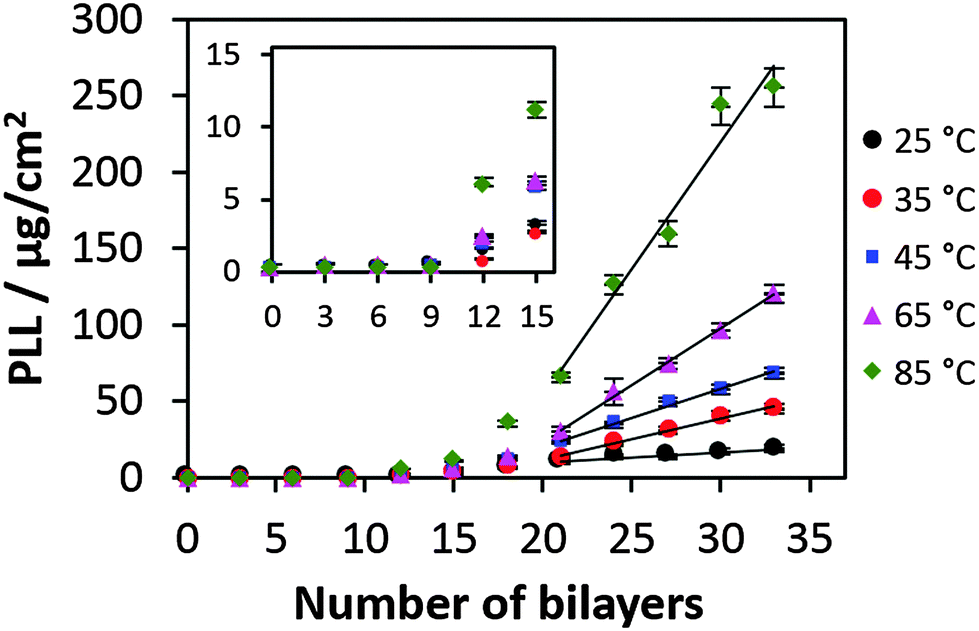 | ||
| Fig. 1 Mass coverage of PLL in the PLL/HA film produced at different temperatures. The inset shows the enlarged growth profile until 15 bilayers. | ||
The effect of the temperature on the film growth is remarkable and very prominent at high temperatures (Fig. 1). No visible film growth can be identified till 10 bilayers that may be related to the diffusion-mediated mechanism of the film growth: the film would start to grow much faster as soon as the film thickness is much above the polymer dimensions. The inset in Fig. 1 shows the exponential build-up regime for the first 18 bilayers prepared at different temperatures. The result of the linear fits of the growth curves shows that after the deposition of 21 bilayers all the films made at various temperatures grow in the linear regime (Fig. 1). Both the exponential and linear growth profiles are significantly affected by the increase of the preparation temperature. Further we focus on the switch from exponential to linear growth regimes or the so-called transition point.
3.2 Determination of the transition point
The transition point, at which the exponential growth turns to the linear one, has attracted strong scientific interest. Porcel and co-authors have concluded that the transition point of PLL/HA films always takes place at about 12 bilayers and does not depend on the preparation conditions such as the deposition time and the molecular weight of polymers.38,39 However, the way of determination of the transition point has not been specified.In recent work it is assumed that the transition point is caused by the onset of diffusion limitation of the polymer into the film.40 During film growth the molecules of one or both polymers can diffuse through the film in order to compensate free charges of the previously adsorbed (oppositely charged) polymer. In the beginning of the film growth the deposition time is enough to saturate the whole film with diffusing polymer molecules and thus free charges of the previously adsorbed polymer are fully compensated. Therefore, the film growth is exponential because an amount of diffused or adsorbed polymer depends on the film thickness (the thicker is the film, the more polymer it can host). The growth switches to a linear one as soon as the number of diffused polymer molecules is less than that necessary for complete charge compensation. After this (transition point) the number of polymer molecules, which enters the film and is involved in the film build-up, is constant and is defined by deposition time. In short, the transition point takes place when the polymer adsorption is below the saturation value for the film.
The independence of the transition point from the deposition time and the polymer weight38,39 seems to be in contradiction with an assumption that the film growth mechanism is based on the polymer diffusion in and out of the film.38,39,44 It is evident that a variation in the deposition time or the polymer molecular weight should affect the amount of polymer molecules diffusing into the film. This will obviously change the position of the transition point. To probe if the transition point is affected by the polymer diffusion we have examined the influence of the deposition temperature (the parameter that should affect the polymer diffusion rate) on the position of the transition point.
To determine the transition point we have fitted datasets of PLL content in the film versus bilayer number measured at various deposition temperatures with both exponential and linear functions at the same time. The expression for fitting is:
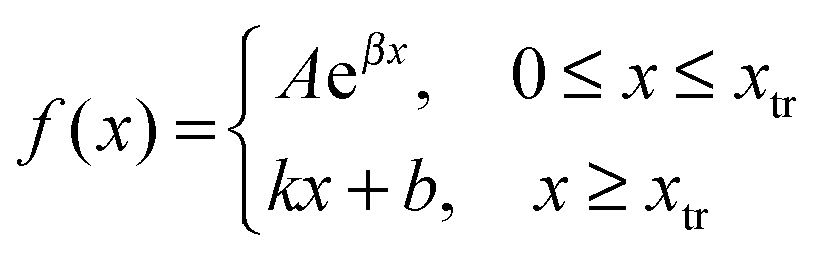 | (1) |
| k = Aβeβxtr; b = Aeβxtr(1 − βxtr) |
An example of the fitting is shown in Fig. 2a for the film prepared at 35 °C. The fitting process was performed using Mathematica 7.0 script (Wolfram Research, Champaign, IL), more details can be obtained from the ESI.† A similar procedure can also be performed using the Excel program (Fig. S2, ESI†). Fig. 2b shows the coefficient of determination (R2) obtained for exponential and linear fitting of experimental data. One can see that R2 can also be effectively used for a straightforward transition point determination. It was found to be equal to 18 bilayers for the film prepared at 35 °C.
