
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater-mediated influence of a crowded environment on internal vibrations of a protein molecule†
Anna
Kuffel
and
Jan
Zielkiewicz
*
Faculty of Chemistry, Gdansk University of Technology, Narutowicza 11/12, 80–233 Gdansk, Poland. E-mail: anna.kuffel@pg.gda.pl; jaz@chem.pg.gda.pl
First published on 25th January 2016
Abstract
The influence of crowding on the protein inner dynamics is examined by putting a single protein molecule close to one or two neighboring protein molecules. The presence of additional molecules influences the amplitudes of protein fluctuations. Also, a weak dynamical coupling of collective velocities of surface atoms of proteins separated by a layer of water is detected. The possible mechanisms of these phenomena are described. The cross-correlation function of the collective velocities of surface atoms of two proteins was decomposed into the Fourier series. The amplitude spectrum displays a peak at low frequencies. Also, the results of principal component analysis suggest that the close presence of an additional protein molecule influences the high-amplitude, low-frequency modes in the most prominent way. This part of the spectrum covers biologically important protein motions. The neighbor-induced changes in the inner dynamics of the protein may be connected with the changes in the velocity power spectrum of interfacial water. The additional protein molecule changes the properties of solvation water and in this way it can influence the dynamics of the second protein. It is suggested that this phenomenon may be described, at first approximation, by a damped oscillator driven by an external random force. This model was successfully applied to conformationally rigid Choristoneura fumiferana antifreeze protein molecules.
Introduction
There is increasing awareness of the importance of the so called protein quinary structures.1,2 This is the highest level of protein organization. Although the quinary interactions are relatively weak (up to several kJ mol−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3,4), they are numerous and they can significantly influence the biological activity. They are especially significant in a crowded interior of a cell, where bulk water is, essentially, not present.5,6 Macromolecules occupy from 5% to 40% of the total volume of a cell. As a consequence, even a larger volume fraction is unavailable to an additional macromolecule.7 This has got implications to the folding of proteins, the formation of complexes and reducing the rate of diffusion.7 It has been observed, for example, that the stability of a protein molecule can be increased by other proteins present in the neighborhood.8–10 This increase may not only be due to an excluded volume effect, but can also be a result of the changes in the properties of solvation water, such as density and viscosity. To add to the problem, it should be stressed that two surfaces of proteins are sometimes able to modify the properties of interfacial water more deeply than a single one, depending on the distance between them.11,12
3,4), they are numerous and they can significantly influence the biological activity. They are especially significant in a crowded interior of a cell, where bulk water is, essentially, not present.5,6 Macromolecules occupy from 5% to 40% of the total volume of a cell. As a consequence, even a larger volume fraction is unavailable to an additional macromolecule.7 This has got implications to the folding of proteins, the formation of complexes and reducing the rate of diffusion.7 It has been observed, for example, that the stability of a protein molecule can be increased by other proteins present in the neighborhood.8–10 This increase may not only be due to an excluded volume effect, but can also be a result of the changes in the properties of solvation water, such as density and viscosity. To add to the problem, it should be stressed that two surfaces of proteins are sometimes able to modify the properties of interfacial water more deeply than a single one, depending on the distance between them.11,12
It is generally acknowledged that the fluctuations of hydration water and the inner motions of proteins are closely coupled.5,13–22 The solvent influences the inner protein motions and the durability of the tertiary structure related to these motions.23–25 As a result of long-term investigations conducted mostly on myoglobin, two kinds of protein motions have been described, i.e. large-scale ones connected to the bulk solvent fluctuations, and inner motions connected to the fluctuations in the hydration shell.17–20 Thus, we can say that each protein has its own signature dynamic pattern, which depends not only on its conformation and amino acid composition but also on dynamics of surrounding water. For these reasons, we can expect that a change in the dynamics of solvation water between two proteins will have an impact on the inner motions of both of them. This may be followed by an assumption that the dynamics of the two protein molecules will become interdependent to some extent.
In our previous paper26 we were able to confirm this effect using molecular dynamics. We described a weak correlation of the movement of surface atoms belonging to proteins separated by a layer of water – a kinesin molecule and a tubulin dimer. We also observed that the presence of the tubulin dimer influenced the movement of the kinesin molecule as a whole.
This behavior should not be specific only to the kinesin–tubulin duo. Herein, we would like to investigate another set of proteins and describe the possible physical mechanism underlying the phenomenon in question. It is detected mainly for low-frequency oscillations, which are especially important for the specific biological activity of proteins.27 The model proposed by us connects the changes of the dynamics of solvation water with the changes of the inner protein dynamics in the low-frequency range.
The synchronization of oscillators is not, generally speaking, a unique phenomenon, though it has not really been discussed in the context of the separate proteins. One example that we were able to find, very loosely connected with the subject of this paper, was an article by Rheinstädter et al.28 who depicted long-range protein–protein interactions in purple membranes as springs connecting the proteins.
Also, not only water mediation can make it possible for one protein to feel the presence of another protein from a distance. These long-range forces can be electrostatic interactions,29,30 depending on the net charge, dipole or multipole moment, the polarity of surface residues and the shape of the molecule.6 Another way of interaction between spatially separated proteins might be via an electromagnetic field,31 though this subject is poorly examined.
