
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Interaction of Cu+ with cytosine and formation of i-motif-like C–M+–C complexes: alkali versus coinage metals†
Juehan
Gao
a,
Giel
Berden
a,
M. T.
Rodgers
b and
Jos
Oomens
*ac
aRadboud University, Institute for Molecules and Materials, FELIX Laboratory, Toernooiveld 7c, 6525 ED Nijmegen, The Netherlands. E-mail: joso@science.ru.nl; Tel: +31 24 3653950
bDepartment of Chemistry, Wayne State University, Detroit, MI 48335, USA
cVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
First published on 9th February 2016
Abstract
The Watson–Crick structure of DNA is among the most well-known molecular structures of our time. However, alternative base-pairing motifs are also known to occur, often depending on base sequence, pH, or the presence of cations. Pairing of cytosine (C) bases induced by the sharing of a single proton (C–H+–C) may give rise to the so-called i-motif, which occurs primarily in expanded trinucleotide repeats and the telomeric region of DNA, particularly at low pH. At physiological pH, silver cations were recently found to stabilize C dimers in a C–Ag+–C structure analogous to the hemiprotonated C-dimer. Here we use infrared ion spectroscopy in combination with density functional theory calculations at the B3LYP/6-311G+(2df,2p) level to show that copper in the 1+ oxidation state induces an analogous formation of C–Cu+–C structures. In contrast to protons and these transition metal ions, alkali metal ions induce a different dimer structure, where each ligand coordinates the alkali metal ion in a bidentate fashion in which the N3 and O2 atoms of both cytosine ligands coordinate to the metal ion, sacrificing hydrogen-bonding interactions between the ligands for improved chelation of the metal cation.
1. Introduction
DNA base pairing in motifs other than the well-known Watson–Crick structure have been under thorough study in recent years, where in particular the G-quadruplex and i-motif structures have received considerable attention. The i-motif, occurring frequently in cytosine-rich regions of telomeric DNA and in extended trinucleotide repeats, is formed by refolding of one of the strands of double-stranded DNA upon itself.1–3 The structure is stabilized by intercalated pairs of cytosine bases, each pair sharing an excess proton. Mass spectrometric studies using ion spectroscopy have shown that the hemiprotonated cytosine dimer forms readily upon electrospray ionization (ESI) of an acidic solution of cytosine (C). The base pairing energy of the hemiprotonated C dimer was experimentally determined to be about 170 kJ mol−1.4,5Formation of structures resembling the i-motif under neutral pH conditions, where the shared proton is replaced by an Ag+ ion, was recently reported.6 This and other studies have reported on the fluorescent properties of Ag+-containing solutions of DNA. More recent ion spectroscopy studies showed that a solution of C with added silver salt indeed leads to the formation of a C–Ag+–C dimer with a structure reminiscent of that of the hemiprotonated C dimer, C–H+–C.7
In the protonated C–H+–C dimer, the proton finds itself in a shallow double-well potential and can be localized on the N3 atom of either of the C nucleobases. The barrier to transfer was computed at 6.7 kJ mol−1.5 The hemiprotonated dimer is further stabilized by two HNH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds between the amino and carbonyl groups of each of the cytosine bases. For the C–Ag+–C dimer, the metal ion localizes at a position symmetric between the two N3-atoms. However, the larger ionic radius of the Ag+ ion forces the dimer to adopt a structure in which the two HNH⋯OC hydrogen bonds have clearly unequal bond lengths of about 3.5 and 5.5 Å (measured from the amino N to carbonyl O atom).8
C hydrogen bonds between the amino and carbonyl groups of each of the cytosine bases. For the C–Ag+–C dimer, the metal ion localizes at a position symmetric between the two N3-atoms. However, the larger ionic radius of the Ag+ ion forces the dimer to adopt a structure in which the two HNH⋯OC hydrogen bonds have clearly unequal bond lengths of about 3.5 and 5.5 Å (measured from the amino N to carbonyl O atom).8
The primary question that we address in this study is whether the i-motif like structure of C–Ag+–C is unique to the silver ion, or whether other 1+ cations induce a similar structure of the C-dimer. In particular, we employ ion spectroscopy in combination with quantum-chemical calculations to study the structure of dimeric complexes of C with the Cu+ ion and the alkali metal ions Li+, Na+ and K+. The copper ion has a 3d10-electronic configuration analogous to the 4d10 configuration of the silver ion, but has a smaller ionic radius than Ag+ such that it may ‘fit’ better within the central cavity formed by the two cytosine residues and thus form a more symmetric structure with nearly equivalent HNH⋯OC hydrogen bonds in analogy to the hemi-protonated cytosine base pair. The ionic radii of the alkali metal ions studied here (Li+, Na+, and K+) span those of the Cu+ and Ag+ ions.9 Mass spectrometric studies have revealed a high propensity of forming M+C2 complexes from ESI solutions of cytosine and alkali metal salts,10 but the geometries of the complexes were not investigated in detail.
Gas-phase IR photodissociation spectroscopy of charged species in ion trapping devices has been applied frequently in recent years to reveal the tautomeric structure of protonated and metalated nucleobases.7,11–21 We present here the first IR spectra of mass selected C–M+–C complexes, where M = Cu, Li, Na and K, and derive their coordination geometries by comparison with computed spectra. For Cu+, dimeric complexes with other N-donor ligands have received ample attention, e.g. in a series of threshold collision-induced dissociation (TCID) studies by the group of Rodgers.22,23 Analysis of the TCID data combined with computational investigations established substantial hybridization of the occupied 3d and empty 4s atomic orbitals of the Cu+ center, giving these dimeric complexes a typical linear N–Cu+–N bond.
Before discussing the C–M+–C complexes, we will discuss the spectra and structure of complexes of Cu+ with a single cytosine ligand. Various studies have addressed the structures of monomeric cytosine complexed to metal ions. Rodgers and coworkers studied the series of alkali metal ions establishing the binding energies and coordination structures using TCID4,24,25 and infrared ion spectroscopy11 combined with quantum-chemical calculations. The gas-phase IR spectrum of monomeric Ag+–C was very recently reported by Pino et al.14
2. Experimental section
2.1 Experiment
IRMPD spectra of copper–cytosine complexes were recorded in the electrospray ionization Fourier-transform ion cyclotron resonance mass spectrometer (ESI-FTICR-MS),26 coupled to the beam line of the FELIX free electron laser.27,28 General experimental methods have been described in detail previously.27,29 The same ESI-FTICR-MS was also used in combination with a pulsed Nd:YAG pumped optical parametric oscillator (OPO, LaserVision, Bellevue, WA) to obtain IR spectra in the hydrogen stretching region of the spectrum at wavelengths around 3 μm. To enhance the IR induced dissociation, a cw CO2-laser was used to (non-resonantly) post-excite the ions after interaction with the OPO laser.30 While this procedure enhances the IRMPD efficiency allowing us to also observe weaker transitions (see Fig. S1 of the ESI†), one also notes an expected slight line-broadening and red-shifting of bands as a result of the higher total IR fluence.31,32ESI solutions were prepared in pure acetonitrile (MeCN) containing 0.35–0.5 mM CuCl and 0.5–1 mM cytosine. Cu+(Cytosine)1,2 and (MeCN)Cu+(Cytosine) complexes were generated using a modified Waters Z-Spray ESI source and were accumulated in a linear hexapole ion trap before being injected into the ICR cell via an octopole rf ion guide. The precursor ions were mass-isolated by a stored-waveform inverse Fourier-Transform (SWIFT26) excitation pulse and subsequently irradiated by the FEL radiation (up to 40 mJ pulse energy, ∼5 μs pulse duration, ∼0.5% bandwidth). A series of mass spectra were recorded with the FEL radiation being tuned over the frequency range from 600 to 1800 cm−1 and also with the IR-OPO being tuned from 3300 to 3700 cm−1 (up to 20 mJ pulse energy, ∼6 ns pulse duration, ∼3 cm−1 bandwidth). When resonant with an allowed vibrational transition, the ion undergoes fragmentation as a consequence of IR multiple photon dissociation (IRMPD).27 An IR spectrum of the ion was reconstructed from the mass spectra by plotting the fragmentation ion yield, defined here as  , as function of IR frequency. Here, Iprecursor is the sum of the intensities of the precursor ions (for 63Cu and 65Cu) and Ifragments is the sum of the intensities of all fragment ions. The yield is linearly corrected for frequency dependent variations in laser pulse energy.
, as function of IR frequency. Here, Iprecursor is the sum of the intensities of the precursor ions (for 63Cu and 65Cu) and Ifragments is the sum of the intensities of all fragment ions. The yield is linearly corrected for frequency dependent variations in laser pulse energy.
Because the preferred coordination number of Cu+ is two, the Cu+(Cytosine)2 complex at m/z = 285 and 287 and the (MeCN)Cu+(Cytosine) complex at m/z = 215 and 217 are produced readily from the ESI source. To prepare the singly coordinated complex Cu+(Cytosine), the Cu+(Cytosine)2 complex was isolated and subsequently irradiated with a 30 W cw CO2 laser for 1.3 seconds, generating the Cu+(Cytosine) complex at m/z 174 and 176. IRMPD of Cu+(Cytosine)2 and (MeCN)Cu+(Cytosine) induces the neutral losses of 111 mass units (neutral cytosine) and 43 mass units (corresponding to HNCO); for Cu(Cytosine)+ neutral loss of 43 mass units is the only dissociation channel observed. These fragmentation patterns are analogous to those observed for the Ag+ complexes of cytosine.7,14 IRMPD spectra of three complexes, Cu+(Cytosine), (MeCN)Cu+(Cytosine) and Cu+(Cytosine)2 are presented in Fig. 1–3, respectively.
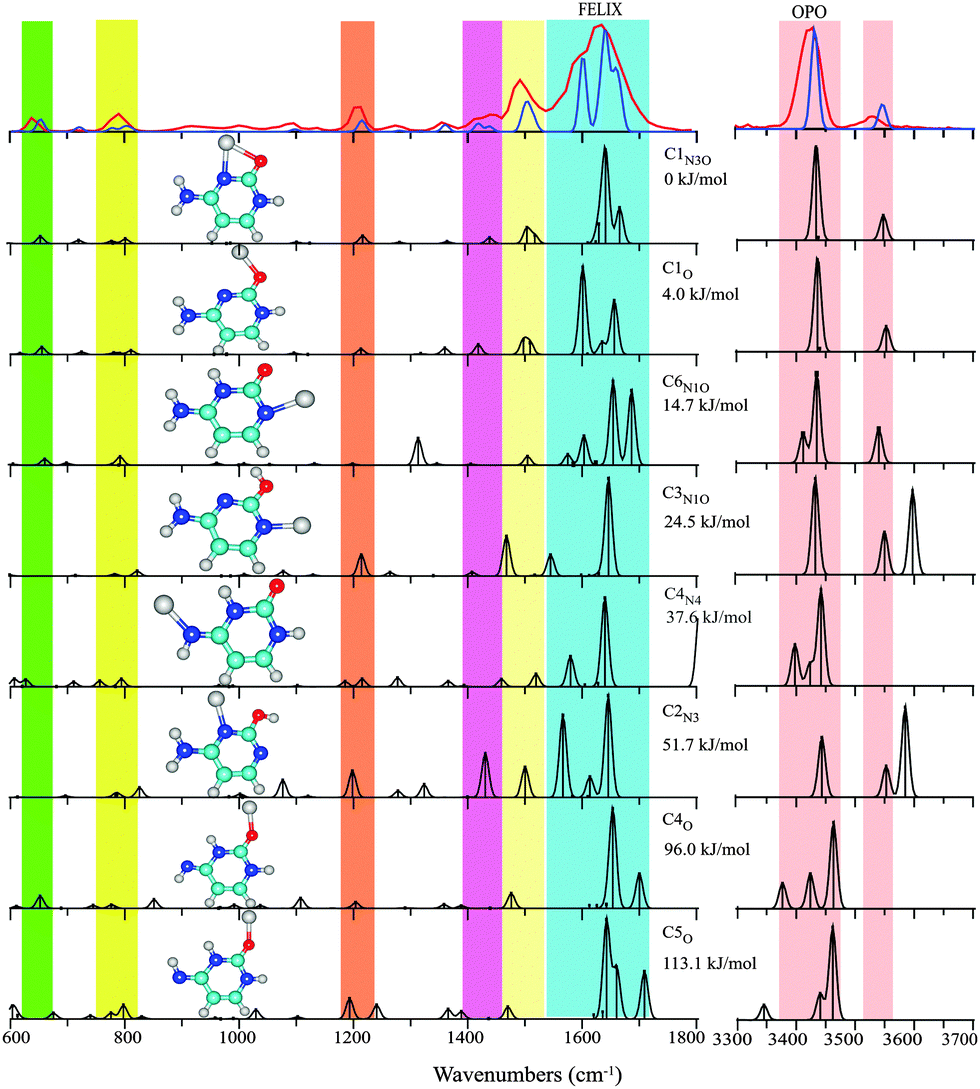 | ||
| Fig. 1 Experimental IRMPD spectrum (red trace) of Cu+(Cytosine) between 600–1800 cm−1 and 3300–3700 cm−1 compared with the computational spectra (black traces) for eight structures ordered by their relative energies at the B3LYP level (see Table 1 and text for nomenclature). The blue trace, which provides a close match to the experimental spectrum, represents a one-to-one mixture of the spectra computed for C1N3O and C1O. Colored vertical bars indicate regions of main IR activity in the experimental spectrum in the top panel. | ||
 | ||
Fig. 2 Experimental IRMPD spectrum of the MeCN–Cu+–C complex between 600–1800 cm−1 and 3300–3700 cm−1 (red trace) compared with the computed spectra of five low-energy isomers (black traces). Colored vertical bars indicate spectral regions of the most prominent experimental bands; from these, contributions to the experimental spectrum from the three higher energy isomers can be readily discarded. The computed spectrum due to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the two lowest-energy isomers is overlaid as the blue trace onto the experimental spectrum in the top panel. :1 mixture of the two lowest-energy isomers is overlaid as the blue trace onto the experimental spectrum in the top panel. | ||
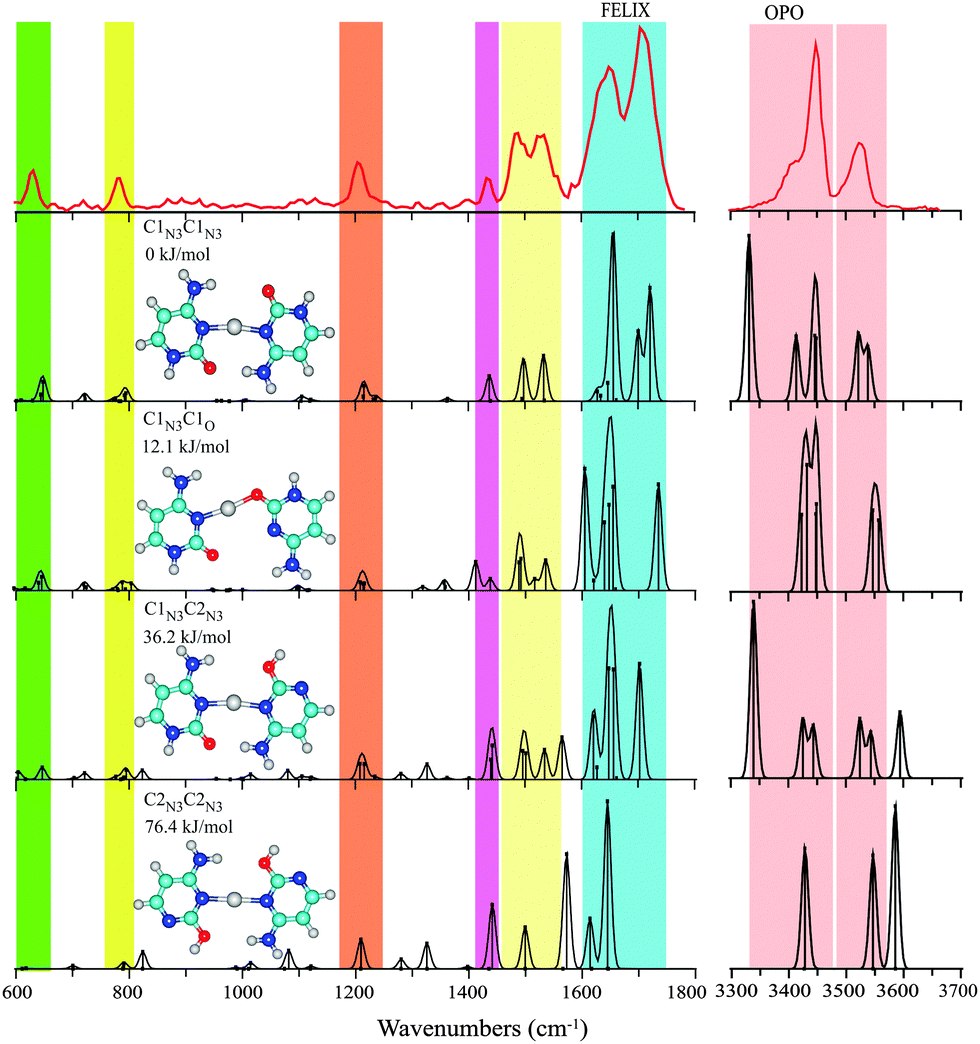 | ||
| Fig. 3 Experimental IRMPD spectrum of C–Cu+–C between 600–1800 cm−1 and 3300–3700 cm−1 (red trace) compared with computational spectra of four low-energy isomers. The computed spectrum for the structure resembling the i-motif, C1N3C1N3, is seen to provide the best agreement with the experimental spectrum, if one ignores the intense feature predicted near 3330 cm−1, which is due to the strongly hydrogen-bonded NH stretching mode (see text). | ||
Dimeric cytosine alkali metal complexes are produced using solutions of 0.5–1 mM of the alkali metal chloride salts and 0.5–1 mM cytosine in an approximately 50:50 MeOH/H2O mixture. IRMPD of Li+(Cytosine)2, Na+(Cytosine)2, and K+(Cytosine)2 results in the loss of a neutral cytosine monomer. The IRMPD spectra of the three dimeric cytosine alkali metal complexes are presented in Fig. 4 and Fig. S4 (ESI†).
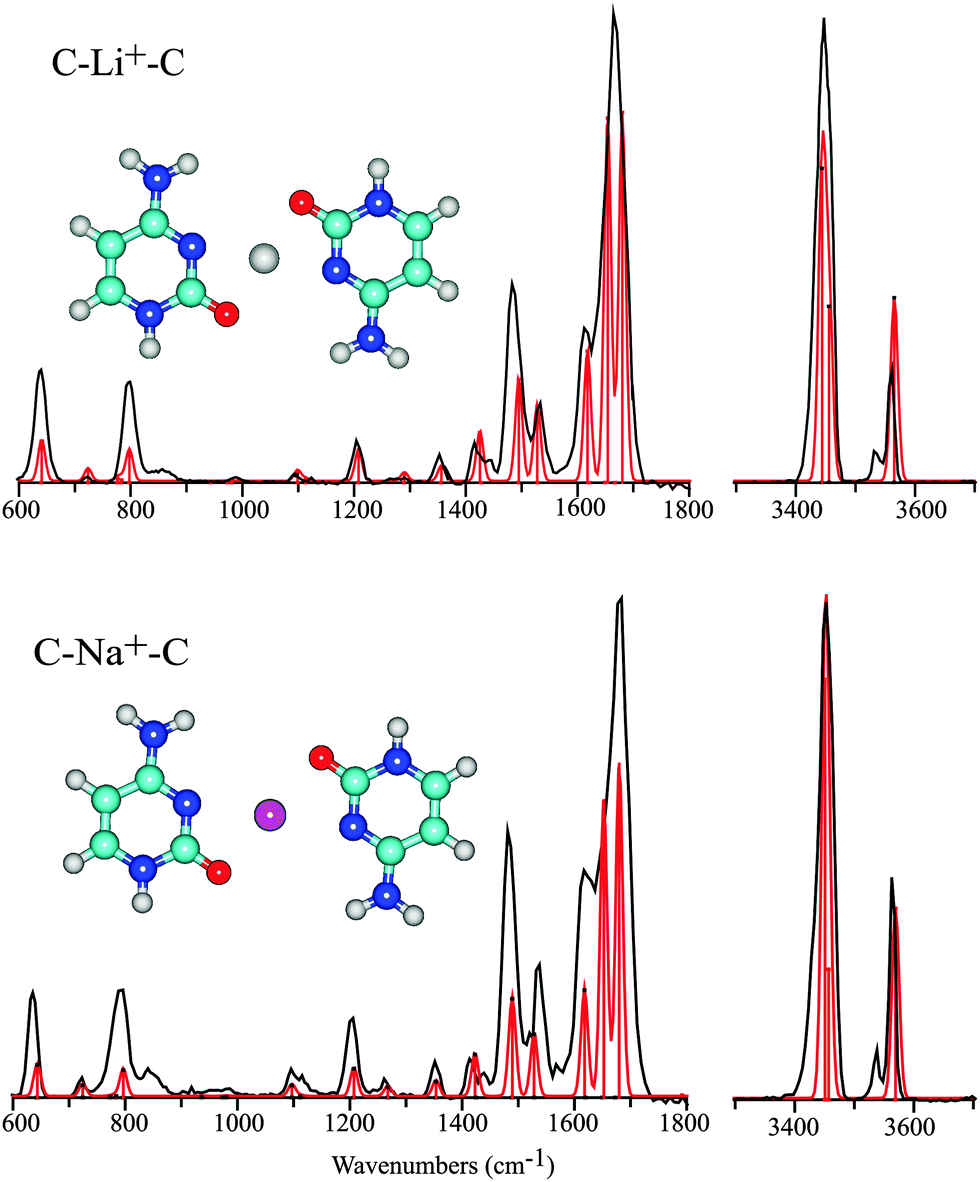 | ||
| Fig. 4 Experimental IRMPD spectra of C–Li+–C and C–Na+–C in the 600–1800 cm−1 and hydrogen stretching regions (black trace), compared with the computed spectra for the C1N3OC1N3O isomers (red trace) of these complexes. | ||
2.2 Computational
Optimized geometries and harmonic vibrational spectra for the complexes of interest were computed using density functional theory (DFT) with the B3LYP functional and the 6-311+G(2df,2p) basis set as implemented in the Gaussian09 suites of programs. Only singlet electronic states were considered. Electronic energies were corrected for zero-point energy (ZPE) and are reported as Gibbs free energies at 298 K. In addition, single-point MP2 energies have been computed at the B3LYP/6-311+G(2df,2p) optimized structure. For the complexes, counterpoise corrections have been applied to correct for basis set superposition errors (BSSE). Partial charges and effects of orbital hybridization were investigated on the basis of natural population analyses (NPA) of the systems. Dispersion corrections for the B3LYP energies were computed using Grimme's D3 empirical correction implemented in Gaussian09.Harmonic vibrational frequencies were scaled by a factor of 0.98, except for the H-stretching modes, which were scaled by 0.956. The computed stick spectra were convoluted using a 15 cm−1 full-width-at-half-maximum (FWHM) Gaussian line shape for easy comparison with experimental spectra.
Numerous studies have reported computed data for complexes of cytosine with metal ions. Low-energy tautomeric conformations of cytosine and its complexes with alkali metal ions were reported among others by Rodgers and coworkers11,24 and Hobza and coworkers.33 Computational investigations of coordination to coinage metals, especially Cu+ and Ag+, have also been reported.7,34–36 As far as we are aware, no computations have been published for dimeric C–M+–C complexes, except for the silver ion complex, where the B3LYP functional with the 6-311G++(d,p) basis set was employed for all atoms except Ag, which was described by the SDD effective core potential.7
3. Results and discussion
Nomenclature
Cytosine can adopt various tautomeric forms, depending on the keto (K) or enol (E) form of the oxygen atom and the amine (A) or imine (I) form of the N4 substituent (see Fig. S2 in the ESI† for atom numbering). Although a nomenclature based on these labels is appealing,7,37,38 it does not uniquely identify the tautomers (e.g. there are two tautomers of KA type), and we shall adopt the C1–C6 labels used by Rodgers and coworkers as well as others.24,34 The correspondence is shown in Table 1. For each of the tautomeric forms of the nucleobase, coordination of the metal ion can occur at different sites; we shall identify the anchor(s) as a subscript to the tautomer label. We only considered σ-bonding of the metal ion to one of the hetero-atoms of cytosine; π-complexes were not considered.| Isomer | Isomera | B3LYPb | MP2c | |
|---|---|---|---|---|
| a Alternative nomenclature. Note that keto/enol and amine/imine labels do not uniquely define the molecular structure. KA and KA′ are different tautomers having a proton on either N1 or N3. Structures labeled with a * have the proton on the enol or imine group in the E/Z isomeric configuration. b Values in brackets: after Grimme's D3 empirical dispersion correction. c Single point calculation on B3LYP optimized structure. | ||||
| C1N3O | KAN3O | 0 | (0) | 0 |
| C1O | KAO | 4.0 | (6.1) | 9.4 |
| C6N1O | KAo′ | 14.7 | 16.8 | |
| C3N1O | EAN1O | 24.5 | 19.0 | |
| C4N4 | KIN4 | 37.6 | 40.9 | |
| C2N3 | EAN3* | 51.7 | 45.9 | |
| C4O | KIO | 96.0 | 102.4 | |
| C5O | KIo* | 113.1 | 121.8 | |
In isolated neutral cytosine, the canonical keto-amino C1 tautomer is very close in energy to the enol-amino tautomer C2; which of the two is lower in energy depends on the level of theory that is used.11 Experimentally, both tautomers have been shown to co-exist.37,38 Alkali metal ion binding strongly stabilizes the C1 tautomer relative to C2 (44 kJ mol−1 for Na+),11,24,39 and a similar stabilization (53 kJ mol−1) was computed for Cu+ coordination in the C1N3O configuration.34
Cu+–Cytosine
In line with previous computations on Cu+(Cytosine) complexes,34,35 we found the structure with the base in the C1 keto tautomeric form and the Cu ion coordinating to the N3 and O atoms to be lowest in energy (C1N3O). This structure is analogous to the minimum energy structures for alkali metal ion complexes of cytosine.11,24,39 Only slightly higher in energy, at 4 kJ mol−1, we identified an apparently unreported local minimum in which the ion coordinates predominantly to the O atom of the C1 tautomer (C1O) with a ∠CO–Cu angle of 114°, suggestive of covalent rather than electrostatic interaction.40 The alternative keto-amine tautomer C6 allows for Cu+-coordination to the N1 atom (C6N1O), forming a complex at about 15 kJ mol−1 from the global minimum C1N3O. Complexes with the nucleobase in the enol or in the imine tautomer forms are at least 24 kJ mol−1 higher in energy. Computed structures, relative energies and scaled harmonic vibrational spectra for representative structures are shown in Fig. 1.
The experimental IR spectrum of Cu+(Cytosine) is shown in the top panel of Fig. 1 along with the computed IR spectra for the structures of Table 1. The broad, double-peaked experimental feature extending from about 1550 to 1700 cm−1 suggests that the spectrum cannot be explained solely by the global minimum C1N3O structure. This suggestion is further enforced by the clear observation of two features between 1400 and 1450 cm−1, where only one band is predicted for C1N3O. Interestingly, the spectrum predicted for the C1O complex, which is only slightly higher in energy, shows IR activity exactly at the frequencies near 1420 and 1600 cm−1 that cannot be explained by C1N3O. The remaining features in the predicted spectra for the two structures, including the hydrogen stretching region, are virtually identical and match excellently with the experimental spectrum. A one-to-one sum of the predicted spectra of C1N3O and C1O is overlaid onto the experimental spectrum in Fig. 1 to showcase the excellent agreement.
The colored vertical bars in Fig. 1 identify the main experimental features. Intense computed IR activity outside these colored regions can be taken as indication for the non-presence of particular isomers. Based on this quick test, we can exclude the presence of the next higher isomers: C6N1O has a fairly intense in-plane N3H bending mode near 1300 cm−1; C3N1O has an OH stretch near 3600 cm−1; C4N4 has its CO stretch near 1800 cm−1; C2N3 can also be excluded based on its high-frequency OH stretch; the imine tautomers C4O and C5O can be excluded based on their spectrum in the H-stretching region.
Hence, from the IR spectral data we conclude that cytosine adopts a C1 tautomeric structure upon Cu+ binding, as in the alkali metal11,24,39 and silver41 ion complexes. However, in contrast to the alkali metal ion complexes, two slightly different Cu+ binding motifs, C1N3O and C1O, appear to coexist. We speculate that the increased covalent character of the Cu–O bond as compared to the alkali metal ions, stabilizes the C1O structure with its 114° ∠CO–Cu angle, sacrificing the stabilization by coordination to the N3 atom. The co-existence of C1N3O and C1O is not unreasonable based on the computed energy difference of 4 kJ mol−1, although the MP2 single-point calculation favors C1N3O more strongly (see Table 1). Existence of the C1O motif was not reported for the Ag+ complex.14
MeCN–Cu+–C complex
Abundant formation of Cu+(Cytosine)(MeCN) complexes occurs under our ESI conditions. The d10-configuration of Cu+ suggests a linear arrangement of the two N-donor ligands around the copper center.42 Computations indeed indicate that the complexes adopt a structure in which each of the ligands coordinates in a monodentate fashion, with MeCN coordinating through its N-atom and cytosine through one of its N or O anchors; bidentate coordination of cytosine, as in the lowest energy C1N3O isomer of Cu+(Cytosine), is not encountered. With the nucleobase in its keto form, coordination of cytosine through its O-atom gives the lowest energy isomer; the ∠CO–Cu bond angle optimizes at 126°, again suggestive of substantial covalent bond character. Coordination through the N3 atom leads to a structure 6 kJ mol−1 higher in energy (see Table 2). MP2 single point calculations reverse the energetic ordering of these isomers, favoring C1N3MeCN slightly over C1OMeCN. Computations for complexes with cytosine in the enol or imine tautomeric forms also lead to monodentate structures with a linear arrangement around the copper center, but they are higher in energy by at least 15 kJ mol−1, although the MP2 calculation places the C3N1MeCN isomer considerably closer in energy, at only 1 kJ mol−1 from the global minimum. Structures are shown in Fig. 2 along with their relative energies and computed vibrational spectra.
The experimental spectrum of MeCN–Cu+–C is compared with the computed spectra of five low-energy isomers. Neither the computed spectrum of C1OMeCN alone nor that of C3N1MeCN alone can explain the three features in the 1600–1800 cm−1 region of the observed spectrum. In contrast, a mixture of the two computed spectra satisfactorily reproduces these experimental features, as well as the remainder of the spectrum. An analysis guided by the colored vertical bars as was done for the Cu+(Cytosine) complex shows that the higher energy isomers, C3N1MeCN, C4N4MeCN and C2N3MeCN, can be excluded based on predicted bands around 3600 cm−1, 1800 cm−1 and 3600 cm−1, respectively. The spectroscopic results therefore appear to be more in line with the energetic ordering suggested by the B3LYP calculations than the MP2 single point calculations.
C–Cu+–C complex
Computations indicate that the C–Cu+–C complex prefers a structure reminiscent of the i-motif structure of the hemiprotonated cytosine dimer, C–H+–C, with both cytosine nucleobases in their keto form, similar to that found for the C–Ag+–C complex.7 In contrast to C–Cu+–MeCN, coordination to the cytosine N atoms, rather than to the O atom, now leads to the lowest energy structure, C1N3C1N3. In this conformation, presented in Fig. 3, the complex enjoys additional stabilization of two NH⋯OC hydrogen bonds between the amino and carbonyl moieties of the two cytosine subunits. The complex is somewhat less symmetric than the hemiprotonated cytosine dimer, as the larger size of the cation causes the two cytosine units to “hinge” apart, giving two NH⋯OC hydrogen bonds of unequal length. The computed N–O distances are 3.24 and 4.44 Å, which may be compared with 3.3 and 5.3 Å for the analogous, but still more asymmetric, C–Ag+–C complex.7 These values are 2.699 and 2.973 Å in the hemiprotonated cytosine dimer.
The next higher-energy structure C1N3C1O is one with both cytosine subunits also in their keto-amino form, though one of them is now coordinating through its O-atom. As found consistently for O-coordinated structures, the ∠C–O–Cu+ angle (121°) optimizes to a value close to that expected for a covalent bond. Based on the structures identified for Cu+–C and MeCN–Cu+–C above involving coordination through the O-atom, one might have suspected that O-coordination would have been preferred for C–Cu+–C as well. However, for this dimeric species it comes at the cost of sacrificing the hydrogen-bond stabilization. The computations place the C1N3C1O isomer 12 kJ mol−1 higher in energy than C1N3C1N3. The MP2 computation consistently places O-coordinated complexes higher in relative energy than B3LYP computations, and this is also observed here (see Table 3). The difference in energies can be traced back to the neglect of dispersion interactions in the DFT computations; applying an empirical dispersion correction (Grimme D3) to the B3LYP energies brings the DFT values closer to the MP2 values for all complexes studied here (see values in brackets for the lowest energy isomers in Tables 1–3). We note that the C1N3C1O isomer was not considered for C–Ag+–C by Berdakin et al.7
The other two C–Cu+–C structures explicitly considered here have either one or both cytosine nucleobases in the C2 enol-amine configuration, while both coordinating to the Cu ion via the N3 atom. The isomer C1N3C2N3 is further stabilized by an NH⋯OH and an NH⋯OC hydrogen bond, whereas C2N3C2N3 is stabilized by two NH⋯OH hydrogen bonds. The relative energies of these isomers, 36 and 76 kJ mol−1, respectively, are in the range of those reported for the analogous C–Ag+–C complexes (32 and 62 kJ mol−1).7
The experimental spectrum of C–Cu+–C is shown in the top panel of Fig. 3. Unlike the two complexes discussed above, the experimental bands in the 600–1800 cm−1 range of the spectrum can be rationalized by a single isomer, that of the global minimum C1N3C1N3 “i-motif like” structure. Note that even fine details of the experimental spectrum are well reproduced by the theoretical prediction, such as the triple-band structure between 1400 and 1570 cm−1 and the 1206 cm−1 feature with its faint blue shoulder. A contribution from the next higher isomer C1N3C1O cannot be entirely excluded, but is believed to be minor at most, judging from the intense predicted bands near 1605 and 1735 cm−1, which overlap only marginally with the broad partially resolved experimental feature between 1600 and 1740 cm−1. The higher energy isomers involving enol tautomers cannot be excluded entirely based on their computed spectra in the 600–1800 cm−1 range, but the spectrum in the hydrogen stretching range provides more compelling evidence for their absence. The strong OH stretching modes diagnostic for these tautomers are predicted around 3590 cm−1 and are clearly not observed.
On the other hand, it may be argued that the intense feature predicted near 3330 cm−1 in the spectrum of the attributed C1N3C1N3 isomer is clearly not present in the experimental spectrum. Visual inspection of the normal modes reveals that this band corresponds to the NH stretching of the hydrogen bonded NH. Severe broadening as well as red-shifting of such strongly hydrogen-bonded NH (and OH) stretches in IRMPD spectra are a well-known phenomenon12,13,43 and we suspect that the absence of this band is another manifestation of this effect. Note also that harmonic calculations may not yield reliable frequencies and intensities for these strongly hydrogen-bonded modes, as inferred from comparisons with linear absorption spectra.44 To verify our assumption, we recorded the 3 μm IRMPD spectrum for the C–Ag+–C complex (see Fig. S3 in the ESI†), which was also assigned to the C1N3C1N3 structure on the basis of its 1100–1800 cm−1 spectrum.7 Comparison with the calculated spectrum again shows that the hydrogen-bonded NH stretching mode is not observed in the experimental spectrum.
We conclude that the C–Cu+–C complex adopts (a slightly asymmetric version of) an i-motif structure, analogous to that identified for the C–Ag+–C complex. The IR spectra of the Cu+–C and MeCN–Cu+–C complexes indicate that Cu+ binding to the carbonyl oxygen and to the N3-nitrogen provide very similar stabilization, such that both binding motifs occur in roughly equal abundances. Note that this is consistent with the behavior for the protonated form of C, dCyd and Cyd.45 In the C–Cu+–C complex, coordination to the carbonyl O-atom is given up and traded in for additional stabilization via hydrogen-bonding. In the remainder of this paper, we investigate whether this structure is generic for C–M+–C complexes, or whether it is unique for the coinage metals, Cu and Ag.
C–M+–C complexes, where M = Li, Na, K
Various experimental and theoretical studies have established the structure of mono-ligated M+–C complexes for the alkali metal ions including Li, Na and K.11,24 These studies have consistently identified the keto-amino tautomer C1 with the alkali metal ion attached to the O-atom as the lowest energy structure. Alkali metal ion coordination leads to a considerable stabilization of the C1 tautomer with respect to C2, which is very close in energy in absence of metal ion coordination. Unlike the coinage metals, the alkali metals coordinate to the carbonyl in an approximately linear CO–M+ geometry11,24 (although alternative structures have also been suggested33). Metal ion binding energies have been reported to be around 235 kJ mol−1 for Li+ (C3 tautomer) and 210 kJ mol−1 for Na+.24,25 Our computations for the dimeric C–Li+–C and C–Na+–C complexes starting from the optimized structure for C–Cu+–C relax to a planar parallel-displaced structure. In this structure, the alkali metal ion is 4-fold coordinated, binding to both the N3 and O-atoms of each of the cytosine nucleobases in an approximately square-planar fashion (see Fig. 4). The hydrogen bonds between the two cytosine ligands are sacrificed to allow for bidentate coordination of the metal ion.
The spherical nature of the alkali metal ions in combination with electron pair repulsion arguments may suggest tetrahedral coordination, with the planes of the two cytosine nucleobases being approximately perpendicular. However, a relaxed potential energy surface scan (shown in Fig. 5) varying the angle between the planes of the two cytosine residues (using the N3–C2–N3′–C2′ dihedral angle as the redundant coordinate) indicates that an anti-parallel planar geometry with approximately square-planar coordination is favored over tetrahedral coordination by approximately 5 kJ mol−1. Moreover, a planar parallel structure is further disfavored, lying about 15 kJ mol−1 above the anti-parallel geometry, suggesting that despite the lack of formal H-bonding interactions, additional stabilization from interaction between the two ligands, leading to a highly symmetric structure and cancellation of the local dipole moments, is still present.
 | ||
| Fig. 5 Relaxed potential energy surface scan for the C–Na+–C complex connecting the planar anti-parallel global minimum structure, C1N3OC1N3O, with the planar parallel structure (both exhibiting approximately square-planar metal ion coordination), traversing the perpendicular structure with an approximate tetrahedral coordination geometry. | ||
Fig. 4 shows the experimental spectra of the C–Li+–C and C–Na+–C complexes with their computed counterparts for the parallel-displaced planar anti-parallel structure overlaid. The spectrum for C–K+–C is of slightly lower quality in terms of the signal to noise ratio, but is otherwise analogous and shown in Fig. S4 in the ESI.† The spectra of the Na+ and Li+ complexes are almost identical, and moreover match the computed spectra very closely. All predicted bands are observed in the experimental spectra, with band positions coinciding within about 10 cm−1 and relative intensities in good qualitative agreement. Both experimental spectra consistently show two weak features, at 850 and 3540 cm−1, that are not accounted for in the computed spectra.
The very symmetric geometry of the Na+ and Li+ complexes has interesting implications for their computed vibrational spectra. Most of the vibrational modes appear in near-degenerate pairs, corresponding to symmetric and antisymmetric combinations of the same normal mode on each of the cytosine nucleobases. The vibrationally induced dipole moments of the two subunits can then either cancel or add, such that the computed intensity of one of the degenerate modes is zero.
Structure properties
The IR spectra in combination with the computational investigations clearly point out that the structure of the C–M+–C complex is different for M = Ag and Cu than for M = Li, Na and K. In all complexes, the metal ion is coordinated to the N3 atoms of both cytosine ligands. Additional coordination of the metal ion to the O atom competes with hydrogen bonding between the two cytosine ligands. Our experiments clearly show that the alkali metal ions compete more strongly for oxygen coordination than the coinage metal ions. The formation of two inter-ligand hydrogen bonds instead of metal–ion–oxygen coordination gives the coinage metal complexes a structure reminiscent of the hemiprotonated cytosine dimer.Qualitatively, the differences in bonding can be explained as a manifestation of sd-hybridization occurring in the coinage metal ions having a d10 electron configuration, but not in the alkali metal ions. In the Cu+ ion, sd-hybridization can be qualitatively understood as the mixing of the empty 4s-orbital with an occupied 3d-orbital, as depicted in Fig. S5 (ESI†). Each of the two sd hybrid atomic orbitals that are formed has two main lobes at 180° (of the same phase). The two sd-hybrids are mutually perpendicular. One of the hybrid orbitals is empty and accepts electron density from ligand based n-orbitals at 180°. The other sd-hybrid orbital – as well as the other unhybridized 3d-orbitals – is filled and is strongly repulsive to filled n-orbitals on the ligands. For Ag+, an analogous hybridization of the occupied 4d and empty 5s-orbitals occurs. Ligand arrangement around the coinage metal thus occurs at 180° with no ligand binding in the orthogonal directions. No hybridization occurs on the alkali metal ions, with s0 electron configurations, which are therefore spherical and the bonding is merely electrostatic in nature. A natural population analysis (NPA) supports the sd-hybridization and gives for the Cu+-ion in the C–Cu+–C complex an electron configuration of 3d9.804s0.484p0.10, close to that found for other bis-ligated N-donor complexes of Cu+.23 The NPA analyses suggest negligible hybridization in the C–M+–C alkali metal complexes, giving valence s-orbital occupancies of 0.11, 0.08 and 0.05 for Li+, Na+ and K+, respectively.
Siu and coworkers studied the binding energy of Ag+ to a range of small oxygen and nitrogen containing ligands and compared these energies with those of Na+.46,47 These studies established that the coinage metal ion prefers binding at nitrogen over oxygen, in contrast to the alkali metal ion. Qualitatively, this was explained within the framework of the hard and soft acids and bases (HSAB) hypothesis, where the coinage metal ions and nitrogen are the softer Lewis acid/base pair and the alkali metal ions and oxygen are considered as hard Lewis acids/bases. These findings are in qualitative agreement with our observations that the Cu+ and Ag+ ions coordinate to the ligand nitrogen atoms rather than to the oxygen atoms.
Having spectroscopically determined the structures of the C–M+–C complexes, we review some of the salient structural features that the computations provide (Table 4). As to the electronic structure, the bonding between the C ligands and the Cu+ ion exhibits partial covalent character, as is for instance shown by the mixing of metal d-orbitals with ligand based AOs in many of the valence molecular orbitals (see Fig. S6, ESI†). Visualization of the MOs for the alkali metal ion complexes shows no mixing between metal and ligand AOs, suggesting predominantly electrostatic binding. More quantitatively, this effect is shown by the partial charges on the metal ion in each of the complexes. The natural population analysis gives the lowest partial charge to the Cu+ ion (+0.710 e), whereas those for the alkali metal ions range between +0.731 e for Li+ to +0.886 e for K+. Only for the hemiprotonated cytosine dimer, where the proton is covalently bound to one of the ligands, does the NPA analysis give a lower partial charge on the proton.
| C–M+–C | Ionic radius (Å)9 | M+ partial charge | Δ(N⋯O)a | ΔEb | ΔEc |
|---|---|---|---|---|---|
| a Difference in distance between pairs of the amino N and carbonyl O atoms (in Å) as a measure of the asymmetry of the complex. b Computed dissociation energy (in kJ mol−1) for the loss of a neutral ligand: C–M+–C → M+–C + C. c Computed dissociation energy after counterpoise correction. d Note that the computation for the potassium complex was carried out using a smaller basis set, resulting in a larger BSSE correction. e Note that the proton resides asymmetrically between the two cytosine molecules, in contrast to the metal ions. | |||||
| Li+ | 0.70 | +0.731 | 0.000 | 203 | 199 |
| Na+ | 0.98 | +0.867 | 0.000 | 171 | 167 |
| K+ | 1.33 | +0.886 | 0.142 | 149 | 132d |
| Cu+ | 0.96 | +0.710 | 1.188 | 285 | 281 |
| Ag+ | 1.26 | 2.07 | |||
| H+ | — | +0.494 | 0.277e | 197 | 194 |
Regarding the geometric structure of the dimeric coinage metal complexes, the structures become more asymmetric as the ionic radius of the metal ion increases. The cavity formed by the two C-ligands is too small to truly fit the Cu+ and Ag+ metal ions, so that the dimer tends to “hinge” open. The degree of asymmetry can be expressed as the difference in N⋯O distance for the two pairs of hydrogen-bonded amino and carbonyl groups, Δ(N⋯O). The difference increases from 1.2 to 2.0 Å going from Cu+ to Ag+,7 whereas the cage is approximately symmetric for H+ (which itself sits in a more asymmetric position within the dimer than the metal ions do). For the alkali metal ions, the asymmetry can also be expressed in terms of this difference Δ(N⋯O), although values are not easily comparable with those for the coinage metal ions due to the different structures.
4. Conclusions
In summary, we have spectroscopically compared the binding modes of alkali and coinage metal monocations to cytosine. For the Cu+–C monomer complex, cytosine adopts the C1 tautomeric form as in alkali metal ion coordination. In contrast to the alkali metal ion, however, our spectra clearly show that a bidentate (N3 and O atoms) and a monodentate (O-atom) coordination motif occur in coexistence for copper. Concurrent coordination of an ancillary MeCN ligand induces solely monodentate coordination of the cytosine ligand; our spectra again show that two motifs – one with coordination to the O-atom and with coordination to the N-atom – coexist in roughly equal abundances.For C–M+–C dimeric structures, different structures are established for complexes bound by coinage versus alkali metal ions. The copper ion in the 1+ oxidation state, analogous to the previously investigated Ag+ ion, forms a structure reminiscent of the hemiprotonated cytosine-dimer, referred to as the i-motif structure. The strong preference for this structure can be explained by sd-hybridization of the metal ion atomic orbitals, inducing bis-coordination of the metal ion at 180°. In contrast, alkali metal ions induce a parallel displaced structure, where the alkali ion is tetra-coordinated to the N3 and O atoms of both cytosine ligands, sacrificing the two hydrogen bonds between the ligands for improved chelation of the metal ion.
Acknowledgements
We gratefully acknowledge the FELIX staff for the skillful technical support. This work is sponsored by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) Chemical Sciences (CW) as part of the Dutch Astrochemistry Network and under VICI grant No. 724.011.002; it was further supported by NWO Physical Sciences (EW) for the use of the facilities at the SurfSARA Supercomputer Center (grants MP-264-14 and SH-260-14). This work is also sponsored by the National Science Foundation, Grants PIRE-0730072 and CHE-1409420 (MTR). JO thanks the Stichting Physica. This work is part of the research program of FOM, which is financially supported by NWO.References
- K. Gehring, J. L. Leroy and M. Gueron, Nature, 1993, 363, 561 CrossRef CAS PubMed.
- H. A. Day, P. Pavlou and Z. A. E. Waller, Bioorg. Med. Chem., 2014, 22, 4407 CrossRef CAS PubMed.
- M. Guéron and J. L. Leroy, Curr. Opin. Struct. Biol., 2000, 10, 326 CrossRef.
- B. Yang, R. R. Wu and M. T. Rodgers, Anal. Chem., 2013, 85, 11000 CrossRef CAS PubMed.
- S. Y. Han and H. B. Oh, Chem. Phys. Lett., 2006, 432, 269 CrossRef CAS.
- H. A. Day, C. Huguin and Z. A. Waller, Chem. Commun., 2013, 49, 7696 RSC.
- M. Berdakin, V. Steinmetz, P. Maitre and G. A. Pino, J. Phys. Chem. A, 2014, 118, 3804 CrossRef CAS PubMed.
- M. Berdakin, G. Féraud, C. Dedonder-Lardeux, C. Jouvet and G. A. Pino, J. Phys. Chem. Lett., 2014, 5, 2295 CrossRef CAS PubMed.
- R. G. Wilson and G. R. Brewer, Ion beams: with applications to ion implantation, Wiley, New York, 1973 Search PubMed.
- K. J. Koch, T. Aggerholm, S. C. Nanita and R. G. Cooks, J. Mass Spectrom., 2002, 37, 676 CrossRef CAS PubMed.
- B. Yang, R. R. Wu, N. C. Polfer, G. Berden, J. Oomens and M. T. Rodgers, J. Am. Soc. Mass Spectrom., 2013, 24, 1523 CrossRef CAS PubMed.
- B. Yang, R. R. Wu, G. Berden, J. Oomens and M. T. Rodgers, J. Phys. Chem. B, 2013, 117, 14191 CrossRef CAS PubMed.
- E. A. L. Gillis, K. Rajabi and T. D. Fridgen, J. Phys. Chem. A, 2009, 113, 824 CrossRef CAS PubMed.
- M. Berdakin, V. Steinmetz, P. Maitre and G. A. Pino, Phys. Chem. Chem. Phys., 2015, 17, 25915 RSC.
- A. A. Power, O. Y. Ali, M. B. Burt and T. D. Fridgen, Int. J. Mass Spectrom., 2012, 330–332, 233 CrossRef CAS.
- J.-Y. Salpin, S. Guillaumont, J. Tortajada, L. MacAleese, J. Lemaire and P. Maitre, ChemPhysChem, 2007, 8, 2235 CrossRef CAS PubMed.
- C. M. Kaczan, A. I. Rathur, R. R. Wu, Y. Chen, C. A. Austin, G. Berden, J. Oomens and M. T. Rodgers, Int. J. Mass Spectrom., 2015, 378, 76 CrossRef CAS.
- K. T. Crampton, A. I. Rathur, Y.-w. Nei, G. Berden, J. Oomens and M. T. Rodgers, J. Am. Soc. Mass Spectrom., 2012, 23, 1469 CrossRef CAS PubMed.
- J. Oomens, A. R. Moehlig and T. H. Morton, J. Phys. Chem. Lett., 2010, 1, 2891 CrossRef CAS.
- Y.-w. Nei, T. E. Akinyemi, J. D. Steill, J. Oomens and M. T. Rodgers, Int. J. Mass Spectrom., 2010, 297, 139 CrossRef CAS.
- Y.-w. Nei, T. E. Akinyemi, J. D. Steill, J. Oomens and M. T. Rodgers, Int. J. Mass Spectrom., 2011, 308, 191 CrossRef CAS.
- N. S. Rannulu and M. T. Rodgers, Phys. Chem. Chem. Phys., 2005, 7, 1014 RSC.
- N. S. Rannulu and M. T. Rodgers, J. Phys. Chem. A, 2007, 111, 3465 CrossRef CAS PubMed.
- Z. Yang and M. T. Rodgers, Phys. Chem. Chem. Phys., 2012, 14, 4517 RSC.
- B. Yang and M. T. Rodgers, Phys. Chem. Chem. Phys., 2014, 16, 16110 RSC.
- A. G. Marshall, C. L. Hendrickson and G. S. Jackson, Mass Spectrom. Rev., 1998, 17, 1 CrossRef CAS PubMed.
- N. C. Polfer and J. Oomens, Phys. Chem. Chem. Phys., 2007, 9, 3804 RSC.
- D. Oepts, A. F. G. Van Der Meer and P. W. Van Amersfoort, Infrared Phys. Technol., 1995, 36, 297 CAS.
- J. J. Valle, J. R. Eyler, J. Oomens, D. T. Moore, A. F. G. van der Meer, G. Von Helden, G. Meijer, C. L. Hendrickson, A. G. Marshall and G. T. Blakney, Rev. Sci. Instrum., 2005, 76, 023103 CrossRef.
- M. Almasian, J. Grzetic, J. van Maurik, J. D. Steill, G. Berden, S. Ingemann, W. J. Buma and J. Oomens, J. Phys. Chem. Lett., 2012, 3, 2259 CrossRef CAS PubMed.
- P. Parneix, M. Basire and F. Calvo, J. Phys. Chem. A, 2013, 117, 3954 CrossRef CAS PubMed.
- A. M. Rijs and J. Oomens, Top. Curr. Chem., 2015, 364, 1 CrossRef PubMed.
- M. Kabelác and P. Hobza, J. Phys. Chem. B, 2006, 110, 14515 CrossRef PubMed.
- N. Russo, M. Toscano and A. Grand, J. Mass Spectrom., 2003, 38, 265 CrossRef CAS PubMed.
- M. V. Vazquez and A. Martínez, J. Phys. Chem. A, 2008, 112, 1033 CrossRef CAS PubMed.
- J. Sponer, M. Sabat, J. V. Burda, J. Leszczynski, P. Hobza and B. Lippert, JBIC, J. Biol. Inorg. Chem., 1999, 4, 537 CrossRef CAS PubMed.
- M. Szczesniak, K. Szczepaniak, J. S. Kwiatkowski, K. KuBulat and W. B. Person, J. Am. Chem. Soc., 1988, 110, 8319 CrossRef CAS.
- J. L. Alonso, V. Vaquero, I. Pena, J. C. Lopez, S. Mata and W. Caminati, Angew. Chem., Int. Ed., 2013, 52, 2331 CrossRef CAS PubMed.
- N. Russo, M. Toscano and A. Grand, J. Am. Chem. Soc., 2001, 123, 10272 CrossRef CAS PubMed.
- A. Günther, P. Nieto, G. Berden, J. Oomens and O. Dopfer, Phys. Chem. Chem. Phys., 2014, 16, 14161 RSC.
- M. Berdakin, V. Steinmetz, P. Maitre and G. A. Pino, Phys. Chem. Chem. Phys., 2015, 17, 25915 RSC.
- C. E. Holloway and M. Melnik, Rev. Inorg. Chem., 1995, 15, 147 CrossRef CAS.
- D. Scuderi, J. M. Bakker, S. Durand, P. Maitre, A. Sharma, J. K. Martens, E. Nicol, C. Clavaguera and G. Ohanessian, Int. J. Mass Spectrom., 2011, 308, 338 CrossRef CAS.
- A. Abo-Riziq, L. Grace, E. Nir, M. Kabelac, P. Hobza and M. S. de Vries, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 20 CrossRef CAS PubMed.
- R. R. Wu, B. Yang, C. E. Frieler, G. Berden, J. Oomens and M. T. Rodgers, J. Phys. Chem. B, 2015, 119, 5773 CrossRef CAS PubMed.
- H. El Aribi, C. F. Rodriquez, T. Shoeib, Y. Ling, A. C. Hopkinson and K. W. M. Siu, J. Phys. Chem. A, 2002, 106, 8798 CrossRef CAS.
- H. El Aribi, T. Shoeib, Y. Ling, C. F. Rodriquez, A. C. Hopkinson and K. W. M. Siu, J. Phys. Chem. A, 2002, 106, 2908 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cp00234j |
| This journal is © the Owner Societies 2016 |