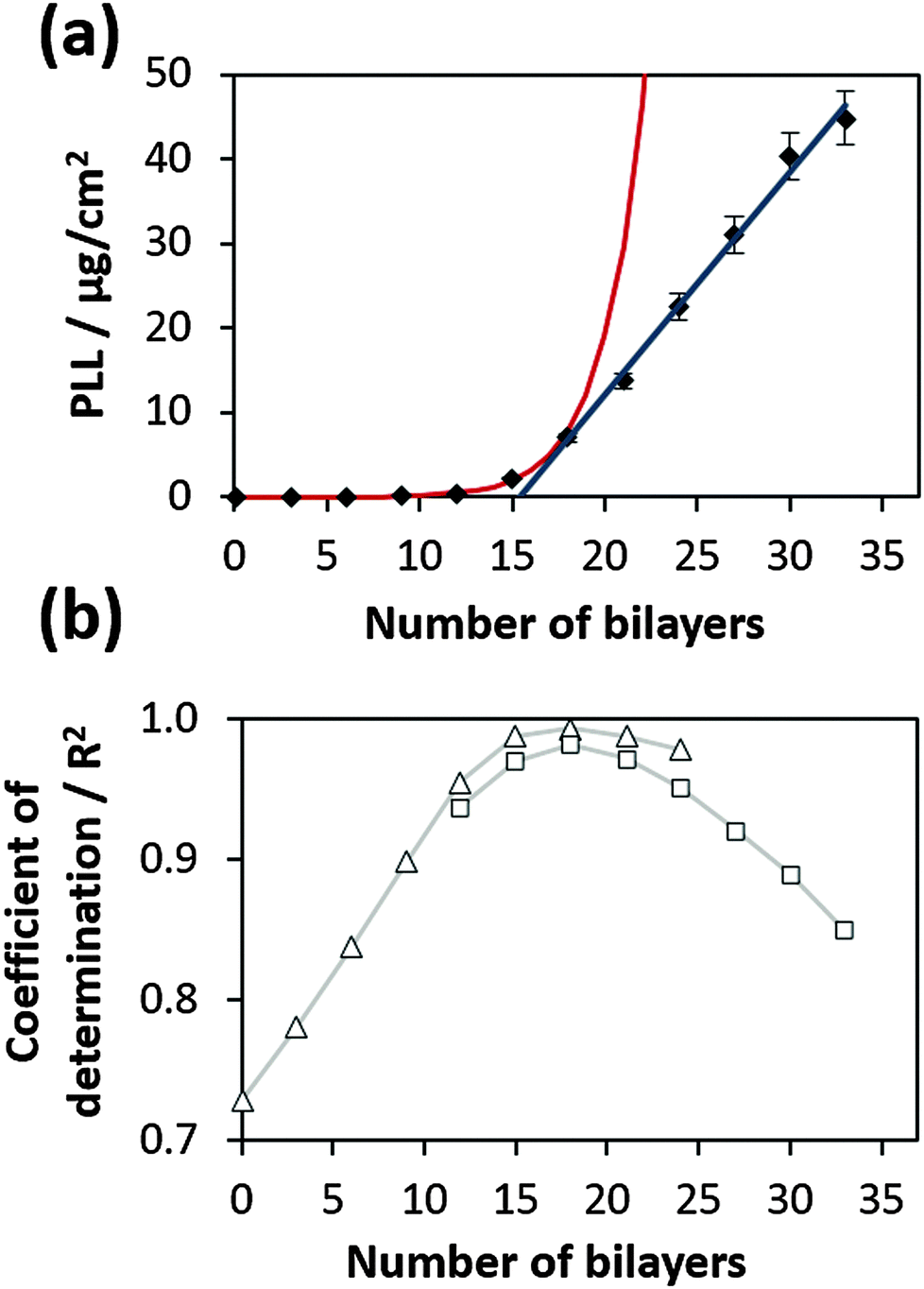 | ||
| Fig. 2 (a) Switch of exponential to linear growth at the transition point for a PLL/HA film prepared at 35 °C. Red and blue curves represent the fits of the experimental data based on eqn (1) describing both exponential and linear growth regimes. (b) R2-values calculated for exponential (open squares) and linear (open triangles) fits of the data points presented in (a). Two maxima determined for both fitting procedures coincide at the transition point corresponding to 18 bilayers. | ||
The calculated transition points are shown in Table 1 as a function of the preparation temperature. A significant and reliable shift from 12 to 21 bilayers is found as the temperature increases from 25 to 85 °C. Despite some transition points being similar (for 35 and 45 °C as well as for 65 and 85 °C), the overall trend shows that at higher temperature the number of bilayers at the transition point is higher. The exponential and linear fittings for additional temperatures are shown in Fig. S2 (ESI†). Our results confirm the concept that film growth is determined by the polymer transport into the film. In this concept the dependence of the transition point on temperature is a result of the temperature dependence of diffusive processes which eventually govern the polymer transport.
| T (°C) | Number of bilayers at the transition point |
|---|---|
| 25 | 12 |
| 35 | 18 |
| 45 | 18 |
| 65 | 21 |
| 85 | 21 |
3.3 Effect of the temperature change during the film build-up
Further we have studied if a temperature variation during the deposition process affects the film build-up rate. For this purpose, the temperature during the film preparation was switched from 65 °C to 25 °C or vice versa at 21 bilayers and then the deposition was continued to 33 bilayers (Fig. 3).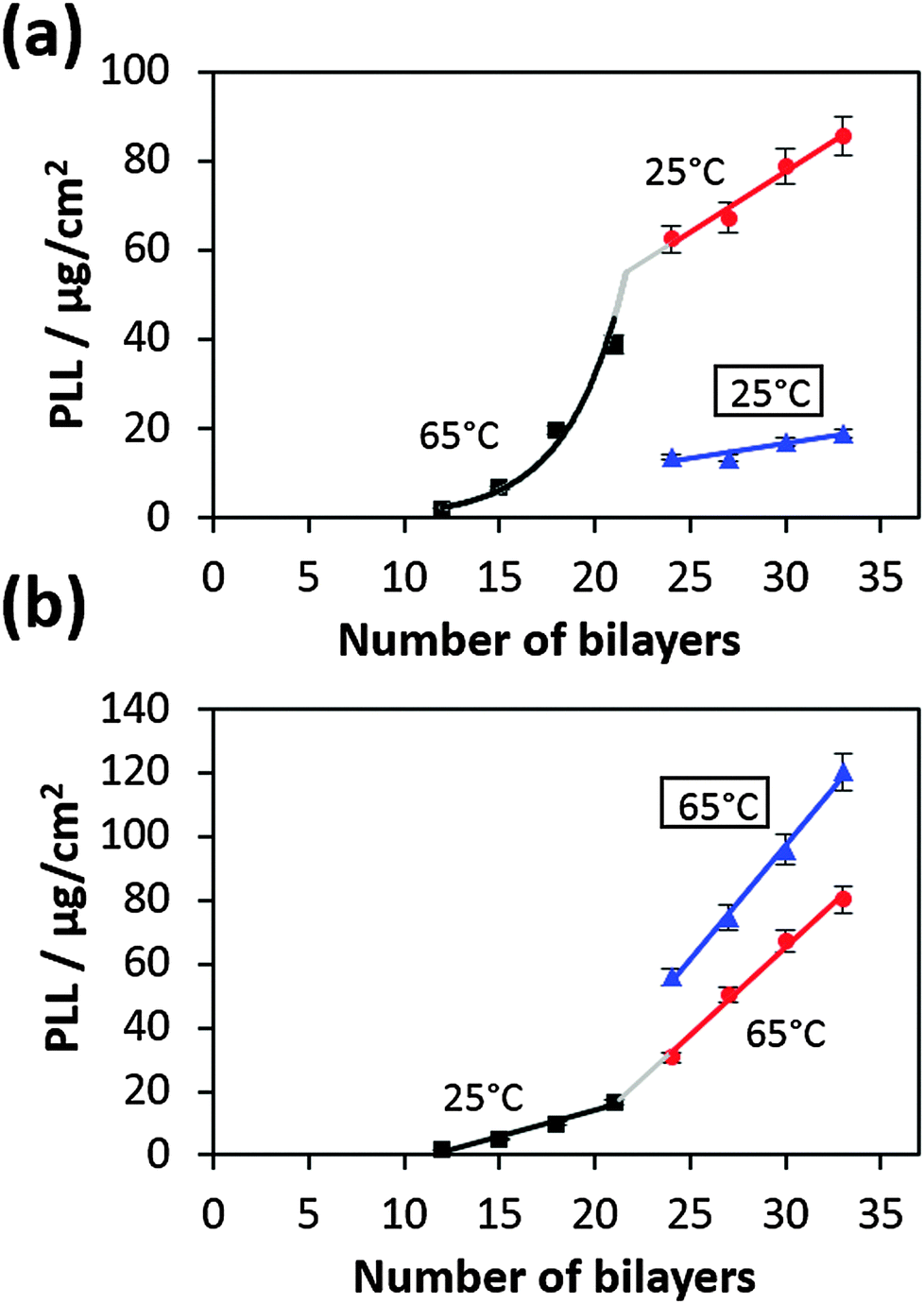 | ||
| Fig. 3 Temperature change from 25 °C to 65 °C and vice versa (in a and b, respectively) at the 21st bilayer. The blue triangles represent film preparation at 25 °C and 65 °C without the temperature change after 21 bilayers. | ||
For better comparison we just consider the slopes of growth profiles in the linear regime after the transition point. Growth profiles of films entirely prepared at a constant temperature (either at 25 °C or at 65 °C) are also shown for comparison (Fig. 3, blue lines). If the temperature was reduced during preparation (from 65 °C to 25 °C), the film growth is faster compared to the film fully grown at 25 °C (Fig. 3a). In the inverse case (increase of temperature from 25 °C to 65 °C), the film growth is slower than if it would have grown at 65 °C from the beginning (Fig. 3b).
These results demonstrate that the film structure most probably depends on the deposition temperature. At higher deposition temperatures the film is more permeable for polymers, and allows their faster diffusion (more porous structure) and, as a result, a faster film growth.
Further we have tested if the film structure can be affected by a temperature change after the film is fabricated. For that the film thickness has been analysed. The fluorescence intensity of the PLL/HA film (30 bilayers, prepared at 85 °C) was measured in the Z-slicing mode in three selected regions (Fig. S4, ESI,† shows the fluorescence reconstruction images from Z-stacking). The intensity profiles were further taken at 45 °C and 25 °C upon cooling down of the film. Typical profiles obtained at 45 °C are shown in Fig. 4. The thickness of the film was determined as the width of the fluorescence profile between inflexion points. The film thickness was not substantially changed upon cooling down from 45 °C (thickness 11.0 ± 1.0 μm) to 25 °C (thickness 11.3 ± 0.8 μm). Our previous findings based on Fourier transform infrared spectroscopy have shown that the film structure does not significantly change if the fabricated film is heated from 25 to 45 °C.53
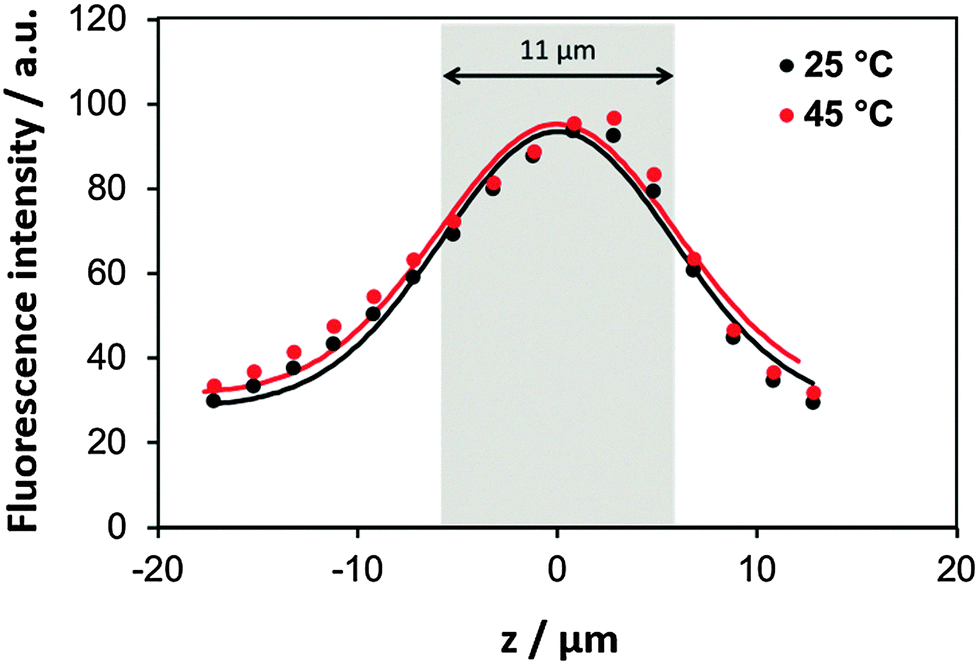 | ||
| Fig. 4 Average fluorescence intensity as a function of the z position of the focal plane using confocal microscopy. A film of (PLL/HA)30 was prepared at 85 °C. Subsequently, the film was cooled down to 45 °C (red circles) and then to 25 °C (black circles). The grey area indicates the average thickness of both films. | ||
These results suggest that the film structure is most likely dependent on the preparation temperature and higher preparation temperature results in a loose structure and at lower preparation temperature the structure is more rigid (Fig. 3). However, once the film is prepared it is not sensitive to the temperature variation anymore. This may indicate that the interconnected polymer network is equilibrated after the film preparation. Below we consider factors, which can influence the film growth as a function of the deposition temperature.
3.4 Arrhenius plot
Arrhenius plots allow the evaluation of the temperature dependence of the rate of a chemical reaction. In the case of multilayer growth, the HA–PLL inter-polymer complex formed during the multilayer growth can be considered as the molecular reaction between the polymers. For a single rate-limited reaction the Arrhenius equation is:| kf = Be−Ea/RT | (2) |
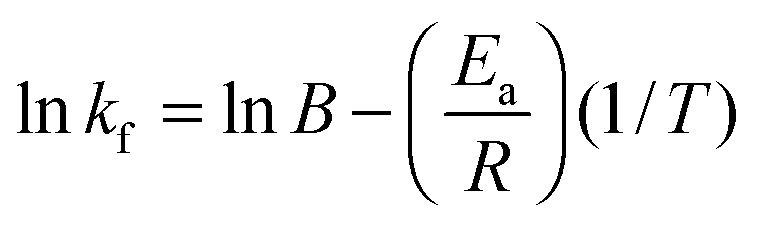 | (3) |
In Fig. 5 the ln of the PLL mass increase in the linear growth regime for a certain growth temperature is plotted versus the inverse absolute growth temperature.
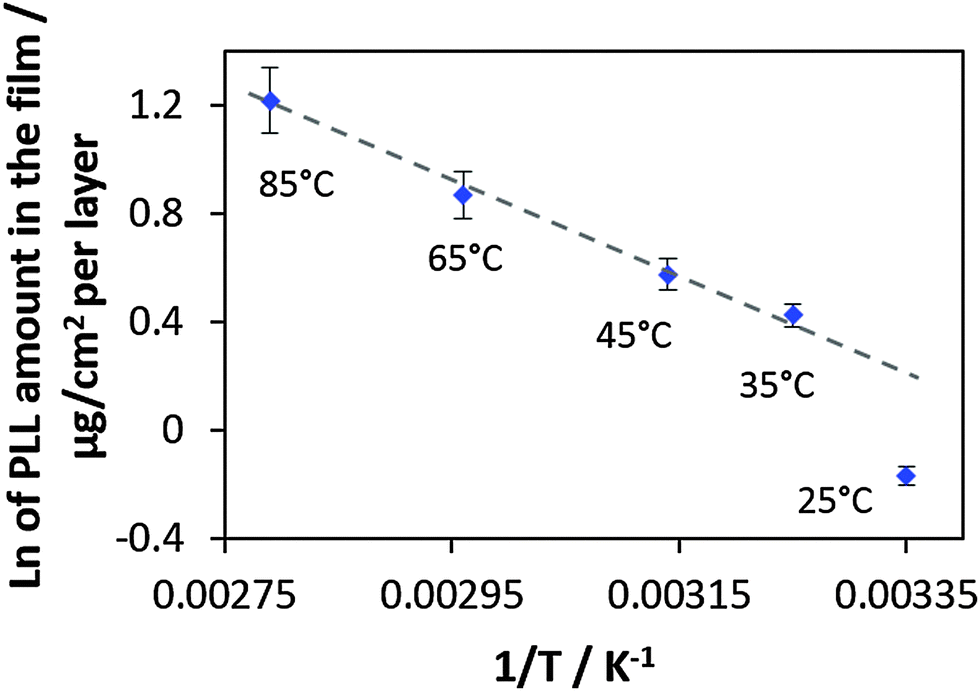 | ||
| Fig. 5 The ln of the amount of PLL in the film (in μg cm−2 per layer) vs. 1/T (K−1) and the linear fit (dashed gray line) through the four experimental points corresponding to the preparation temperatures from 35 to 85 °C. | ||
The quality of the linear fit applied to the experimental points from the films prepared in the temperature range of 35–85 °C is very good showing a R2 value of 0.993. This indicated that the film growth follows the Arrhenius equation (eqn (2) and (3)). The single data point (not used for fitting) in the graph corresponding to the film prepared at the lowest temperature (25 °C) does not fit to the common linear trend. If this point would be included in the linear fit, the R2 value becomes worse (0.925). We believe that the experimental point corresponding to a preparation temperature of 25 °C can be explained by its closeness to a phase transition temperature of PLL/HA multilayers. The phase transition temperature is rather low and should be below room temperature because the multilayers behave as a fluid at room temperature (as indicated by a high PLL mobility).
Furthermore, the average activation energy Ea has been calculated from the Arrhenius plot using the simple relation presented in eqn (3). The slope is then −Ea/R. The calculated Ea has been found to be 42.6 kJ mole per PLL molecule used in this study. This corresponds to an Ea value of 318 J mol−1 for a monomer. Activation energies of the same order of magnitude have been reported for inter-polymer interactions between carboxymethyl cellulose and chitosan56 and linearly growing PSS with PAH.57 The chemical structure of used polymers and the corresponding activation energies are presented in Table S1 (ESI†). The authors of these publications56,57 calculated the activation energies by either thermal decomposition or considering temperature-mediated diffusion of polymer chains within the multilayer film. The difference between Ea found in this work and Ea given in Table S1 (ESI†) might be explained by different methods used, the use of different polymers as well as different numbers of ion pairs formed in the multilayers of different nature. The number of ion pairs in the multilayers is expected to vary and is hardly predictable on the basis of the polymer chemistry.
3.5 Factors affecting the multilayer growth
Earlier literature on the LbL films suggests that solvent quality defines the interaction between polymer groups and solvent molecules, and thus plays a crucial role in the temperature-related variation of multilayer growth.58,59 However, experimental observations of the polymer layer thickness at different temperatures showed that the solvent–solute interaction may not be responsible for these variations and rather the internal structure of films may play a role.60 It is nowadays strongly believed that increased fluidity in the film promotes faster film growth at higher temperature; however, there is no commonly accepted mechanism. Therefore, in order to elucidate the results of this study, we consider three main factors, which can have an impact on the film growth as a result of variations of preparation temperature:(i) Polymer diffusion
(ii) Polymer conformation
(iii) Inter-polymer interactions
In the next paragraphs we describe each of these factors and evaluate their potential impact on the temperature-dependent film growth. Without doubts, polymer diffusion plays a crucial role in the growth of LbL assembled films. The growth of PLL/HA films is driven by the diffusion of free polymer chains (PLL weakly bound to the film) in and out of the film.44 PLL molecules in the film present as at least two populations with diffusion coefficients of about 1 and 0.2 μm2 s−1.61 The free PLL molecules diffuse fast into the film during deposition of PLL and diffuse out followed by complexation with HA at the film top during the deposition step of HA. This complexation results in newly formed polymer complexes that lead to an increment of film growth. Thus, the diffusive transport of PLL will significantly contribute to the growth rate. It is known that the diffusion coefficient of molecules depends on temperature:62
| D = D0e−EaD/RT | (4) |
According to eqn (4) an increase of the temperature during the build-up process will result in higher mobility of free polymer chains in the film that should lead to more PLL diffusing into the film and, as a result, faster film growth.
Polymer conformation in solution can also affect the film growth rate. The secondary structure of PLL and polyglutamic acid (PGA) in PLL/PGA films corresponds to the structure of these polymers in solution.63,64 Other studies also show the correlation of the polymer structure in solution and in the film. Studies on PAH/PSS multilayers65 show that with the increase of the preparation temperature, thickening of multilayers takes place. At higher temperatures, the more pronounced interpenetration of the fresh polymer into the film was suggested. The internal order (conformation) of the polyelectrolytes was expected to undergo a transition from an extended structure to a coiled form, as suggested by the results of neutron reflectivity measurements. This structural transition is due to intramolecular changes, i.e. weaker repulsion between the charges within the polymer backbone at higher temperature.
Thus, the literature findings above suggest that upon increasing the preparation temperature the film structure will be changed according to the changes of the structure of the polymer molecules in solution. With respect to the PLL/HA films, in solution PLL undergoes a temperature dependent conformational change from a random coil to the α-helix or β-sheet.66 This may explain why the HA/PLL film structure depends on the preparation temperature (Fig. 3a and b). The changes of the polymer conformation in solution are related to repulsive interactions on the polymer backbone. This without doubts will also affect inter-polymer (HA–PLL) interactions taking place during the multilayer growth as described below.
The contribution of the inter-polymer interactions by ion pairing to the rate of the film growth is, however, of dual character. The temperature increase promotes the dissociation of ionic contacts making the inter-polymer interaction weaker and thus increasing the polymer mobility in the multilayers. From one point of view, this will increase polymer diffusion into the film and accelerates the film growth as discussed above. However, the increase of the temperature should also make the films less permeable due to annealing.60 The increased polymer mobility allows polymer chains to realize a maximum number of ion pairs for the desired stoichiometric interaction. This reduces a number of defects in the polymer network (annealing) and makes the network more compact and less permeable for diffusing polymers.
It is hard to discriminate between the impacts of the effect of polymer conformation and the effect of strength of inter-polymer interactions on the film growth rate because they are interrelated. For instance, more coiled polymer conformation will affect both the multilayer structure and the number of ion pairs due to sterical hindrance for one polymer chain to form ion pairs with another oppositely charged one.
4. Conclusions and perspectives
We have demonstrated that the growth of PLL/HA films significantly depends on the temperature during the film build-up. The multilayer growth is non-linear or so-called exponential-like. It is the first exponential showing switch (transition point) from the exponential to linear growth regime at 10–21 bilayers. An increase of temperature promotes the enhancing of the growth rate in both the exponential and linear growth regimes. The transition point is shifted to a larger number of bilayers with higher deposition temperatures. Both findings are related to an increase of polymer diffusion in the film at elevated temperatures. We assume that more permeable films are formed at higher preparation temperature as concluded by the experiment when the preparation temperature has been switched during the film growth. The higher is the initial temperature, the faster the film growth after the temperature switch. The as-prepared multilayers are, however, insensitive to temperature changes in the range of 25–45 °C. The average activation energy calculated from the Arrhenius plot has been found to be about 0.3 kJ mol−1 for ion pairing assuming stoichiometric molar interaction between HA and PLL. Due to the dependence of the film assembly rate on the polymer diffusion, polymer conformation, and inter-polymer interactions which in turn all depend on the temperature, it is hard to evaluate the impact of these factors separately.Due to the highly mutual nature of the relation between the factors affecting the film growth rate as described above, the introduction of temperature insensitive molecules into the film might be an approach to entangle the role of various factors. At the same time, fast growth of micrometre-sized HA/PLL films demonstrates strong interest to utilize them as matrices for loading of a large amount of biologically active compounds such as proteins. This opens new avenues for the use of the multilayers as artificial extracellular matrices. Up to now the analysis of the protein–multilayer interactions has been done at room temperature67–69 but not at a physiologically relevant temperature (37 °C). The mechanism of the protein–multilayer interaction is still unclear and first attempts to understand it showed that the PLL diffusion into the film may be responsible for the protein diffusion,67 however, a deeper understanding will be concerned in our future studies.
In addition to this, in future we also plan to evaluate the effect of deposition conditions such as the deposition time and the polymer molecular weight on the film growth and on the position of the transition point for the film build-up. For this we will employ microfluidics as it provides a better control over polymer transport to the film.70 An interesting issue to be considered is the effect of the temperature on processes taking place on the multilayer–solution interface such as polymer chain rearrangement.71 The polymer diffusion through a temperature insensitive porous membrane may shed light on the polymer diffusion through matrices with pore size comparable to polymers. Mesoporous carbonate microspheres are good candidates for such a study and also for applications towards tailor-made templating of polymer-based particles (capsules and beads) with well-defined internal structures.72–79
Acknowledgements
This work has been supported by Alexander von Humboldt Foundation (Sofja Kovalevskaja Program) and DFG (project VO 1716/2-1). PS would like to acknowledge the Erasmus program of Oulu University for financial support.References
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- Y. J. Wang, L. Hosta-Rigau, H. Lomas and F. Caruso, Phys. Chem. Chem. Phys., 2011, 13, 4782–4801 RSC.
- K. Ariga, J. P. Hill and Q. Li, Phys. Chem. Chem. Phys., 2007, 9, 2319–2340 RSC.
- S. H. Yang, E. H. Ko and I. S. Choi, Macromol. Res., 2011, 19, 511–514 CrossRef CAS.
- B. P. Timko, T. Dvir and D. S. Kohane, Adv. Mater., 2010, 22, 4925–4943 CrossRef CAS PubMed.
- K. Ariga, Q. M. Ji, J. P. Hill and A. Vinu, Soft Matter, 2009, 5, 3562–3571 RSC.
- V. Gribova, R. Auzely-Velty and C. Picart, Chem. Mater., 2011, 24, 854–869 CrossRef PubMed.
- S. Pavlukhina and S. Sukhishvili, Adv. Drug Delivery Rev., 2011, 63, 822–836 CrossRef CAS PubMed.
- D. L. Elbert, C. B. Herbert and J. A. Hubbell, Langmuir, 1999, 15, 5355–5362 CrossRef CAS.
- X. H. Liu, L. Smith, G. B. Wei, Y. J. Won and P. X. Ma, J. Biomed. Nanotechnol., 2005, 1, 54–60 CrossRef CAS.
- G. Mehta, M. J. Kiel, J. W. Lee, N. Kotov, J. J. Linderman and S. Takayama, Adv. Funct. Mater., 2007, 17, 2701–2709 CrossRef CAS.
- M. Y. Zhao, L. H. Li, C. R. Zhou, F. Heyroth, B. Fuhrmann, K. Maeder and T. Groth, Biomacromolecules, 2014, 15, 4272–4280 CrossRef CAS PubMed.
- N. Aggarwal, N. Altgarde, S. Svedhem, K. Zhang, S. Fischer and T. Groth, Colloids Surf., B, 2014, 116, 93–103 CrossRef CAS PubMed.
- N. Aggarwal, N. Altgarde, S. Svedhem, K. Zhang, S. Fischer and T. Groth, Langmuir, 2013, 29, 13853–13864 CrossRef CAS PubMed.
- S. Schweizer, T. Schuster, M. Junginger, G. Siekmeyer and A. Taubert, Macromol. Mater. Eng., 2010, 295, 535–543 CrossRef CAS.
- A. Dierich, E. Le Guen, N. Messaddeq, J. F. Stoltz, P. Netter, P. Schaaf, J. C. Voegel and N. Benkirane-Jessel, Adv. Mater., 2007, 19, 693–697 CrossRef CAS.
- N. Madaboosi, K. Uhlig, S. Schmidt, M. S. Jager, H. Mohwald, C. Duschl and D. V. Volodkin, Lab Chip, 2012, 12, 1434–1436 RSC.
- A. A. Antipov, G. B. Sukhorukov and H. Mohwald, Langmuir, 2003, 19, 2444–2448 CrossRef CAS.
- J. Irigoyen, L. Han, I. Llarena, Z. W. Mao, C. Y. Gao and S. E. Moya, Macromol. Rapid Commun., 2012, 33, 1964–1969 CrossRef CAS PubMed.
- W. J. Tong, C. Y. Gao and H. Mohwald, Colloid Polym. Sci., 2008, 286, 1103–1109 CAS.
- Y. F. Pan, H. N. Xiao, G. L. Zhao and B. H. He, Polym. Bull., 2008, 61, 541–551 CrossRef CAS.
- D. V. Volodkin, N. Madaboosi, J. Blacklock, A. G. Skirtach and H. Mohwald, Langmuir, 2009, 25, 14037–14043 CrossRef CAS PubMed.
- D. Volodkin, A. Skirtach, N. Madaboosi, J. Blacklock, R. von Klitzing, A. Lankenau, C. Duschl and H. Mohwald, J. Controlled Release, 2010, 148, e70–e71 CrossRef CAS PubMed.
- D. V. Volodkin, M. Delcea, H. Mohwald and A. G. Skirtach, ACS Appl. Mater. Interfaces, 2009, 1, 1705–1710 CAS.
- A. G. Skirtach, D. V. Volodkin and H. Mohwald, ChemPhysChem, 2010, 11, 822–829 CrossRef CAS PubMed.
- A. G. Skirtach, A. M. Javier, O. Kreft, K. Kohler, A. P. Alberola, H. Mohwald, W. J. Parak and G. B. Sukhorukov, Angew. Chem., Int. Ed., 2006, 45, 4612–4617 CrossRef CAS PubMed.
- Z. Tang, Y. Wang, P. Podsiadlo and N. A. Kotov, Adv. Mater., 2006, 18, 3203–3224 CrossRef CAS.
- T. Boudou, T. Crouzier, K. Ren, G. Blin and C. Picart, Adv. Mater., 2010, 22, 441–467 CrossRef CAS PubMed.
- K. Glinel, C. Dejugnat, M. Prevot, B. Scholer, M. Schonhoff and R. V. Klitzing, Colloids Surf., A, 2007, 303, 3–13 CrossRef CAS.
- H. Ai, S. A. Jones and Y. M. Lvov, Cell Biochem. Biophys., 2003, 39, 23–43 CrossRef CAS PubMed.
- D. Volodkin, A. Skirtach and H. Mohwald, in Bioactive Surfaces, ed. H. G. Borner and J. F. Lutz, Springer-Verlag Berlin, Berlin, 2011, vol. 240, pp. 135–161 Search PubMed.
- D. Volodkin, A. Skirtach and H. Mohwald, Polym. Int., 2012, 61, 673–679 CrossRef CAS.
- P. Lavalle, J. C. Voegel, D. Vautier, B. Senger, P. Schaaf and V. Ball, Adv. Mater., 2011, 23, 1191–1221 CrossRef CAS PubMed.
- D. Volodkin, R. von Klitzing and H. Mohwald, Polymers, 2014, 6, 1502–1527 CrossRef.
- J. Irigoyen, N. Politakos, R. Murray and S. E. Moya, Macromol. Chem. Phys., 2014, 215, 1543–1550 CrossRef CAS.
- D. Gregurec, M. Olszyna, N. Politakos, L. Yate, L. Dahne and S. E. Moya, Colloid Polym. Sci., 2015, 293, 381–388 CAS.
- S. E. Moya, J. J. I. Ramos and I. Llarena, Macromol. Rapid Commun., 2012, 33, 1022–1035 CrossRef CAS PubMed.
- C. Porcel, P. Lavalle, G. Decher, B. Senger, J. C. Voegel and P. Schaaf, Langmuir, 2007, 23, 1898–1904 CrossRef CAS PubMed.
- C. Porcel, P. Lavalle, V. Ball, G. Decher, B. Senger, J.-C. Voegel and P. Schaaf, Langmuir, 2006, 22, 4376–4383 CrossRef CAS PubMed.
- D. Volodkin and R. von Klitzing, Curr. Opin. Colloid Interface Sci., 2014, 19, 25–31 CrossRef CAS.
- N. Laugel, C. Betscha, M. Winterhalter, J.-C. Voegel, P. Schaaf and V. Ball, J. Phys. Chem. B, 2006, 110, 19443–19449 CrossRef CAS PubMed.
- J. E. Wong, F. Rehfeldt, P. Hanni, M. Tanaka and R. V. Klitzing, Macromolecules, 2004, 37, 7285–7289 CrossRef CAS.
- C. Picart, P. Lavalle, P. Hubert, F. J. G. Cuisinier, G. Decher, P. Schaaf and J.-C. Voegel, Langmuir, 2001, 17, 7414–7424 CrossRef CAS.
- P. Lavalle, C. Picart, J. Mutterer, C. Gergely, H. Reiss, J.-C. Voegel, B. Senger and P. Schaaf, J. Phys. Chem. B, 2004, 108, 635–648 CrossRef CAS.
- D. T. Haynie, E. Cho and P. Waduge, Langmuir, 2011, 27, 5700–5704 CrossRef CAS PubMed.
- N. Hoda and R. G. Larson, J. Phys. Chem. B, 2009, 113, 4232–4241 CrossRef CAS PubMed.
- D. Kovacevic, S. van der Burgh, A. de Keizer and M. A. C. Stuart, J. Phys. Chem. B, 2003, 107, 7998–8002 CrossRef CAS.
- K. Buscher, K. Graf, H. Ahrens and C. A. Helm, Langmuir, 2002, 18, 3585–3591 CrossRef.
- M. Westwood, A. P. Gunning and R. Parker, Macromolecules, 2010, 43, 10582–10593 CrossRef CAS.
- M. Salomäki, I. A. Vinokurov and J. Kankare, Langmuir, 2005, 21, 11232–11240 CrossRef PubMed.
- J. S. Shi, F. Hua, T. H. Cui and Y. M. Lvov, Chem. Lett., 2003, 32, 316–317 CrossRef CAS.
- P. Nazaran, V. Bosio, W. Jaeger, D. F. Anghel and R. v. Klitzing, J. Phys. Chem. B, 2007, 111, 8572–8581 CrossRef CAS PubMed.
- D. V. Volodkin, Y. Arntz, P. Schaaf, H. Mohwald, J.-C. Voegel and V. Ball, Soft Matter, 2008, 4, 122–130 RSC.
- T. Crouzier and C. Picart, Biomacromolecules, 2009, 10, 433–442 CrossRef CAS PubMed.
- C. Üzüm, J. Hellweg, N. Madaboosi, D. V. Volodkin and R. von Klitzing, Beilstein J. Nanotechnol., 2012, 3, 778–788 CrossRef PubMed.
- C. Rosca, M. I. Popa, G. Lisa and G. C. Chitanu, Carbohydr. Polym., 2005, 62, 35–41 CrossRef CAS.
- T. H. Silva, V. Garcia-Morales, C. Moura, J. A. Manzanares and F. Silva, Langmuir, 2005, 21, 7461–7467 CrossRef CAS PubMed.
- K. Glinel, A. Moussa, A. M. Jonas and A. Laschewsky, Langmuir, 2002, 18, 1408–1412 CrossRef CAS.
- S. T. Dubas and J. B. Schlenoff, Macromolecules, 1999, 32, 8153–8160 CrossRef CAS.
- H. L. Tan, M. J. McMurdo, G. Q. Pan and P. G. Van Patten, Langmuir, 2003, 19, 9311–9314 CrossRef CAS.
- L. Jourdainne, S. Lecuyer, Y. Arntz, C. Picart, P. Schaaf, B. Senger, J. C. Voegel, P. Lavalle and T. Charitat, Langmuir, 2008, 24, 7842–7847 CrossRef CAS PubMed.
- D. R. Lide, T. J. Bruno and W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, Colorado, USA, 2010 Search PubMed.
- F. Boulmedais, P. Schwinte, C. Gergely, J.-C. Voegel and P. Schaaf, Langmuir, 2002, 18, 4523–4525 CrossRef CAS.
- D. T. Haynie, S. Balkundi, N. Palath, K. Chakravarthula and K. Dave, Langmuir, 2004, 20, 4540–4547 CrossRef CAS PubMed.
- M. Gopinadhan, H. Ahrens, J. U. Gunther, R. Steitz and C. A. Helm, Macromolecules, 2005, 38, 5228–5235 CrossRef CAS.
- L. Rodriguez-Maldonado, A. Fernandez-Nieves and A. Fernandez-Barbero, Colloids Surf., A, 2005, 270, 335–339 CrossRef.
- K. Uhlig, N. Madaboosi, S. Schmidt, M. S. Jager, J. Rose, C. Duschl and D. V. Volodkin, Soft Matter, 2012, 8, 11786–11789 RSC.
- C. Vogt, V. Ball, J. Mutterer, P. Schaaf, J. C. Voegel, B. Senger and P. Lavalle, J. Phys. Chem. B, 2012, 116, 5269–5278 CrossRef CAS PubMed.
- D. Sustr, C. Duschl and D. Volodkin, Eur. Polym. J., 2015, 68, 665–670 CrossRef CAS.
- N. Madaboosi, K. Uhlig, M. S. Jäger, H. Mohwald, C. Duschl and D. V. Volodkin, Macromol. Rapid Commun., 2012, 33, 1775–1779 CrossRef CAS PubMed.
- A. S. Vikulina, S. T. Aleed, T. Paulraj, Y. A. Vladimirov, C. Duschl, R. von Klitzing and D. Volodkin, Phys. Chem. Chem. Phys., 2015, 17, 12771–12777 RSC.
- S. Schmidt and D. Volodkin, J. Mater. Chem. B, 2013, 1, 1210–1218 RSC.
- M. Behra, S. Schmidt, J. Hartmann, D. V. Volodkin and L. Hartmann, Macromol. Rapid Commun., 2012, 33, 1049–1054 CrossRef CAS PubMed.
- D. Volodkin, Adv. Colloid Interface Sci., 2014, 207, 306–324 CrossRef CAS PubMed.
- N. G. Balabushevich, A. V. Lopez de Guerenu, N. A. Feoktistova and D. Volodkin, Phys. Chem. Chem. Phys., 2015, 17, 2523–2530 RSC.
- M. A. Pechenkin, H. Mohwald and D. V. Volodkin, Soft Matter, 2012, 8, 8659–8665 RSC.
- I. Marchenko, A. Yashchenok, T. Borodina, T. Bukreeva, M. Konrad, H. Mohwald and A. Skirtach, J. Controlled Release, 2012, 162, 599–605 CrossRef CAS PubMed.
- B. G. De Geest, R. E. Vandenbroucke, A. M. Guenther, G. B. Sukhorukov, W. E. Hennink, N. N. Sanders, J. Demeester and S. C. De Smedt, Adv. Mater., 2006, 18, 1005–1009 CrossRef CAS.
- A. Szarpak, I. Pignot-Paintrand, C. Nicolas, C. Picart and R. Auzely-Velty, Langmuir, 2008, 24, 9767–9774 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Graphs for FITC–PLL calibration, transition points for film growth at different temperatures, CSLM Z-stack for the 30 bilayer film at 45 and 25 °C, and a table with activation energy for various polymer pairs. See DOI: 10.1039/c6cp00345a |
| This journal is © the Owner Societies 2016 |