Herein, we use a hyperactive antifreeze protein from Choristoneura fumiferana (CfAFP). Antifreeze proteins are produced by many organisms that live in the cold environment to protect them against freezing. It is believed that the changes in the properties of solvation water are important for the functioning of AFPs. For example, they are said to facilitate the binding of these proteins to the surface of ice.32–36 However, the general picture is not entirely clear – there are some inconsistencies between the conclusions reached by various authors. These differences in opinion concern, among other things, the dynamic properties of solvation water. The importance of the properties of water between AFP molecules was acknowledged indirectly, when discussing the concentration dependence of antifreeze activity. For example Ebbinghaus et al.37,38 hypothesized that the modified properties of water can be important for maintaining the high antifreeze activity at lower concentrations thanks to the long range of changes. It should be mentioned, however, that their conclusions were partially questioned by Halle39 and the problem of the extent of the hydration shell of antifreeze agents is under debate. Some papers indicate that the dynamic properties of water around antifreeze proteins do not differ very significantly from the ones around proteins that do not interact with ice.40,41 Yet another result was obtained by Nutt and Smith, who investigated CfAFP solvation water by computer simulations and observed increased mobility of the solvent at some distance from the two protein surfaces which do not interact with ice.42 These conclusions were not entirely in accordance with ours, described in a previous paper.43
The selected AFP is a convenient choice for our study because of its structure. This molecule comprises three well-defined planes, therefore the proteins could be arranged so that the planes face each other. We hoped that such an arrangement would possibly emphasize any correlations of the movement of the protein atoms. The molecule as a whole is quite stiff – its prism-like structure is held together with disulfide bonds. However, on the two β-sheet planes of the molecule amino acids with quite long and mobile side chains can be found. These features of the CfAFP molecule make it more suitable for the present model-development studies than the more flexible and more complex kinesin molecule investigated previously.26
Methods
The results were obtained using computer simulations, with the molecular dynamics package Amber10,44 and ff03 force field, suitable for proteins.45System setup
Systems with one, two and three CfAFP molecules were constructed (see Fig. 1). The initial coordinates of the CfAFP molecule were taken from the Protein Data Bank (PDB ID: 1L0S). The original file contained one iodated tyrosine (not present in natural protein), which was changed into tyrosine. Moreover, the counterions (chloride anions, Cl−), and all missing residues and atoms were added in the LEaP program, which is a part of Amber package. Amino acids with charged side chains were Arg, Asp, Glu and Lys. Finally, the single protein was placed inside a truncated octahedral box, while two and three proteins were placed inside a cubic box. The boxes were solvated with SPC/E water, with a minimal distance between the protein and the box walls equal to 2.5 nm.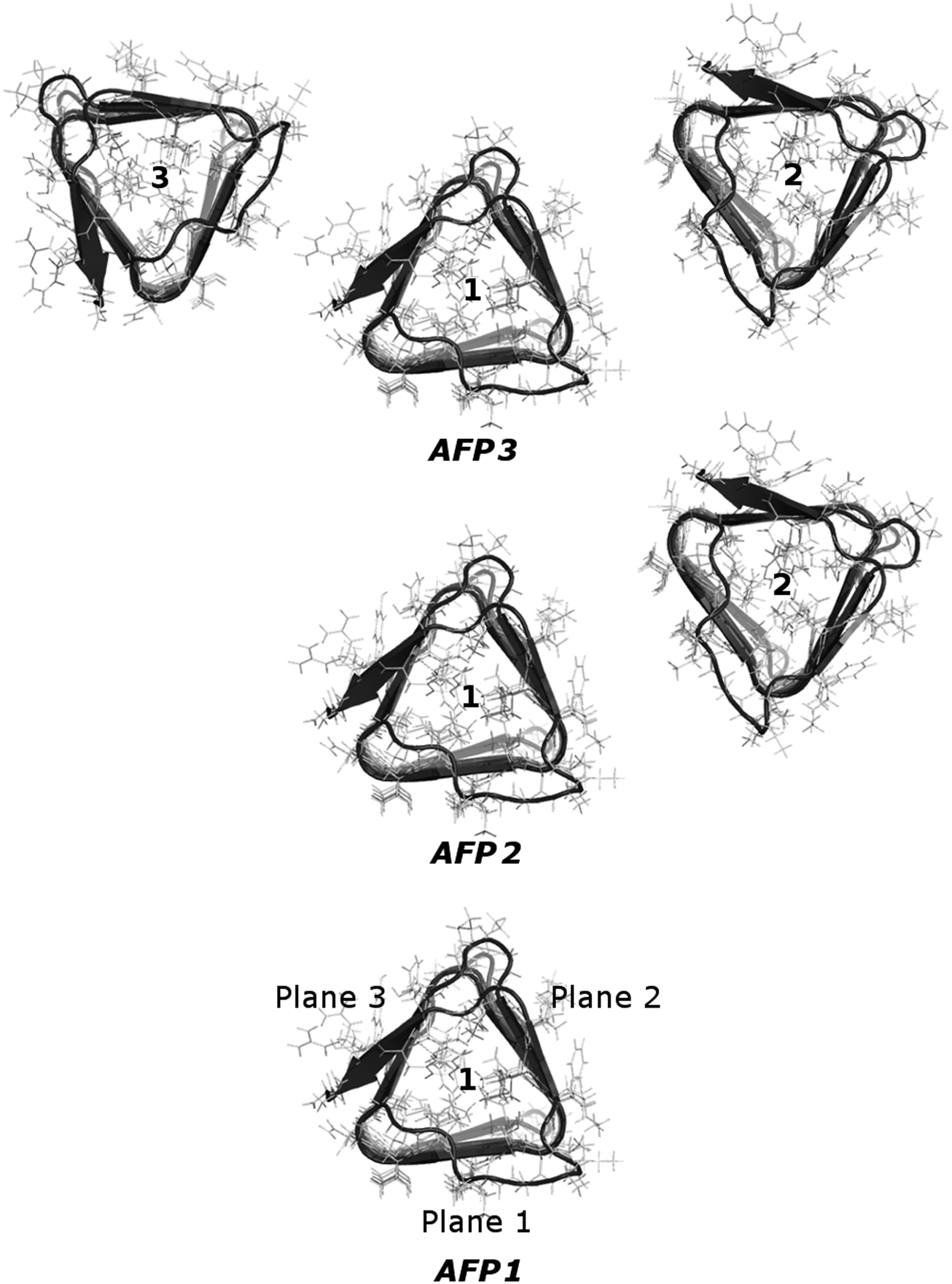 | ||
| Fig. 1 Three studied systems, consisting of one CfAFP molecule, two CfAFP molecules and three CfAFP molecules. The molecule for which the calculations were performed is molecule number 1. It was called AFP1, AFP2 or AFP3, depending on the number of proteins in the simulation box. In the bottom picture, the three planes of the prism-shaped protein were signed. The ice-binding plane was called plane 1. | ||
Simulation procedure
A preliminary period of equilibration under NPT conditions lasted for about 2.0 ns. The temperature (300 K) was kept constant by the weak coupling to an external bath (τT = 1.0 ps) using a Berendsen thermostat.46 The pressure (1 bar) was kept constant by the weak coupling method (τp = 1.0 ps). The particle-mesh Ewald method was used for electrostatic interactions, and the lengths of chemical bonds involving hydrogen atoms were fixed using SHAKE. A cutoff of 1.2 nm for nonbonding interactions was used. The equilibrated systems were simulated under NVE conditions, using a time step equal to 2 fs. Trajectories were saved every step. The summed length of all trajectories used for calculations was equal to about 48 ns (PCA analysis) or 96 ns (cross-correlation functions of the collective velocities of the surface protein atoms) for each system. More details regarding the simulation procedure can be found in the ESI.†Calculation of diffusion coefficients
The translational diffusion coefficients DT were calculated from the velocity auto-correlation function, as it was described previously,47 using the Green–Kubo relation:48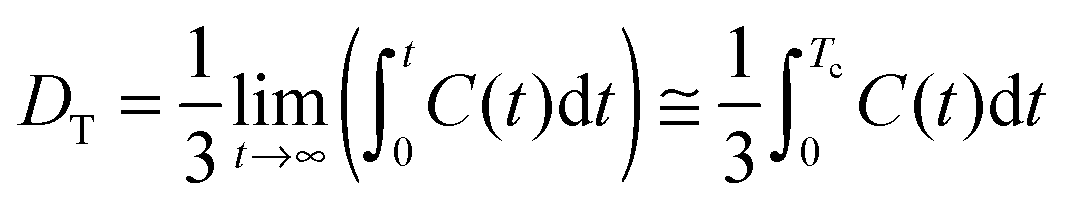 | (1) |
Definition of solvation layers
We used a commonly accepted definition based on the distance between water molecules and the protein surface. The distance used in the calculation was equal to 0.5 nm. The choice of this distance may be justified by the paper of Chen et al.50 According to them, the water–protein spatial distribution function reaches near-zero values for a water–protein distance greater than 4.5 Å. A similar definition of the solvation shell was used by us in our previous articles on the CfAFP,43,49 though the distance was then 0.4 nm. Xu et al.51 also used a cut-off distance equal to 0.5 nm to analyze the solvation of CfAFP.To ascribe a particular molecule to the solvation shell, the following procedure was adopted. For a selected water molecule, its mean space position was determined over a 4 ps-long time interval. On the basis of this mean position we ascribed the water molecule to the appropriate solvation layer of CfAFP.
Covariance matrix and principal component analysis
To calculate the principal components, a covariance matrix of internal atomic displacements was calculated (the coordinates of each atom were weighted by its mass):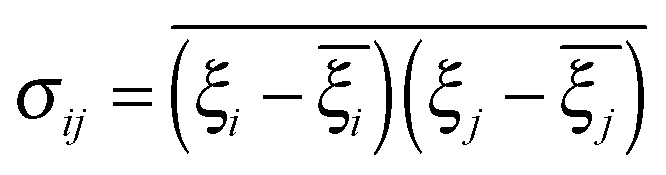 | (2) |
Results
Previously, for a kinesin and a tubulin dimer, we were able to find a correlation of the motion of surface atoms of proteins separated by a layer of water. Moreover, the presence of the tubulin dimer caused the change in the principal components characterizing the inner motions of the whole kinesin head.To investigate this phenomenon further, we now take into consideration three different systems: one comprised of a single CfAFP molecule immersed in water, one comprised of two CfAFP molecules and one comprised of three CfAFP molecules. The proteins were not in direct contact – they were separated by a layer of water. The planes of the prism-shaped proteins faced each other. The arrangement of the molecules is depicted in Fig. 1. To capture the influence of the presence of one or two additional protein molecules in a close neighborhood, the dynamics of the single protein (AFP1) was compared with the dynamics of the protein with one (AFP2) or two (AFP3) neighboring molecules.
The discussion of the results was divided into several parts and is presented below.
(1) The correlation of vectors of collective velocity of surface atoms from two separate protein molecules. We used the method described in our previous paper.26,49 We investigated the cross-correlation function of the vectors of collective velocity of atoms located at the surfaces of the two neighboring AFP molecules. The details of the method are as follows.
Let us assume that at time t some surface atom of the protein molecule number 1 (AFP2 or AFP3 in Fig. 1) has got the coordinate r01. The closest surface atom on the second protein has got the coordinate r02. The distance between the atoms is d. For these two atoms the vectors of the collective velocities of the surface atoms are calculated as a weighted arithmetic mean. The weights are from the Gaussian distribution. The closest atoms contribute the most to the final result. The equation is as follows:
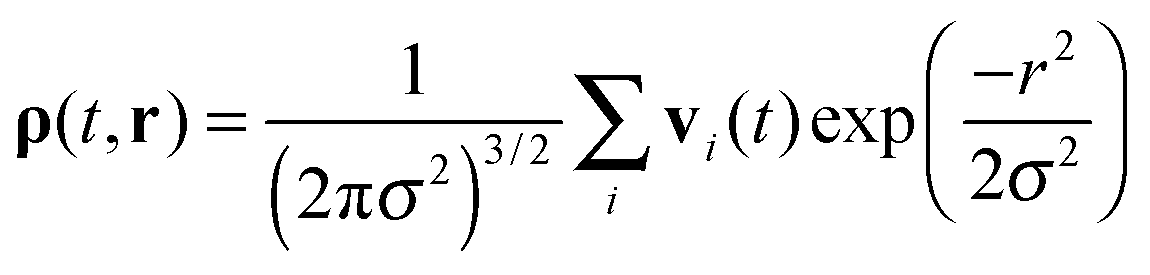 | (3) |
The cross-correlation function between the collective velocities of both surface areas is defined as:53
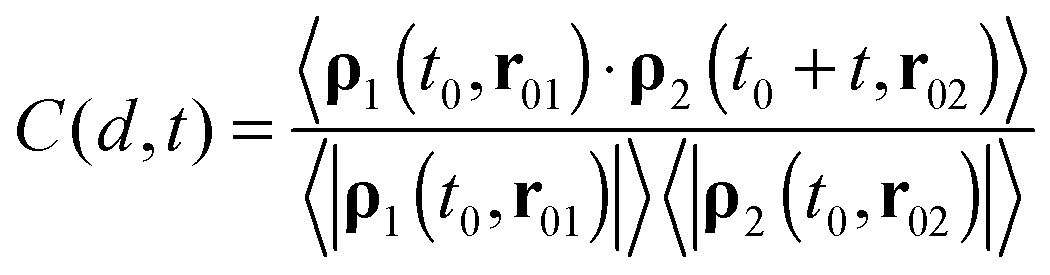 | (4) |
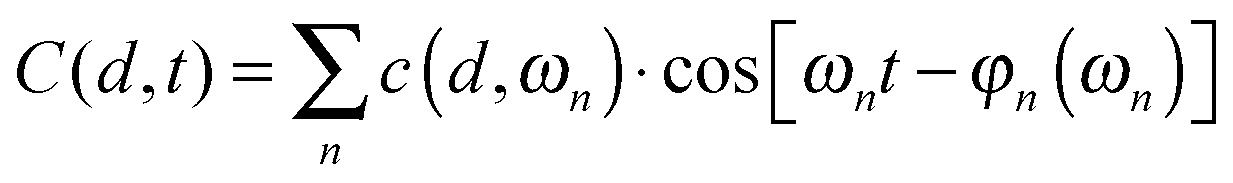 | (5) |
The distance between the proteins could change slightly during the simulation. The average value of d was equal to 1.1 nm. Therefore, in Fig. 2 we present the amplitude spectra and the phase spectra calculated for this value.
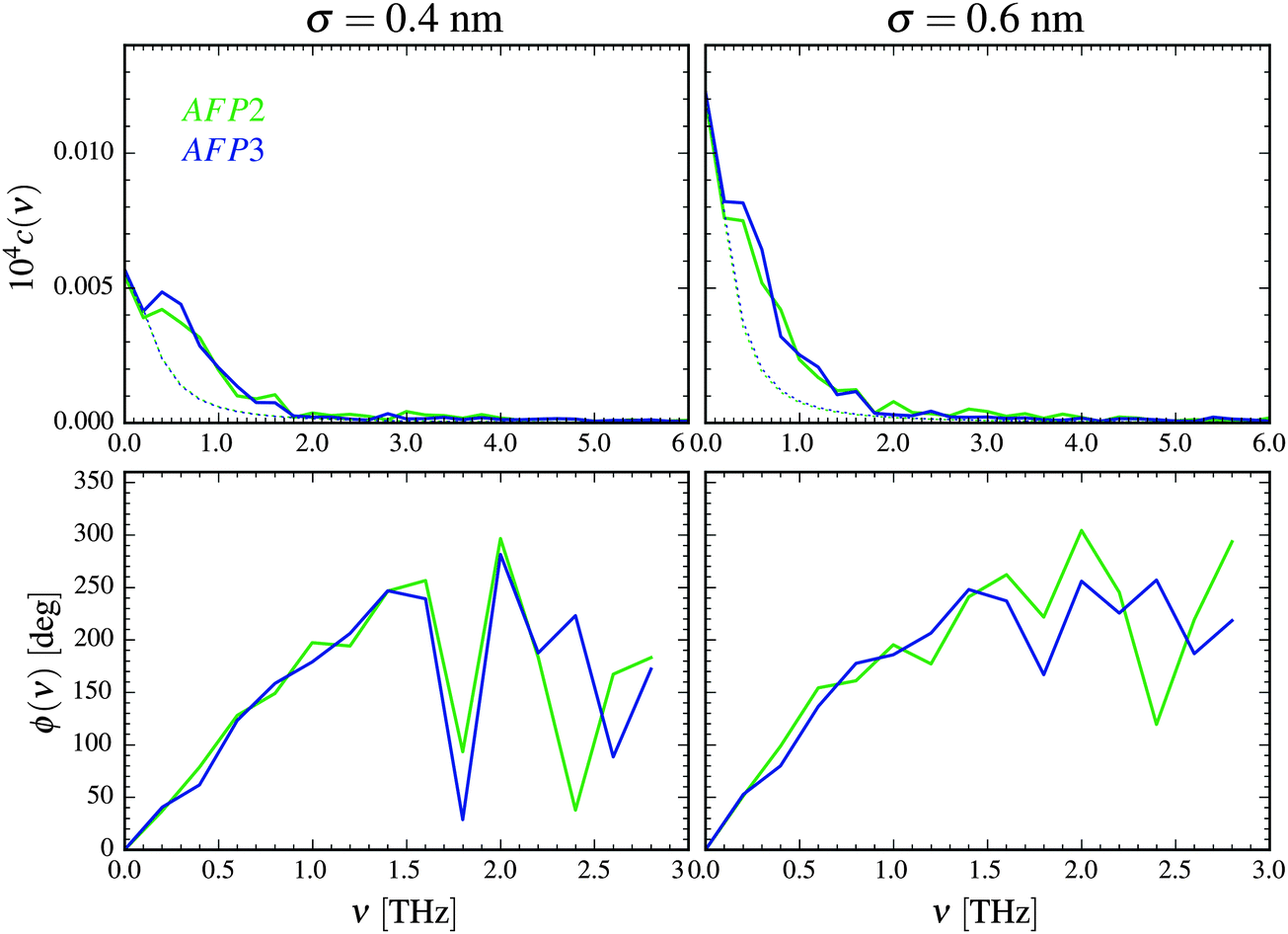 | ||
| Fig. 2 Amplitude spectra (top) and phase spectra (bottom) of the collective velocity correlation function of the AFP molecule 1 and 2 calculated according to eqn (1)–(3) for σ = 0.4 nm and σ = 0.6 nm. The distance between the atoms belonging to different proteins is d = 1.1 nm. The estimation of the purely diffusional part of the spectra is presented as a dotted line. | ||
The correlation of motion is most visible for the frequencies lower than about 2 THz and diminishes for the frequencies higher than 5 THz. As we discussed in the paper,49 the high values for the near-zero frequencies may originate from the concerted diffusion motion of the investigated molecules. The influence of this factor can be estimated, following the concept of Lin et al.54 We have to stress, however, that this estimation is very rough. As it is presented in Fig. 2, although the influence of the concerted diffusion on the low-frequency part of the amplitude spectra is significant, it cannot fully explain the obtained intensities – especially the asymmetric band below 1 THz.
There are three most important features of these spectra. The first one is the similarity of the spectra denoted as AFP2 and AFP3. The additional molecule (number 3) may slightly increase the correlation between the motions of the surface atoms of the two investigated proteins (number 1 and 2). However, overall, there is not any strong influence of the presence of the third protein molecule on the amplitude spectra measured for the first and the second molecule.
The second feature is the small values of the correlation coefficients – the order of magnitude is 10−6. The third feature is a linear growth of the phase shift with increasing frequency. Following the suggestion of Heyden and Tobias,55 the correlation may be explained by a sound wave propagation between the protein surfaces. The wave travels through the interfacial solvent and this fact is a reasonable explanation of the small values of the correlation coefficients.
(2) The influence of the neighboring protein molecules on the inner motions of the protein molecule. To investigate the influence of the close presence of neighbors on the inner motions of the protein molecule, the principal component analysis56 (PCA) was performed. It is one of the methods used to obtain information on the dynamics of proteins.57–59
To calculate the principal components, a covariance matrix of internal atomic displacements was calculated, after removing the translation and rotation of the molecule as a whole. Let us assume, very roughly, that the oscillations of the atoms are harmonic. In this case, the eigenvalues are directly related to the mean square displacements from the equilibrium, and it is possible to evaluate the frequency of the oscillator (from the equipartition theorem). We have to note, however, that this assumption is almost certainly not true for the whole range of frequencies – especially for the first few modes with very low frequencies.60
To check how the inner motions of the protein are affected by the presence of one or two neighbors, the frequencies of the oscillations were estimated for the molecule with no neighbors (AFP1) and accompanied with one (AFP2) or two (AFP3) additional proteins.
The relative changes of the estimated frequencies are depicted in Fig. 3. The differences in the frequencies of the consecutive modes are divided by the frequency of that mode estimated for the single protein. The frequency of the oscillations is undoubtedly changed when there are additional protein molecules present in the system.
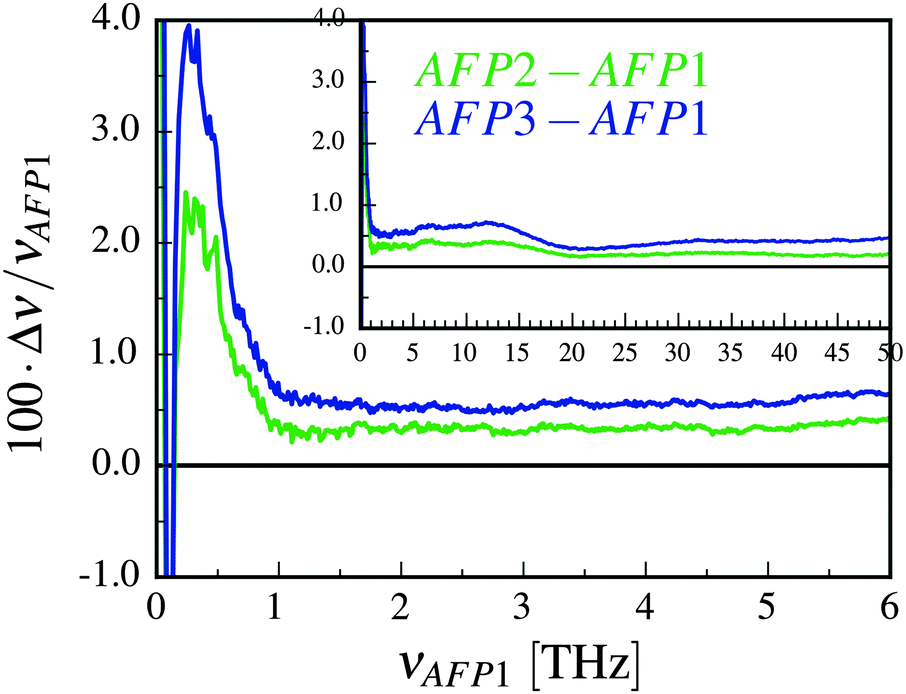 | ||
| Fig. 3 The relative change of the frequencies of the normal modes of the AFP molecule in the presence of one or two neighboring protein molecules. The relative change is plotted as a function of the frequencies of the protein without any neighbors (AFP1). The change Δν is equal to νAFPX–νAFP1, where X = 2 or X = 3. The inset depicts the same results over the greater frequency range. | ||
Because the assumption of harmonicity may be regarded as dubious, we decided to evaluate the conformational changes of the protein also in a different way. As a measure of conformational changes, we used the temporal changes of the dipole moment of the protein. The results are described in the ESI† and in our opinion they confirm the conclusions reached after the PCA analysis.
There are at least two causes that can explain the changes in the dynamic pattern of the protein. The first cause may be the water-mediated interactions between the neighboring protein molecules. The second and probably more important one is the change of the properties of the interfacial solvent (its viscosity, diffusion coefficient, density, etc.).
Let us discuss the significance of these two factors separately.
(a) The water-mediated interactions between the neighboring protein molecules. As we can see in Fig. 3, for the frequencies lower than 1 THz there is a band that illustrates a significant relative increase of the frequencies (and decrease of the amplitude). It is the low-frequency mode that differs the most between the single AFP molecule (AFP1) and the molecule with one or two neighbors (AFP2 and AFP3). This range of frequencies agrees well with the range of frequencies for which the correlation of motion of surface protein atoms was observed for the molecules 1 and 2, what was described above. Although this may suggest that the correlation is responsible for modifying the frequencies of the modes, this hypothesis does not seem to be probable. As we have mentioned, the correlation coefficients are very low (∼10−6) – they would be even lower if we excluded the influence of the concerted diffusive motion of the whole molecules. Therefore, it does not seem to be plausible that such a subtle effect can trigger considerably larger (∼10−2) relative changes of the frequencies depicted in Fig. 3. Therefore, we turn to the second proposed factor that, possibly, underlies the changes of the frequencies, which is the change of the properties of water.
(b) The change of the properties of solvation water. Let us start with a commonly used parameter characterizing the properties of water, which is a translational diffusion coefficient. This choice is well grounded since there is a confirmed interconnection between the dynamics of the solvent and dynamics of the protein, as it was mentioned in the Introduction.
The diffusion coefficients were calculated for the first solvation shells of the three planes of the analyzed protein. The results can be found in Table 1 and they agree with the previously published data.49,51 As we could have expected, the mean mobility of water molecules diminishes with the increasing number of neighbors. The presence of the additional protein molecule may cause deeper changes in the solvation water properties – deeper than a single protein is able to induce. The diffusion coefficients of solvation water of the AFP molecule drop when the second and the third molecule are introduced.
| Protein | Plane 1 | Plane 2 | Plane 3 | Average |
|---|---|---|---|---|
| AFP1 | 1.51 | 1.56 | 1.59 | 1.65 |
| AFP2 | 1.42 | 1.35 | 1.54 | 1.51 |
| AFP3 | 1.41 | 1.33 | 1.44 | 1.47 |
The lower mobility of the solvent should result in diminishing the amplitude and increasing the frequency of the oscillations of the surface protein atoms. This can be observed in Fig. 3. For the higher frequencies, the relative change of the frequency of the modes stops varying – the plots are shifted by an approximately constant value, which is about 0.4% for the AFP2 and 0.6% for the AFP3. Alternatively, we can say that the fast fluctuations are shifted to longer time scales.61 We believe that this shift is caused by the change of diffusion coefficient/viscosity of neighboring water.62
However, there still remains the problem of how to explain the non-uniformity of the changes – the presence of the high peak below 1.5 THz. Therefore, we are going to examine the dynamic properties of the interfacial solvent further and calculate the velocity power spectra of water molecules in the solvation shell of molecule 1 with one or two neighbors (AFP2 and AFP3). These spectra will be compared to the spectrum of solvation water of a single protein (AFP1).
The oscillations of the protein can be influenced by the translation and rotation of a water molecule.63 The (relatively small) differences between the rotational spectra in Fig. 4 can be seen mostly at about 15–25 THz. Since this is not the range of the biggest changes of the frequencies in Fig. 3, we decided to omit the rotational spectrum in the further discussion.
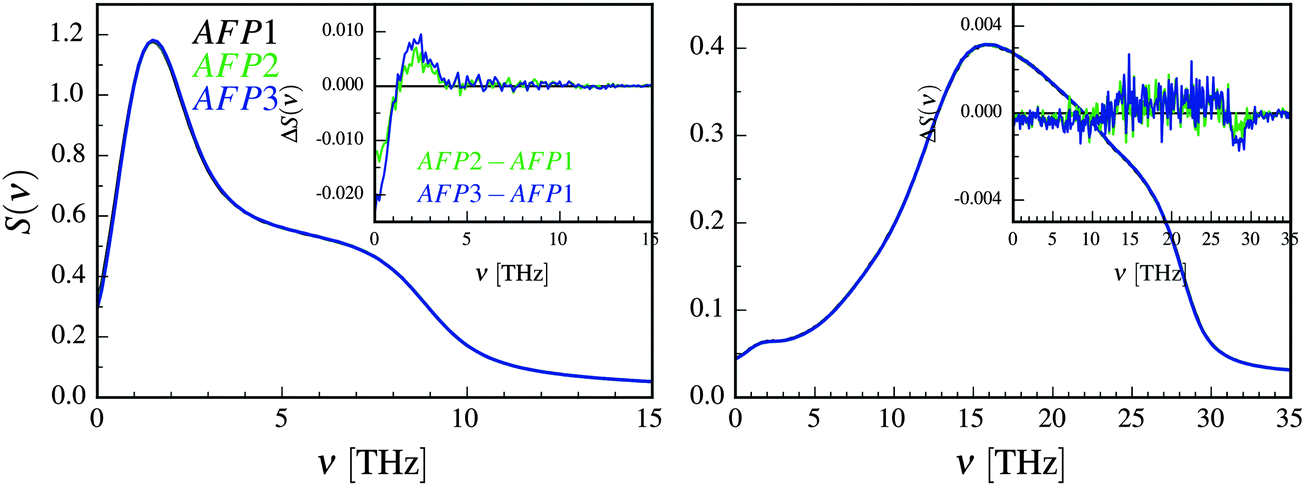 | ||
| Fig. 4 The translational (left) and rotational (right) velocity power spectra of solvation water of molecule 1 with no neighbors (AFP1), one neighbor (AFP2) and two neighbors (AFP3). The inset shows the differential spectra. The change ΔS(ν) is equal to S(ν)AFPX–S(ν)AFP1, where X = 2 or X = 3. | ||
The translational differential velocity spectra in Fig. 4 display high negative values for the lowest frequencies. These are a result of differences in the diffusion coefficients, which increase in the following order: AFP3 < AFP2 < AFP1. There is also a peak at about 2 THz. This frequency is closer to the observed frequency changes in Fig. 3. The relative magnitudes of the changes in protein dynamics (Fig. 3) and in solvation water dynamics (Fig. 4) are comparable.
These observations founded our working hypothesis – that the measured changes in the frequencies of the inner motion of the protein in the presence of the neighbors may be connected with the observed changes in the translational power spectra of water. Thus, the next question is what might be the specific physical explanation of the observed phenomenon of the concerted changes in the frequency characteristic of water and protein motion. We will try to model the protein as a damped harmonic oscillator with many degrees of freedom and subject to an external random force. Our aim is to demonstrate that our model is able to predict the general shape of the distribution of the mean square displacements of each mode if the spectral characteristics of external random force are known. The predicted results may be compared with the eigenvalues of the covariance matrix, obtained from the simulation. The details of the model are presented below.
Model presentation
There are continuous collisions of the protein molecule with the solvent. Therefore, we could try to describe the system as a damped harmonic oscillator with many degrees of freedom subjected to an external random force. The source of the external force is the thermal collisions with the solvent. We also assume that the damping force is the viscous one. Unfortunately, this model does not perfectly represent the actual physical reality in this case. Within the harmonic approximation, all normal modes of the oscillating system are independent. We are aware that, in reality, the inner motions of the protein molecule are anharmonic, which permits (according to the generalized equipartition theorem64) the energy exchange between the modes. However, we will consciously and cautiously accept the harmonic approximation to move forward.Let us denote the function that describes the frequency dependence of the external force by S(ν). In the case of white noise S(ν) = const > 0. Since we assumed that the source of the force is the collisions of water molecules with the protein, we used the translational velocity power spectrum as a function S(ν). The rotation of water molecules is neglected, as it was argued above.
If the frequency dependence of the external force is known, we can calculate the mean square displacement 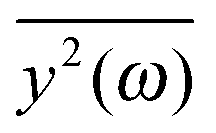 of the normal mode.65 In the below equation, the frequency of the mode is denoted by ω, to differentiate it from the frequency dependence of the force, denoted by ν. The friction force is represented by the coefficient ξ.
of the normal mode.65 In the below equation, the frequency of the mode is denoted by ω, to differentiate it from the frequency dependence of the force, denoted by ν. The friction force is represented by the coefficient ξ.
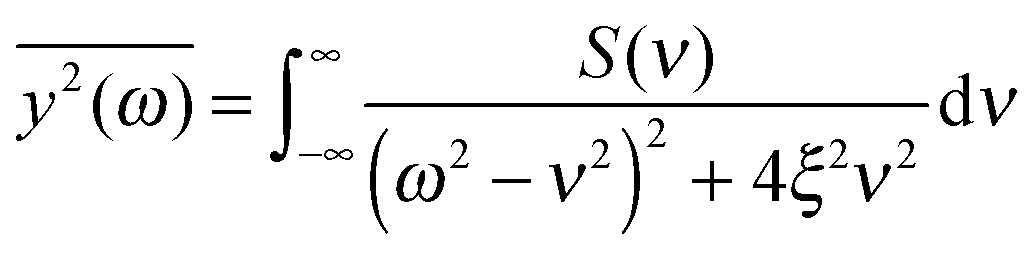 | (6) |
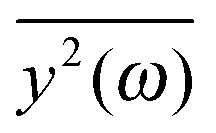 depends on the spectral characteristic of the external force S(ν). The change of
depends on the spectral characteristic of the external force S(ν). The change of 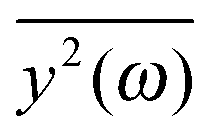 after the change of the frequency dependence of the force can be calculated as:
after the change of the frequency dependence of the force can be calculated as: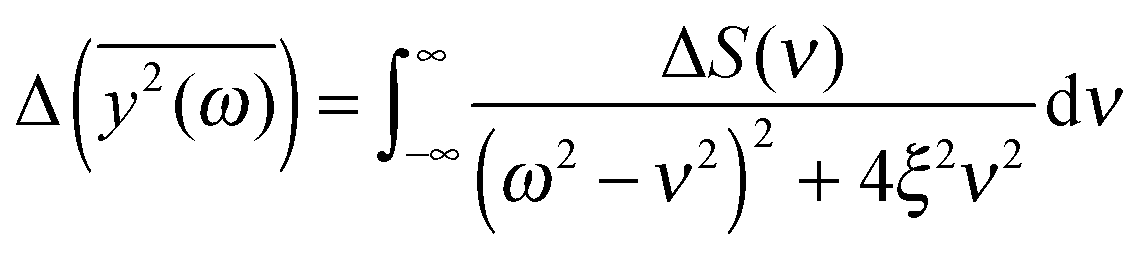 | (7) |
The assumption of the constant value of the friction coefficient may be disputable to some extent. We justify it as follows. Two main factors that contribute to the value of friction coefficient are the viscosity of solvation water and the “internal” viscosity of the protein molecule. The second term seems to be dominant. Because the viscosity of solvation water varies relatively little (below 10%, as can be estimated from the data in Table 1), the error, resulting from the above assumption, should be small. We should also mention that the friction coefficient of water may vary not only from system to system, but also can depend on the frequency.66,67
Another problem is the choice of the value of the friction coefficient. Its value should be small enough to allow for stationary oscillations of all modes of interest. Thus, we place ξ = 0.05 in eqn (7). The comparison of calculated 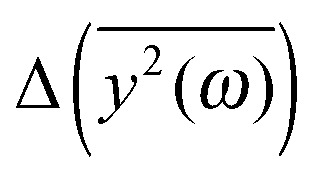 for several different values of ξ can be found in the ESI.†
for several different values of ξ can be found in the ESI.†
There is also a silent assumption that the external random force influences the protein homogeneously (the translational velocity power spectra are calculated for the whole solvation shell). However, the differences in them are mostly caused by the differences in this part of the solvation shell that faces another protein.
Since the eigenvalues obtained from the principal component analysis can be directly related to the mean square displacements, we are now able to compare the theoretical values of 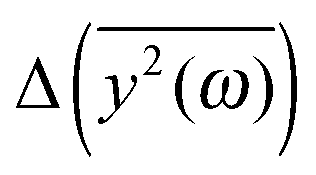 with the results of the principal component analysis. However, the direct comparison of the model with the results is difficult for several reasons. As previously discussed, it seems that all modes of AFP2 and AFP3 are shifted to higher frequencies by a small and approximately constant percent (Fig. 3). We believe that this shift is caused by the difference in the diffusion coefficients. The model does not account for this effect (there is no predicted difference of
with the results of the principal component analysis. However, the direct comparison of the model with the results is difficult for several reasons. As previously discussed, it seems that all modes of AFP2 and AFP3 are shifted to higher frequencies by a small and approximately constant percent (Fig. 3). We believe that this shift is caused by the difference in the diffusion coefficients. The model does not account for this effect (there is no predicted difference of 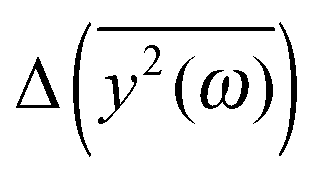 for high ω). Because of that, it would be highly desirable to eliminate this shift. Therefore, as a preliminary step, we tried to remove the difference between the high frequency modes by multiplying all eigenvalues by a constant factor. As a result, the differences between the modes of the highest frequencies are set to about zero. The shifted plots can be found in the ESI.† The troublesome point is that the relative differences between the modes are not strictly constant for all high frequencies. Therefore, we had to arbitrarily (to some extent) choose a specific frequency range that will be set to zero. The influence of this choice on the results can also be found in the ESI.† To obtain the plot in Fig. 5, we shifted to zero the modes for the frequency equal to about 10 THz.
for high ω). Because of that, it would be highly desirable to eliminate this shift. Therefore, as a preliminary step, we tried to remove the difference between the high frequency modes by multiplying all eigenvalues by a constant factor. As a result, the differences between the modes of the highest frequencies are set to about zero. The shifted plots can be found in the ESI.† The troublesome point is that the relative differences between the modes are not strictly constant for all high frequencies. Therefore, we had to arbitrarily (to some extent) choose a specific frequency range that will be set to zero. The influence of this choice on the results can also be found in the ESI.† To obtain the plot in Fig. 5, we shifted to zero the modes for the frequency equal to about 10 THz.
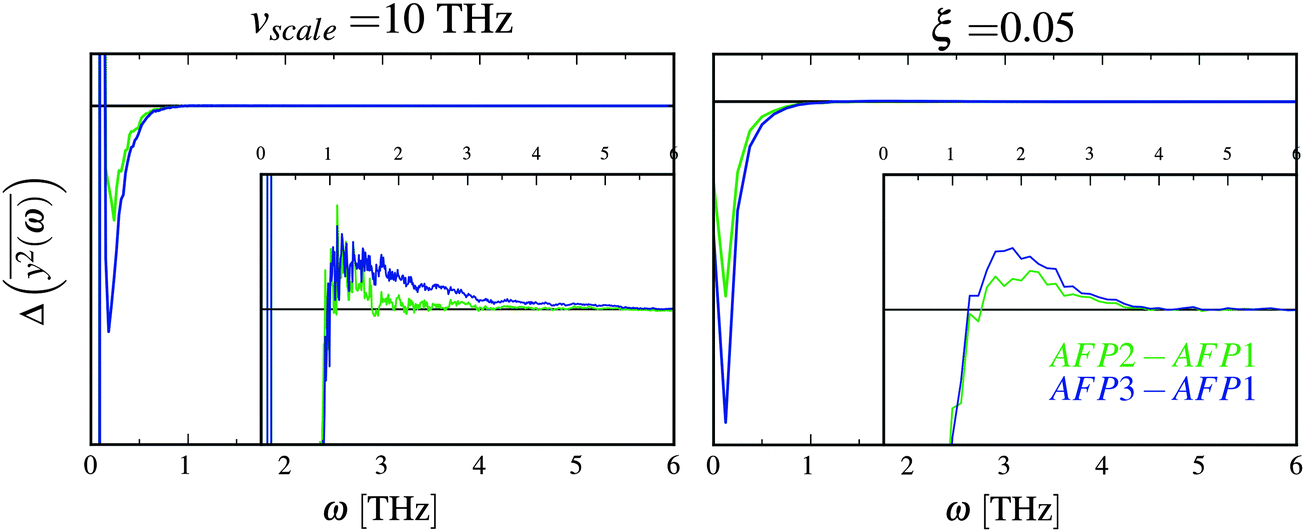 | ||
Fig. 5 The comparison of the differences of the amplitudes 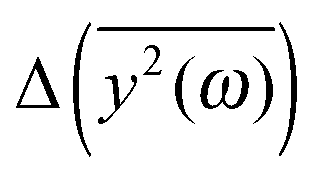 obtained from the simulations (left) and calculated from eqn (7) (right). The inset displays enlarged fragments of the plots. The units of obtained from the simulations (left) and calculated from eqn (7) (right). The inset displays enlarged fragments of the plots. The units of 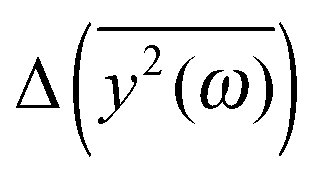 are arbitrary, therefore the values on the axes are not displayed. are arbitrary, therefore the values on the axes are not displayed. | ||
We have to remember that the frequencies are estimated from the eigenvalues. Because of that, the question arises: how to measure the changes between the values of amplitudes with frequency if the latter is calculated from the former. We decided to calculate the differences 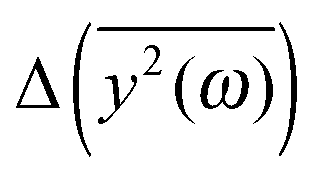 for subsequent eigenvalues and plot them as a function of the estimated frequency for the protein AFP1 (just like in Fig. 3).
for subsequent eigenvalues and plot them as a function of the estimated frequency for the protein AFP1 (just like in Fig. 3).
There are some qualitative similarities between the left and right plot in Fig. 5. This may suggest that the model, despite many severe simplifications, may describe some aspects of the role of water and its influence on the inner dynamics of the protein molecule.
In the discussion, we omit the first three modes (with the greatest amplitudes). The harmonicity assumption is most certainly not valid for them. In the differential diagrams in Fig. 3 and 5, they peak at very low or high values, hence the almost vertical lines at the beginning.
The first similarity of the distributions of 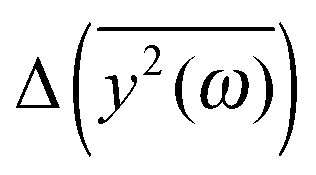 is a significant decrease of the amplitudes for the lowest frequencies (if the friction parameter is small enough). The bulge at about 1–3 THz fully appears when the scaling of the eigenvalues is done for the reference frequency equal to 10 THz. For comparison with differently scaled distributions, see ESI.†
is a significant decrease of the amplitudes for the lowest frequencies (if the friction parameter is small enough). The bulge at about 1–3 THz fully appears when the scaling of the eigenvalues is done for the reference frequency equal to 10 THz. For comparison with differently scaled distributions, see ESI.†
To summarize the discussion, a very simplified model, a damped oscillator with many degrees of freedom driven by an external random force, was used to explain the modification of inner dynamics of the protein molecule in the presence of additional protein molecules. The most affected frequencies are smaller than 1 THz. The changes in the frequency characteristic of translational dynamics of solvation water are best visible below 4 THz, which includes the range of the most modified frequencies of the oscillations of the protein.
As it was mentioned previously in the Introduction, the low-frequency range is crucial for the biological activity of proteins. Thus, the presented model sheds some light on the possible mechanism of the water influence on the functioning of biomolecules. We also have to remember that the discussed effects are not very prominent, and they do not exceed a few percents.
The last issue worth mentioning is the generality of these findings. We believe that the most important factor that makes the model applicable to the CfAFP molecule is its stiffness. The model requires the oscillations to be harmonic and for more stiff molecules this assumption is fulfilled better. To examine the role of flexibility, we prepared a less rigid version of the protein molecule – a CfAFP molecule with artificially removed disulfide bonds. Fig. S5 in the ESI† depicts a relative difference of the frequencies of the oscillations of the single wild-type protein and two or three proteins without the disulfide bonds. The low-frequency differences become negative due to the increased flexibility of the molecule. However, the peak below 1 THz is still visible. The same is generally true for the kinesin–tubulin dimer that originated these calculations.26 However, the interpretation of the results for that system is more complicated because the kinesin molecule is more conformationally labile. Taking this into account, we suspect that the general conclusions may be applicable to a wide range of proteins, though the details will probably be different.
Acknowledgements
The calculations were carried out at the Academic Computer Center (TASK) in Gdańsk. This research was also supported in part by PL-Grid Infrastructure. This work was supported by the Polish National Science Centre within the research grant No. DEC-2013/09/D/NZ1/01087.References
- A. J. Wirth and M. Gruebele, BioEssays, 2013, 35, 984–993 CrossRef CAS PubMed.
- A. Gershenson, J. Mol. Biol., 2014, 426, 4–6 CrossRef CAS PubMed.
- P. Chien and L. M. Gierasch, Mol. Biol. Cell, 2014, 25, 3474–3477 CrossRef PubMed.
- E. H. McConkey, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 3236–3240 CrossRef CAS.
- P. Ball, Chem. Rev., 2008, 108, 74–108 CrossRef CAS PubMed.
- A. P. Minton, J. Biol. Chem., 2001, 276, 10577–10580 CrossRef CAS PubMed.
- R. J. Ellis and A. P. Minton, Nature, 2003, 425, 27–28 CrossRef CAS PubMed.
- Y. Wang, M. Sarkar, A. E. Smith, A. S. Krois and G. J. Pielak, J. Am. Chem. Soc., 2012, 134, 16614–16618 CrossRef CAS PubMed.
- A. C. Miklos, M. Sarkar, Y. Wang and G. J. Pielak, J. Am. Chem. Soc., 2011, 133, 7116–7120 CrossRef CAS PubMed.
- A. C. Miklos, C. Li, N. G. Sharaf and G. J. Pielak, Biochemistry, 2010, 49, 6984–6991 CrossRef CAS PubMed.
- A. Kuffel and J. Zielkiewicz, Phys. Chem. Chem. Phys., 2013, 15, 4527–4537 RSC.
- L. Hua, X. Huang, R. Zhou and B. J. Berne, J. Phys. Chem. B, 2006, 110, 3704–3711 CrossRef CAS PubMed.
- S. Khodadadi, J. H. Roh, A. Kisliuk, E. Mamontov, M. Tyagi, S. A. Woodson, R. M. Briber and A. P. Sokolov, Biophys. J., 2010, 98, 1321–1326 CrossRef CAS PubMed.
- P. Ball, ChemPhysChem, 2008, 9, 2677–2685 CrossRef CAS PubMed.
- S. Hayward, A. Kitao, F. Hirata and N. Go, J. Mol. Biol., 1993, 234, 1207–1217 CrossRef CAS PubMed.
- V. Helms, ChemPhysChem, 2007, 8, 23–33 CrossRef CAS PubMed.
- P. W. Fenimore, H. Frauenfelder, B. H. McMahon and F. G. Parak, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16047–16051 CrossRef CAS PubMed.
- P. W. Fenimore, H. Frauenfelder, B. H. McMahon and R. D. Young, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14408–14413 CrossRef CAS PubMed.
- H. Frauenfelder, P. W. Fenimore, G. Chen and B. H. McMahon, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15469–15472 CrossRef CAS PubMed.
- H. Frauenfelder, G. Chen, J. Berendzen, P. W. Fenimore, H. Jansson, B. H. McMahon, I. R. Stroe, J. Swenson and R. D. Young, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5129–5134 CrossRef CAS PubMed.
- N. Shenogina, P. Keblinski and S. Garde, J. Chem. Phys., 2008, 129, 155105 CrossRef PubMed.
- R. Walser and W. F. van Gunsteren, Proteins, 2001, 42, 414–421 CrossRef CAS.
- D. Vitkup, D. Ringe, G. A. Petsko and M. Karplus, Nat. Struct. Biol., 2000, 7, 34–38 CrossRef CAS PubMed.
- A. L. Tournier, J. C. Xu and J. C. Smith, Biophys. J., 2003, 85, 1871–1875 CrossRef CAS PubMed.
- A. L. Tournier, V. Réat, R. Dunn, R. Daniel, J. C. Smith and J. Finney, Phys. Chem. Chem. Phys., 2005, 7, 1388–1393 RSC.
- A. Kuffel and J. Zielkiewicz, Phys. Chem. Chem. Phys., 2015, 17, 6728–6733 RSC.
- I. Bahar, T. R. Lezon, A. Bakan and I. H. Shrivastava, Chem. Rev., 2010, 110, 1463–1497 CrossRef CAS PubMed.
- M. C. Rheinstädter, K. Schmalzl, K. Wood and D. Strauch, Phys. Rev. Lett., 2009, 103, 128104 CrossRef PubMed.
- M. Ahmad, W. Gu and V. Helms, Angew. Chem., 2008, 47, 7626–7630 CrossRef CAS PubMed.
- G. Schreiber, G. Haran and H.-X. Zhou, Chem. Rev., 2009, 109, 839–860 CrossRef CAS PubMed.
- M. Cifra, J. Z. Fields and A. Farhadi, Prog. Biophys. Mol. Biol., 2011, 105, 223–246 CrossRef CAS PubMed.
- C. Yang and K. A. Sharp, Biophys. Chem., 2004, 109, 137–148 CrossRef CAS PubMed.
- C. Yang and K. A. Sharp, Proteins, 2005, 59, 266–274 CrossRef CAS PubMed.
- N. Smolin and V. J. Daggett, J. Phys. Chem. B, 2008, 112, 6193–6202 CrossRef CAS PubMed.
- J. Cui, K. Battle, A. Wierzbicki and J. D. Madura, Int. J. Quantum Chem., 2009, 109, 73–80 CrossRef CAS.
- D. R. Nutt and J. C. Smith, J. Am. Chem. Soc., 2008, 130, 13066–13073 CrossRef CAS PubMed.
- S. Ebbinghaus, K. Meister, B. Born, A. L. DeVries, M. Gruebele and M. Havenith, J. Am. Chem. Soc., 2010, 132, 12210–12211 CrossRef CAS PubMed.
- K. Meister, S. Ebbinghaus, Y. Xu, J. G. Duman, A. DeVries, M. Gruebele, D. M. Leitner and M. Havenith, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1617–1622 CrossRef CAS PubMed.
- B. Halle, J. Phys. Chem. B, 2014, 118, 10806–10812 CrossRef CAS PubMed.
- E. Doboue-Dijon and D. Laage, J. Chem. Phys., 2014, 141, 22D529 CrossRef PubMed.
- K. Modig, J. Qvist, C. B. Marshall, P. L. Davies and B. Halle, Phys. Chem. Chem. Phys., 2010, 12, 10189–10197 RSC.
- D. R. Nutt and J. C. Smith, J. Am. Chem. Soc., 2008, 130, 13066–13073 CrossRef CAS PubMed.
- A. Kuffel, D. Czapiewski and J. Zielkiewicz, J. Chem. Phys., 2014, 141, 055103 CrossRef PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, M. Crowley, R. C. Walker, W. Zhang, K. M. Merz, B. Wang, S. Hayik, A. Roitberg, G. Seabra, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, L. Yang, C. Tan, J. Mongan, V. Hornak, G. Cui, D. H. Mathews, M. G. Seetin, C. Sagui, V. Babin and P. A. Kollman, AMBER 10, University of California, San Francisco, 2008 Search PubMed.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. M. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. M. Wang and P. Kollman, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- A. Kuffel and J. Zielkiewicz, J. Phys. Chem. B, 2008, 112, 15503–15512 CrossRef CAS PubMed.
- D. Chandler, Introduction to Modern Statistical Mechanics, Oxford University Press, 1987 Search PubMed.
- A. Kuffel, D. Czapiewski and J. Zielkiewicz, J. Chem. Phys., 2015, 143, 135102 CrossRef PubMed.
- X. Chen, I. Weber and R. W. Harrison, J. Phys. Chem. B, 2008, 112, 12073–12080 CrossRef CAS PubMed.
- Y. Xu, R. Gnanasekaran and D. M. Leitner, J. At., Mol., Opt. Phys., 2012, 125071 Search PubMed.
- F. Sittel, A. Jain and G. Stock, J. Chem. Phys., 2014, 141, 014111 CrossRef PubMed.
- M. B. Priestley, