
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational prediction of optimal metal ions to induce coordinated polymerization of muscle-like [c2]daisy chains†
Yan-Ling
Zhao
*a,
Rui-Qin
Zhang
b,
Christian
Minot
ac,
Klaus
Hermann
d and
Michel A.
Van Hove
a
aInstitute of Computational and Theoretical Studies & Department of Physics, Hong Kong Baptist University, Hong Kong, China. E-mail: ylzhao2008@gmail.com
bDepartment of Physics and Materials Science, City University of Hong Kong, Hong Kong, China
cLaboratoire de Chimie Théorique, Université Pierre & Marie Curie - Paris 6, CNRS, UMR7616, case 137, 4 place Jussieu, 75252 Paris Cedex 05, France
dInorganic Chemistry Department, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, 14195 Berlin, Germany
First published on 5th February 2016
Abstract
Recently, a muscle-like organometallic polymer has been successfully synthesized using Fe2+ as a linker atom. The polymer exhibits acid–base controllable muscle-like expansion and contraction on the micrometer scale. Further development could be facilitated by revealing the polymerization mechanism and by searching for optimal linker atoms. In this work, we have examined possible equilibrium and intermediate polymer structures, which consist of [c2]daisy chains linked by divalent transition metal ions (Sc2+, Ti2+, Fe2+, Co2+, Ni2+ or Zn2+) with various hexa-coordination arrangements, based on calculations using density functional theory. We find that the metal linkers in polymers are weaker in acid than in base due to excess positive charges on the polymer, leading to their thermodynamical instability or even decomposition. This can explain the experimental difficulty in improving the degree of polymerization for metal-linked polymers. We also find that the polymers with either Fe2+ or Co2+ are the most favorable, with the latter extending 1.4% longer than with the former. Since Fe2+ has been confirmed experimentally to be a successful linker, Co2+ would function equally well and thus could be used as an alternative choice for polymerization.
1. Introduction
For decades, scientists have synthesized and examined muscle-like functional materials endowed with mechanical expansion and contraction triggered by chemical,1–7 electrochemical,1,8–15 or photochemical10,16–21 stimuli under human control. In 2012, Giuseppone et al.22 successfully synthesized a polymer containing [c2]daisy chains and Fe2+ ions, which is acid–base controllable and micrometer-scale responsive while having muscle-like capability. As revealed by measurements of light scattering and small-angle neutron scattering,22 the unit lengths of the extended and contracted polymer units were found to be 5.4 and 3.2 nm, respectively. The 2.2 nm length difference in each unit is amplified in a linear polymer with about 3000 units to cause a stretch to 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 from 9400 nm, thus realizing muscle-like motion in a quasi-macroscopic regime. This achievement to some extent overcame the challenge of a low degree of polymerization and significantly advanced the technological prospect of practical muscle-like functional materials.23
900 from 9400 nm, thus realizing muscle-like motion in a quasi-macroscopic regime. This achievement to some extent overcame the challenge of a low degree of polymerization and significantly advanced the technological prospect of practical muscle-like functional materials.23
A [c2]daisy chain is a kind of doubly-threaded rotaxane. Each [c2]daisy chain supramolecule consists of two identical rod-and-ring molecules that are threaded to each other; they have the capability of telescoping as well as folding, which enables expansion and contraction (shown in Fig. 1) of each monomer. By adding the linking metal ion Fe2+, polymerization can be induced. As schematically shown in Fig. 1, the polymer unit is composed of one [c2]daisy chain and one Fe2+ ion, which are linked by organometallic hexa-coordination. The number of units (n) in the experimentally synthesized polymer was found to be about 3000 by Giuseppone's group.22 Each half of the [c2]daisy chain is threaded through the other half's dibenzo[24]-crown-8 (DB24C8) ether ring, while the bulky stopper (terpyridine group together with its adjacent ethinyl and phenyl species) prevents dethreading and also serves as a polymerizing linkage to the neighboring [c2]daisy chain by means of a bridging Fe2+. Each half of the [c2]daisy chain contains two recognition sites located in the thread, which serve as “docking stations” for the contracted vs. extended forms of the polymer: one is a triazolium ion (C2N3+) and the other is a neutral amino group (NH) at high pH, which changes to a secondary dialkyl ammonium ion (NH2+) in a low pH environment. The length of each unit varies dynamically as the binding affinity of DB24C8 for the two docking stations changes with pH: in acidic solution (e.g., trifluoroacetic acid), the NH2+ preferentially attaches to the crown ether, resulting in the extended form; in basic solution (e.g., sodium hydroxide), the deprotonated NH has lower affinity for the crown ether, which therefore binds with the triazolium ion, favoring the contracted form. The contraction/extension cycle of the above described single [c2]daisy chain24 supramolecule has been studied in our previous computational simulations25 using the monomer of this muscle-like polymer.22,23 Our systematic quantum chemical and molecular dynamics calculations have revealed that the polymer length change is not only due to the linear telescoping between the two docking stations but also due to the folding/unfolding of the two bulky stoppers,25 which is a striking difference as illustrated in Fig. 1 (top vs. bottom).
 | ||
| Fig. 1 Schematic representations of the polymerized [c2]daisy chain linked with Fe2+ ions (yellow balls). Each half of the [c2]daisy chain consists of a hydrocarbon rod (straight segments) with two docking stations (blue and purple balls), a crown ether ring DB24C8 (red ring), and a terpyridine end group (green arc). Two identical halves are mutually threaded to form a [c2]daisy chain unit, docked (top panel) at the triazolium+ station (in high pH) in the contracted state, or (bottom panel) at the NH2+ station (in low pH) in the extended state. | ||
Following a bottom-up strategy, the experimental quasi-macroscopic muscle-like behavior originates from the uniform change of many identical organometallic units. Thus, extending our prior study of the behavior of an individual [c2]daisy chain, investigations of the characteristics of the polymer containing different transition metal ions carried out in this work can clarify important aspects of this system and shed light on the mechanism of polymerization induced by the metal ions, which in turn can help in the design of optimal linkers.
2. Models and computational details
In this work, we choose Sc2+, Ti2+, Fe2+, Co2+, Ni2+ or Zn2+ to evaluate their effects on the polymer structures as they belong to the same period of elements with 3d1, 3d2, 3d6, 3d7, 3d8 or 3d10 valence electrons, respectively. It is known that the Lewis acidity26,27 of Fe2+, Co2+, Ni2+ and Zn2+ ranges between soft and hard, giving some flexibility to coordinate with soft or hard Lewis bases. Nonetheless, their chemical characteristics may differentiate their role in inducing coordinated polymerization in our case and thus allow optimizing their use in applications. Although Sc2+ and Ti2+ are not the most frequent oxidation states of these elements, they are chosen to track the dependence of polymer energetics on the number of 3d electrons. Since Fe2+ or Zn2+ was used in experiment22 to induce polymerization, we use them as the comparative reference in this work.This work uses the same approach and terminology as our previous study of the monomeric [c2]daisy chain.25 Briefly, the extended and contracted [c2]daisy chains are still named ext4+ and cont2+, respectively. The configuration ext4+ (C116H134O16N14) carries four positive charges localized at two NH2+ and two triazolium cations, while the two positive charges of cont2+ (C116H132O16N14) are localized at two triazolium cations. The acidic environment is represented by two protons attached to the nitrogen atoms of the amino groups and the basic environment is modeled through neutralizing the two protons (of NH2+), which are then left out of the calculation. The metal ions are, before further optimization, inserted at equal distances (∼2 Å) from nearby nitrogen atoms of the terpyridines so as to hexa-coordinate with two [c2]daisy chains, which connect head-to-tail through a metal ion due to a periodic boundary condition. In the dimensions perpendicular to the chain, there are large intermolecular distances to avoid discernible interaction between parallel polymers.
In the experiment by Giuseppone et al.22 the cationic [c2]daisy chains and Fe2+ ions were polymerized in an ionic liquid, where stoichiometric PF6− and Cl− counterions accompany the polymers to neutralize the charge in the environment of organic solvents (acetonitrile CH3CN and chloroform CHCl3 in a 1:1 mixture solution). The neutral polymer unit in acid or base is actually composed of C116H134O16N14Fe(PF6)4Cl2 or C116H132O16N14Fe(PF6)2Cl2. However, we do not include the PF6− and Cl− counterions in our calculations as they usually interact only indirectly with the polymer and float around the cationic polymer in complexes with solvent molecules. We have previously25 presented the stable complexes PF6−(CH3CN)n or PF6−(CHCl3)n (n = 1–8) and verified that these solvents provide a soft negative electrostatic potential environment for [c2]daisy chains, which thus avoids strong electrostatic interactions between the [c2]daisy chains and the surrounding counter-ions while keeping the [c2]daisy chain in a functioning state during the muscle-like motion. To simplify the calculation model, we thus feel justified to study the cationic polymer under vacuum.
Due to the fact that [c2]daisy chains are of large size and contain weak interactions such as hydrogen bonds and/or π–π stacking, the self-consistent charge density-functional tight-binding dispersion (SCC-DFTB-D) method28–35 is first utilized to optimize the molecular and unit cell structure. Dispersion contributions are included via Lennard-Jones potentials, which take the parameters from the universal force field. The Slater–Koster (SK) potentials of metals with C, H, O, N are taken from the parameter sets trans3d36,37 and znorg,38–40 which have been well tested to predict comparably accurate geometries using DFT.
Applying periodic boundary conditions, the lattice component force is calculated together with the interatomic force during each SCC cycle. If the structure reaches the force optimization criterion but the lattice does not, the unit cell is automatically modified to other cell lengths until the lattice force converges. A global search is carried out to find the most favorable equilibrated and intermediate conformations of the polymer units corresponding to lowest total energies.
To verify the reliability of DFTB in predicting geometric structures in our work, we firstly study small complexes with hexa-coordination bonds formed between two terpyridines and one metal ion by SCC-DFTB-D and ωB97XD/6-31G* optimization, as listed in Table S1 (ESI†). The results confirm that SCC-DFTB-D can successfully predict similar hexa-coordination geometries as DFT and can deal with periodic and complicated polymer structures in this study. To obtain reliable molecular orbitals and energetic data, single-point calculations using the geometries derived at the SCC-DFTB-D level of theory are carried out using ωB97XD/6-31G*, a long-range hybrid density functional theory with dispersion corrections, available in the Gaussian 09 package.41 Note that the ωB97XD42 functional yields satisfactory accuracy for thermochemistry, kinetics, and non-covalent interactions.
3. Results and discussion
3.1 Polymer units
The combined optimization of geometry and cell parameters yields polymer structures containing transition metal ions (M2+: Sc2+, Ti2+, Fe2+, Co2+, Ni2+, or Zn2+). Fig. 2(a) and (b) and Fig. S1(a)–(d) (cf. ESI†) display two equilibrium structures and two intermediate polymer structures of three units of an infinite polymer chain for each of the six metal ions. Hereafter, the extended equilibrium and protonated intermediate polymers under acidic conditions are called [ext4+-M2+]n and [proton-cont4+-M2+]n, respectively, while the contracted equilibrium and deprotonated intermediates in base are denoted as [cont2+-M2+]n and [deproton-ext2+-M2+]n, respectively. Here n stands for infinite repetition in the polymers. The muscle-like motion cycles among four conformations follow the generic sequence shown in Scheme 1.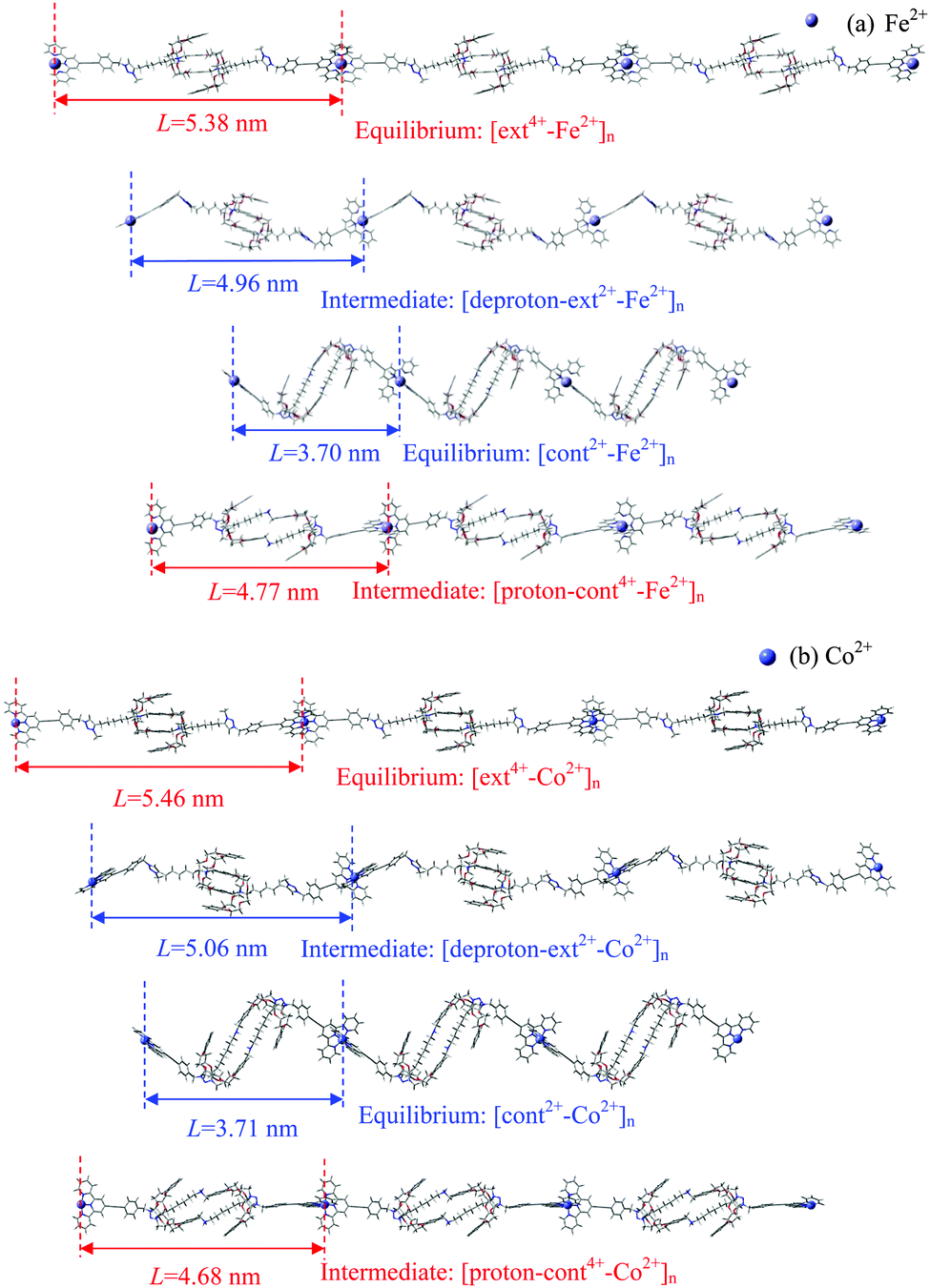 | ||
| Fig. 2 The equilibrium and intermediate polymer structures in three linked units containing (a) Fe2+ or (b) Co2+, from SCC-DFTB-D calculations under vacuum. Gray, white, red, and blue atom colors represent C, H, O, and N, respectively. (Note that the terpyridine and benzene groups in the bulky stoppers are connected by –C≡C– groups, which in this representation look like abnormally long single bonds.) | ||
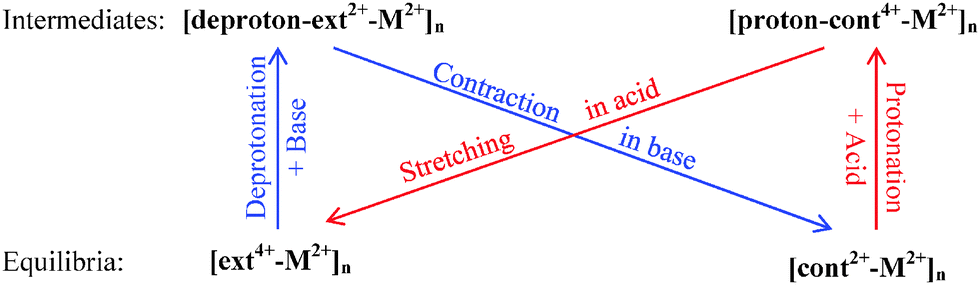 | ||
| Scheme 1 The cyclic process of acid-induced extension and base-promoted contraction among two equilibria and two intermediates in a simplified energy diagram. M2+ ions: Sc2+, Ti2+, Fe2+, Co2+, Ni2+, or Zn2+. | ||
Interestingly, our calculations show that not all of the metal ions can induce stable muscle-like polymers. While polymerization is accomplished when using Sc2+, Fe2+ or Co2+, the use of Ti2+, Ni2+, or Zn2+ induces instabilities in the protonated intermediate polymer, as shown in Fig. S1(b)–(d) (ESI†). In [proton-cont4+-Ti2+]n and [proton-cont4+-Ni2+]n, the N–C bonds near NH2+ break up, fragmenting the [c2]daisy chains and interrupting further extension. For example, the distances between N (of NH2+) and the neighboring C (of CH2) in [proton-cont4+-Ti2+]n are increased to 2.055 and 2.057 Å, indicating bond breaking. In [proton-cont4+-Zn2+]n, two DB24C8 rings are opened considerably and the hydrocarbon rods are no longer kept linear, counteracting the lengthening step. Thus, it appears that the conformations under the acidic conditions become unstable and may even decompose. The N–C bond cleavage phenomenon is worth noticing by the experimentalists as it probably impedes the smooth stretching of the polymer in its practical application.
To quantify the hexa-coordination behavior, we check the distances between the M2+ and the six N atoms to which the M2+ ion is coordinated in the terpyridine groups of [c2]daisy chains, as listed in Table 1. In most conformations, two shorter and four longer M2+–N bond lengths reflect reasonable metal coordination as suggested by the d2–sp3 orbital hybrid. However, one particularly long distance (>2.5 Å) appears in the systems [ext4+-Fe2+]n, [proton-cont4+-Fe2+]n, [proton-cont4+-Co2+]n, [ext4+-Ni2+]n, [proton-cont4+-Ni2+]n, [cont2+-Ni2+]n, and [deproton-ext2+-Ni2+]n. This suggests that the conformations are becoming more distorted and weaker in acid than in base. Ni2+ is not a good linker ion for this muscle-like polymer as it cannot hexa-coordinate well with [c2]daisy chains, as shown by one unusual Ni2+–N distance (∼3 Å) in all its configurations. The more uneven hexa-coordination bonds via Ni2+ will probably make the polymer more easily disordered than other ions so as to fail in undergoing multiple muscle-like cycles. Fe2+–N bond lengths are in the range 1.9–2.1 Å (a distance larger than 2.5 Å is obtained in the unstable intermediate and not considered here), basically consistent with the experimental value of 2.06 Å, as reported in a similar complex.43 This again reflects that SCC-DFTB-D can ensure comparable coordination bond lengths with the experiment.
| Extended polymer | M2+–N distances | Contracted polymer | M2+–N distances |
|---|---|---|---|
| [ext4+-Sc2+]n | 2.349, 2.349, 2.372 | [cont2+-Sc2+]n | 2.342, 2.343, 2.372 |
| 2.373, 2.375, 2.375 | 2.372, 2.373, 2.374 | ||
| [deproton-ext2+-Sc2+]n | 2.325, 2.326, 2.369 | [proton-cont4+-Sc2+]n | 2.349, 2.350, 2.372 |
| 2.369, 2.373, 2.373 | 2.372, 2.375, 2.376 | ||
| [ext4+-Ti2+]n | 1.520, 1.935, 1.981 | [cont2+-Ti2+]n | 1.920, 1.983, 2.075 |
| 2.028, 2.238, 2.304 | 2.120, 2.170, 2.258 | ||
| [deproton-ext2+-Ti2+]n | 2.040, 2.041, 2.118 | [proton-cont4+-Ti2+]n | 1.936, 1.942, 1.990 |
| 2.119, 2.119, 2.121 | 1.991, 2.293, 2.301 | ||
| [ext4+-Fe2+]n | 1.921, 1.971, 1.980 | [cont2+-Fe2+]n | 1.934, 1.935, 2.042 |
| 2.031, 2.032, 2.538 | 2.042, 2.043, 2.043 | ||
| [deproton-ext2+-Fe2+]n | 1.933, 1.935, 2.041 | [proton-cont4+-Fe2+]n | 1.920, 1.970, 1.979 |
| 2.041, 2.042, 2.042 | 2.031, 2.031, 2.538 | ||
| [ext4+-Co2+]n | 1.888, 1.888, 1.950 | [cont2+-Co2+]n | 1.885, 1.886, 1.948 |
| 1.950, 1.950, 1.951 | 1.948, 1.948, 1.948 | ||
| [deproton-ext2+-Co2+]n | 1.882, 1.882, 1.945 | [proton-cont4+-Co2+]n | 1.862, 1.915, 1.923 |
| 1.945, 1.946, 1.946 | 1.928, 1.936, 2.784 | ||
| [ext4+-Ni2+]n | 1.953, 2.026, 2.027 | [cont2+-Ni2+]n | 1.951, 2.022, 2.028 |
| 2.096, 2.109, 3.020 | 2.082, 2.106, 3.000 | ||
| [deproton-ext2+-Ni2+]n | 1.946, 2.018, 2.022 | [proton-cont4+-Ni2+]n | 1.953, 2.023, 2.028 |
| 2.089, 2.099, 3.008 | 2.100, 2.102, 3.006 | ||
| [ext4+-Zn2+]n | 2.080, 2.081, 2.106 | [cont2+-Zn2+]n | 2.074, 2.076, 2.103 |
| 2.107, 2.107, 2.108 | 2.105, 2.112, 2.114 | ||
| [deproton-ext2+-Zn2+]n | 2.064, 2.064, 2.110 | [proton-cont4+-Zn2+]n | 2.081, 2.081, 2.105 |
| 2.110, 2.114, 2.114 | 2.105, 2.108, 2.108 |
To examine the impact of the metal linker on the polymer length, we define the unit length (L) as the distance between two successive M2+ ions, as labeled in Fig. 2 and Fig. S1 (ESI†) and then compared in Fig. 3. The unit length data are all listed in Table S2 (ESI†). Firstly, it should be noted that the theoretical L value of [ext4+-Fe2+]n is found to be 5.38 nm, in good agreement with the corresponding experimental value of 5.4 nm. Furthermore, the muscle-like motion is clearly exhibited by the unit length changes among these four conformations. Starting from the equilibrated state in acid, as the solution is changed from acidic to basic, the [ext4+-M2+]n state (leftmost solid red bars in Fig. 3) converts to the [deproton-ext2+-M2+]n state (next bars toward the right) with the lengths slightly decreasing by 0.3–0.4 nm. This change is induced by the slight folding of the bulky stoppers as displayed in Fig. 2 and Fig. S1 (ESI†). Next, the contraction process from the [deproton-ext2+-M2+]n intermediate to the [cont2+-M2+]n equilibrium (next blue bars toward the right) causes the unit length to greatly decrease by 1.3–1.5 nm. As the solution is changed from basic to acidic, [cont2+-M2+]n becomes [proton-cont4+-M2+]n (rightmost bars) and the unfolding of the bulky stoppers is clearly manifested as the unit length dramatically increases by 0.9–1.0 nm. The cycle is completed when the extension process from the [proton-cont4+-M2+]n intermediate to the [ext4+-M2+]n equilibrium (leftmost solid red bars) shows unit lengths increasing by 0.6–0.8 nm. This indicates that the pathway of the contraction and stretching in the polymer is basically consistent with our analysis for the individual [c2]daisy chain supramolecule and confirms that the folding of the two bulky stoppers occurs in the last phase of contraction while unfolding takes place in the first phase of stretching.25 The response of the polymer to the acid is obvious at first, namely, the initial unfolding process significantly increasing the polymer length. Afterwards, the linear extension in the following process is hampered by a higher barrier and becomes difficult as revealed in our previous study.25 By contrast, the structural response of the polymer to the base is not as apparent as to the acid at first, while the subsequent contraction is smoother than stretching with a lower energy barrier.
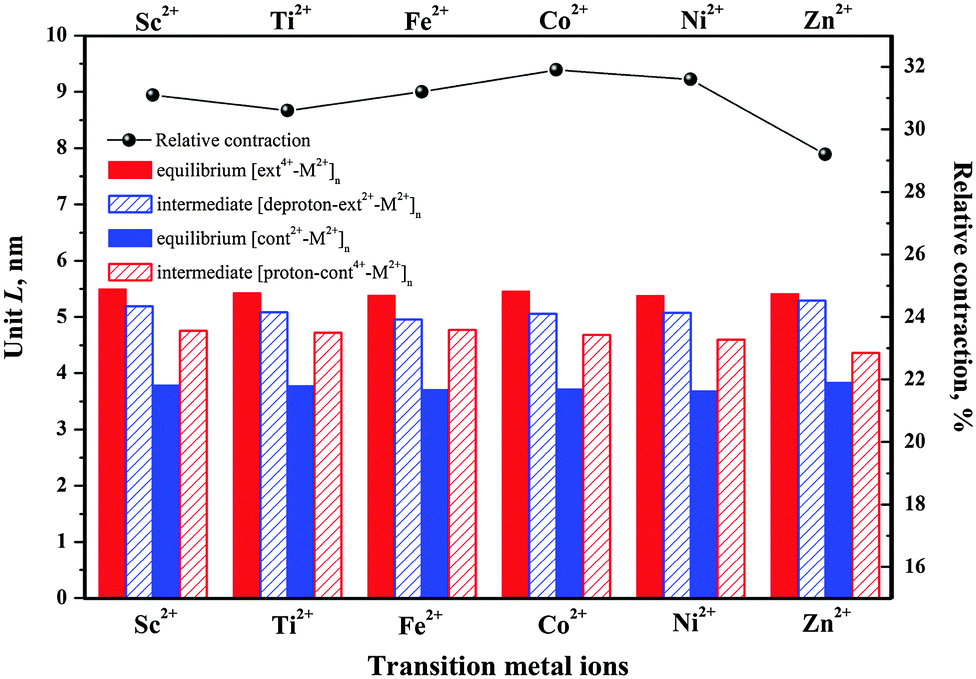 | ||
| Fig. 3 Calculated polymer unit lengths of equilibria and intermediates and relative contraction between two equilibria containing Sc2+, Ti2+, Fe2+, Co2+, Ni2+, or Zn2+, from SCC-DFTB-D calculations under vacuum. | ||
Notably, the unit length difference between [ext4+-M2+]n and [cont2+-M2+]n equilibria plays a key role in determining the relative polymer contraction under acid–base actuation. As shown in Fig. 3, the highest relative contraction, 31.9%, is found in the case of Co2+, while the second and third highest, are 31.6% and 31.2% for Ni2+ and Fe2+. Ni2+ is excluded here due to unsuccessful [proton-cont4+-Ni2+]n configuration. The main advantage of Co2+ is that the [ext4+-Co2+]n is perfectly extended 1.4% longer than [ext4+-Fe2+]n, while [cont2+-Co2+]n remains relatively as short as [cont2+-Fe2+]n. Considering that Fe2+ has been successfully used in experiment, we thus conjecture that the polymer containing Co2+ might function equally well as that containing Fe2+.
3.2 Hexa-coordination of M2+ with [c2]daisy chains
Integrated muscle-like polymerization was realized in experiment by using metal ions to link daisy chains together. A single M2+ connects two [c2]daisy chains by forming the bis(terpyridine)-M2+ coordination complex with coordination of the metal ion to 6 N atoms in 6 pyridine rings. Increasing the degree of polymerization to lengthen the polymer is a key for enabling macroscopic expansion and contraction for large-scale mechanical applications. Hence, it is important to study the coordination of M2+ with rotaxanes at the molecular level.It should be noted here that, compared to the [c2]daisy chain monomers, the polymerization reaction reorients 4 of the 6 pyridine rings in the two adjacent [c2]daisy chains such that 6 N atoms can bond to the linking metal ion: this converts the two adjacent terpyridines from trans to cis configuration. The trans form is a natural and stable state for [c2]daisy chains before polymerization and needs to be activated to the cis form and then to coordinate with M2+ for creating a polymer. This trans to cis conversion deforms the terpyridines moderately. For example, the N–C–C–N dihedral angle highlighted by enlarged balls in the trans and cis forms is shown in Fig. S2 (ESI†) for the four conformers of the monomer. While terpyridine is close to planar in the monomeric trans form, it is tilted by about 20° between two pyridine parts in the monomeric cis form, except for cont2+ which is planar, as shown in Fig. S3 (ESI†). The inevitable deformation will, to some extent, impact the coordination with metal ions in a symmetric coordination field. Nonetheless, the metal ions may try to flatten the terpyridine as they react with the [c2]daisy chains. The energy differences in the trans to cis conversions for the four states range from 151 to 156 kcal mol−1 at the ωB97XD/6-31G* level of theory, cf. Fig. S3 (ESI†). This means that cis-terpyridine will be automatically converted to the trans form once the M2+–N coordination bond is broken and that more input energy is required to recover the polymerization reaction.
The formation of hexa-coordination bonds in an octahedral environment would require the use of the unoccupied 3d, 4s, 4p orbitals of the metal ions to interact with orbitals occupied by twelve electrons of the [c2]daisy chain. This is facilitated by two terpyridines each consisting of three nitrogen atoms each of which has a σ donor electron pair. Fig. S4 (ESI†) shows the twelve electrons localized at two terpyridine groups, confirming that the polymerization reaction occurs at the two ends. The six 3d electrons of Fe2+ fill the 3dxy, 3dxz and 3dyz orbitals, leaving behind six empty 3dz2, 3dx2−dy2, 4s, 4px, 4py, 4pz orbitals pointing at the N ligands. Six fully filled bonding orbitals and six empty anti-bonding orbitals will be formed in the Fe-based hexa-coordination compound. For Co2+, Ni2+ and Zn2+, one, two, or four more 3d electrons should fill the “anti-bonding” (eg*) orbitals, destabilizing more the hexa-coordination compound than Fe2+. For Sc2+ and Ti2+, the only one or two 3d electrons should also fill the “non-bonding” (t2g) orbitals but their relatively large ion radii may be not as favorable as Fe2+ for forming stable hexa-coordination bonds. Thus, Fe2+ is the best choice. On the other hand, the Sc2+ or Co2+ has an unpaired electron as it hexa-coordinates with [c2]daisy chains. Thus, the spin signal from the Sc-based and Co-based polymers may also help in probing some changes from the polar or non-polar environment provided by the ionic liquid.
We have discussed above how the hexa-coordination is impacted by the conformational change. To examine the linking strength, i.e., the thermal stability of the organometallic coordinated polymers, we have calculated the hexa-coordination reaction heats (ΔEhexa) between two [c2]daisy chains and M2+ according to the equation: ΔEhexa = Ehexacoord − EM2+ − 2E[c2]DaisyChain. Here, all terpyridine groups in all hexa-coordination compounds are in cis form, and [c2]daisy chains can be cis-ext4+, cis-deproton-ext2+, cis-cont2+, and cis-proton-cont4+. The structures containing Co2+ are shown in Fig. S5 (ESI†) as an example, where the Co2+ and six coordinated N are highlighted by enlarged balls. The polymerization reaction heats (ΔEhexa) for M2+ linking two [c2]daisy chains in four configurations were calculated at the ωB97XD/6-31G* level of theory based on the SCC-DFTB-D geometries, as shown in Fig. 4. The single-point energy corrections at the higher level of DFT based on the structures predicted by SCC-DFTB-D can guarantee a correct trend of the hexa-coordination thermal stability (see Fig. S6, ESI†). Among the metal ions that we studied, the linking strength is in the order: Fe2+ > Co2+ > Ni2+ > Zn2+ ≈ Ti2+ > Sc2+. The structure with Fe2+ presents the highest stability whereas the one with Sc2+ is the least stable. This leaves Co2+ as the theoretically next favorable choice for being applied in this muscle-like polymer. Our results in fact support the experimental observation23 that, when Zn2+ instead of Fe2+ was used as the transition metal, the partial depolymerization of its extended polymer occurred while the contracted polymer was successfully obtained.
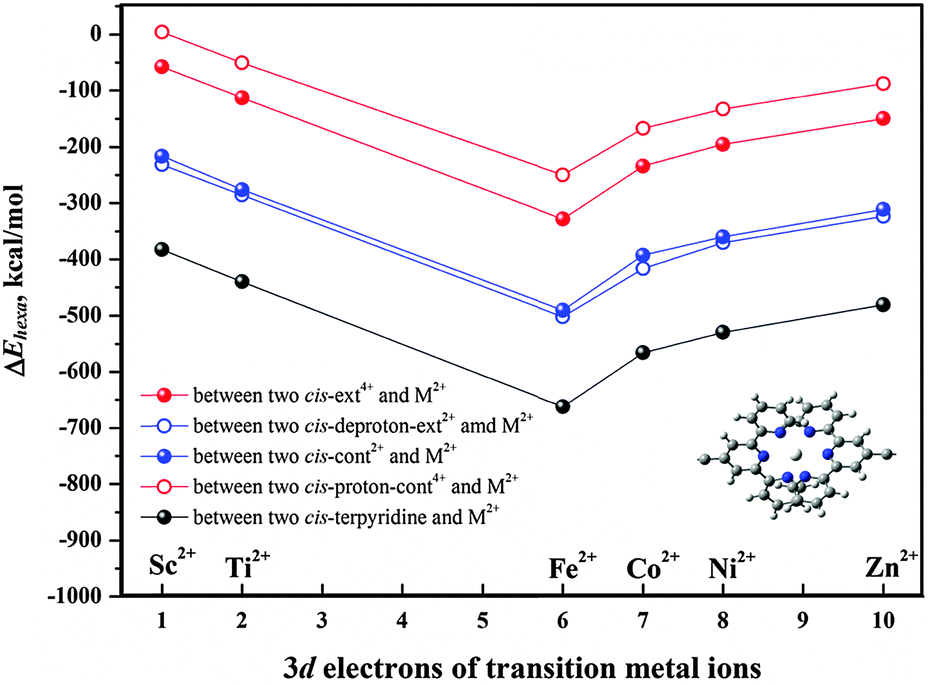 | ||
| Fig. 4 Calculated hexa-coordination reaction heats (ΔEhexa) at the level of ωB97XD/6-31G*. | ||
Moreover, Fig. 4 indicates that the polymerization of [c2]daisy chains with M2+ occurs in the order: deproton-ext2+ > cont2+ > ext4+ > proton-cont4+. The contraction in base is energetically more favorable than the stretching in acid, with the reaction heats for the contracted case being about twice as large as for the extended case. A smaller reaction heat would easily lead to thermal decomposition of the extended polymer, especially for the protonated intermediates, and thus stops expansion during a cyclic motion. It is actually difficult to polymerize cationic monomers, as more positive charges on ext4+ or proton-cont4+ may hinder the polymerization reaction due to a larger electrostatic repulsion between the tetra-cationic [c2]daisy chains and the di-cationic M2+. We have verified this by calculating the reaction heat for two neutral cis-terpyridine molecules coordinating with one M2+, which represents the linking segment in all polymer structures. As shown in Fig. 4, their reaction heats are all increased by the same amount, much larger than for any polymerization. Clearly, in the experiment there are PF6− and Cl− counterions in solution to compensate charged monomers and to help alleviate the Coulomb repulsion from its positive charges and ensure that polymerization occurs. Otherwise the hexa-coordination of M2+ with the [c2]daisy chains would be weakened considerably. However, from a different perspective, counterions are actually counter-productive because this polymer relies on two positive recognition sites to perform contraction and expansion. We believe that this is the main reason why in experiment it has been difficult to further improve the degree of polymerization. We note that Fig. 4 merely shows a general trend of the polymerization ability for a charged [c2]daisy chain monomer as the computational model does not include the accompanying counterions. Nevertheless, it tells us that further improvement of the thermal stability of the polymer by selecting an optimal metal ion such as Fe2+ and Co2+ or moving to neutral [c2]daisy chain supramolecules may help in increasing the degree of polymerization.
To further reflect the interaction strength between the metal ion and the [c2]daisy chain, we additionally calculate the Mayer bond order44–46 (MBO) in each hexa-coordination compound, as shown in Fig. 5. It is surprisingly found that the MBOs from Co2+ are strongest except for coordinating with cis-deproton-ext2+, while those from Zn2+ are weakest. The MBO order is: Co2+ > Ti2+ > Fe2+ > Ni2+ > Sc2+ > Zn2+. It is seen that Co2+ is indeed an option for being applied to the muscle-like [c2]daisy chain polymer.
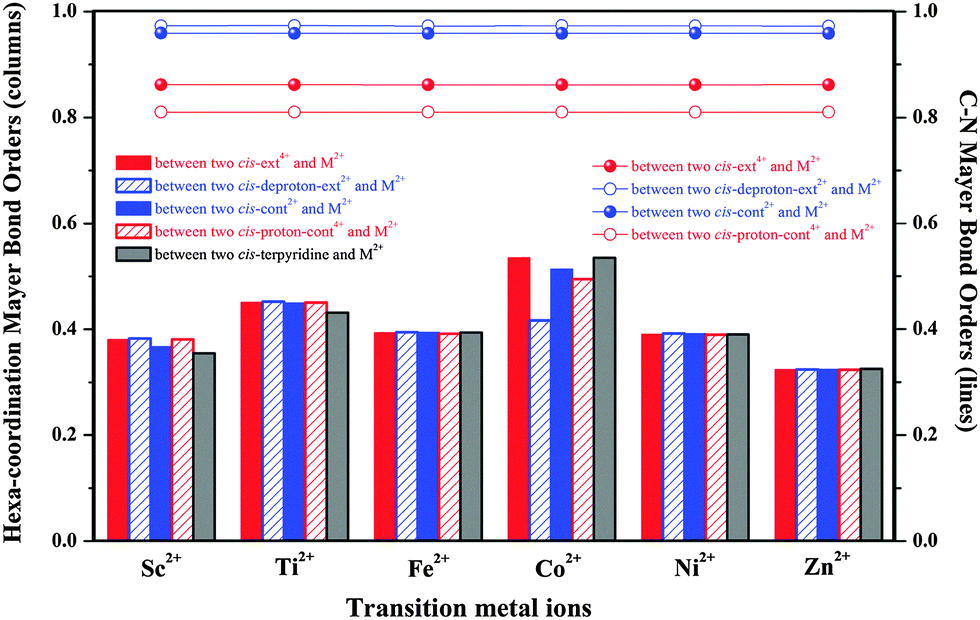 | ||
| Fig. 5 Calculated Mayer bond orders of the hexa-coordination bonds (in columns) and the C–N bonds at the C–N(H/H2+)–C sites (in lines) at the level of ωB97XD/6-31G*. | ||
Lastly, an MBO analysis can help reflect the N–C bond weakening in the C–N(H/H2+)–C sites of all hexa-coordination compounds, as shown in Fig. 5. Under the basic conditions, the MBO values of the NH group with two neighboring alkyl C are 11–20% higher than those of NH2+ under the acidic conditions, especially compared to the protonated intermediates. This indicates that the N–C bond strength is significantly reduced in the acidic environment, analogous to the hexa-coordination thermal stability. Since structural deformation in all hexa-coordination compounds is not as strong as in the realistic periodic polymers, we do not see the breakage of the N–C bond in compounds. However, it is expected from bond order results that the N–C breakage in the polymers will most likely occur in the most heavily distorted [proton-cont4+-M2+]n, which is supported by the N–C cleavages in [proton-cont4+-Ti2+]n and [proton-cont4+-Ni2+]n. The N–C bond order change has no direct relationship with the metal type, see the four almost flat curves in Fig. 5, and will be significantly affected by the acid–base environment change.
4. Conclusions and perspectives
The study of equilibrium and intermediate polymer configurations in acid or base formed by linking muscle-like [c2]daisy chains with various transition metal ions reveals stable polymers linked by Sc2+, Fe2+ and Co2+ instead of Ti2+, Ni2+ and Zn2+. The conformations in acid suffer strong distortions quite easily and can even decompose, especially for the protonated contracted polymer. The bond between N (of NH2+) and its neighbor C (of CH2) is much weakened due to acid catalysis. The positive excess charges on the [c2]daisy chains greatly reduce the reaction heat for polymerization and weaken the hexa-coordination links with metal ions, showing the difficulty to improve the degree of polymerization to macroscale as experienced in experiment. In hexa-coordination compounds, the thermal stability vary with the order: Fe2+ > Co2+ > Ni2+ > Zn2+ ≈ Ti2+ > Sc2+, while the Mayer bond order has the order: Co2+ > Ti2+ > Fe2+ > Ni2+ > Sc2+ > Zn2+. Thus, Fe2+ and Co2+ are both optimal metal ions for polymerization by theoretical predictions. Furthermore, the extended polymer linked by Co2+ is 1.4% longer than that by Fe2+, producing a 0.7% higher relative contraction than with Fe2+. As Fe2+ has been shown in experiment to be a successful linker, Co2+ ions would function equally well and thus could be used as an alternative choice for polymerization.Acknowledgements
We thank the High Performance Cluster Computing Centre, Hong Kong Baptist University, which receives funding from the Research Grants Council, University Grants Committee of the HKSAR and HKBU. This work was supported in part by project no. 9041650 of the Research Grants Council of HKSAR and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, Ministry of Education of China (Grant No. [2013]693). MAVH and YLZ were supported in part by the HKBU Strategic Development Fund. CM acknowledges support from HKBU's Sabbatical Visitor scheme and partial support from the HKBU Strategic Development Fund. KH acknowledges support from HKBU's Distinguished Scholar scheme and partial support from the HKBU Strategic Development Fund.References
- R. A. Bissell, E. Crdova, A. E. Kaifer and J. F. Stoddart, Nature, 1994, 369, 133–137 CrossRef CAS.
- M.-V. Martínez-Díaz, N. Spencer and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1997, 36, 1904–1907 CrossRef.
- P. R. Ashton, R. Ballardini, V. Balzani, I. Baxter, A. Credi, M. C. T. Fyfe, M. T. Gandolfi, M. Gómez-López, M.-V. Martínez-Díaz, A. Piersanti, N. Spencer, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1998, 120, 11932–11942 CrossRef CAS.
- M. C. Jiménez, C. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284–3287 CrossRef.
- J. Wu, K. C. Leung, D. Bentez, J.-Y. Han, S. J. Cantrill, L. Fang and J. F. Stoddart, Angew. Chem., Int. Ed., 2008, 47, 7470–7474 CrossRef CAS PubMed.
- P. G. Clark, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 13631–13633 CrossRef CAS PubMed.
- Z. Zhang, C. Han, G. Yu and F. Huang, Chem. Sci., 2012, 3, 3026–3031 RSC.
- P. Gaviña and J.-P. Sauvage, Tetrahedron Lett., 1997, 38, 3521–3524 CrossRef.
- L. Raehm, J.-M. Kern and J.-P. Sauvage, Chem. – Eur. J., 1999, 5, 3310–3317 CrossRef CAS.
- N. Armaroli, V. Balzani, J.-P. Collin, P. Gaviña, J.-P. Sauvage and B. Ventura, J. Am. Chem. Soc., 1999, 121, 4397–4408 CrossRef CAS.
- R. Ballardini, V. Balzani, W. Dehaen, A. E. Dell'Erba, F. M. Raymo, J. F. Stoddart and M. Venturi, Eur. J. Org. Chem., 2000, 591–602 CrossRef CAS.
- J.-P. Collin, C. Dietrich-Buchecker, P. Gaviña, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477–487 CrossRef CAS PubMed.
- Y. Liu, A. H. Flood, P. A. Bonvallet, S. A. Vignon, B. H. Northrop, H.-R. Tseng, J. O. Jeppesen, T. J. Huang, B. Brough, M. Baller, S. Magonov, S. D. Solares, W. A. Goddard, C.-M. Ho and J. F. Stoddart, J. Am. Chem. Soc., 2005, 127, 9745–9759 CrossRef CAS PubMed.
- S. Saha, A. H. Flood, J. F. Stoddart, S. Impellizzeri, S. Silvi, M. Venturi and A. Credi, J. Am. Chem. Soc., 2007, 129, 12159–12171 CrossRef CAS PubMed.
- G. Fioravanti, N. Haraszkiewicz, E. R. Kay, S. M. Mendoza, C. Bruno, M. Marcaccio, P. G. Wiering, F. Paolucci, P. Rudolf, A. M. Brouwer and D. A. Leigh, J. Am. Chem. Soc., 2008, 130, 2593–2601 CrossRef CAS PubMed.
- A. C. Benniston and A. Harriman, Angew. Chem., Int. Ed. Engl., 1993, 32, 1459–1461 CrossRef.
- A. C. Benniston, A. Harriman and V. M. Lynch, Tetrahedron Lett., 1994, 35, 1473–1476 CrossRef CAS.
- H. Murakami, A. Kawabuchi, K. Kotoo, M. Kunitake and N. Nakashima, J. Am. Chem. Soc., 1997, 119, 7605–7606 CrossRef CAS.
- P. R. Ashton, R. Ballardini, V. Balzani, A. Credi, K. R. Dress, E. Ishow, C. J. Kleverlaan, O. Kocian, J. A. Preece, N. Spencer, J. F. Stoddart, M. Venturi and S. Wenger, Chem. – Eur. J., 2000, 6, 3558–3574 CrossRef CAS.
- A. Harada, Acc. Chem. Res., 2001, 34, 456–464 CrossRef CAS PubMed.
- R. E. Dawson, S. F. Lincoln and C. J. Easton, Chem. Commun., 2008, 3980–3982 RSC.
- G. Du, E. Moulin, N. Jouault, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2012, 51, 12504–12508 CrossRef CAS PubMed.
- C. J. Bruns and J. F. Stoddart, Nat. Nanotechnol., 2013, 8, 9–10 CrossRef CAS PubMed.
- P. R. Ashton, I. Baxter, S. J. Cantrill, M. C. T. Fyfe, P. T. Glink, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 1294–1297 CrossRef CAS.
- Y. L. Zhao, R. Q. Zhang, C. Minot, K. Hermann and M. A. Van Hove, Phys. Chem. Chem. Phys., 2015, 17, 18318–18326 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- G. Seifert, J. Phys. Chem. A, 2007, 111, 5609–5613 CrossRef CAS PubMed.
- B. Aradi, B. Hourahine and T. Frauenheim, J. Phys. Chem. A, 2007, 111, 5678–5684 CrossRef CAS PubMed.
- D. Porezag, T. Frauenheim, T. Köhler, G. Seifert and R. Kaschner, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 12947–12957 CrossRef CAS.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- T. Frauenheim, G. Seifert, M. Elstner, Z. Hajnal, G. Jungnickel, D. Porezag, S. Suhai and R. Scholz, Phys. Status Solidi B, 2000, 217, 41–62 CrossRef CAS.
- M. Elstner, T. Frauenheim, E. Kaxiras, G. Seifert and S. Suhai, Phys. Status Solidi B, 2000, 217, 357–376 CrossRef CAS.
- T. Frauenheim, G. Seifert, M. Elstner, T. Niehaus, C. Köhler, M. Amkreutz, M. Sternberg, Z. Hajnal, A. D. Carlo and S. Suhai, J. Phys.: Condens. Matter, 2002, 14, 3015–3047 CrossRef CAS.
- W. J. Fan, J. Zeng and R. Q. Zhang, J. Chem. Theory Comput., 2009, 5, 2879–2885 CrossRef CAS PubMed.
- A. R. R. Neto and H. W. L. Alves, Phys. Status Solidi C, 2010, 7, 308–311 CrossRef CAS.
- W. Heckel, B. A. M. Elsner, C. Schulz and S. Müller, J. Phys. Chem. C, 2014, 118, 10771–10779 CAS.
- N. H. Moreira, G. Dolgonos, B. Aradi, A. L. da Rosa and T. Frauenheim, J. Chem. Theory Comput., 2009, 5, 605–614 CrossRef CAS PubMed.
- N. H. Moreira, A. L. da Rosa and T. Frauenheim, Appl. Phys. Lett., 2009, 94, 193109 CrossRef.
- A. Domínguez, N. H. Moreira, G. Dolgonos, T. Frauenheim and A. L. da Rosa, J. Phys. Chem. C, 2011, 115, 6491–6495 Search PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- J. F. Berry, E. Bill, E. Bothe, S. D. George, B. Mienert, F. Neese and K. Wieghardt, Science, 2006, 312, 1937–1941 CrossRef CAS PubMed.
- I. Mayer, Chem. Phys. Lett., 1983, 97, 270–274 CrossRef CAS.
- A. J. Bridgeman, G. Cavigliasso, L. R. Ireland and J. Rothery, J. Chem. Soc., Dalton Trans., 2001, 2095–2108 RSC.
- I. Mayer, J. Comput. Chem., 2007, 28, 204–221 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Figures of polymer structures containing Sc2+, Ti2+, Ni2+ or Zn2+, structures and energetic data of individual [c2]daisy chains in -cis and -trans forms, occupied molecular orbitals of [c2]daisy chains, and the table of calculated polymer unit lengths and contraction rate data are included. See DOI: 10.1039/c5cp07772a |
| This journal is © the Owner Societies 2016 |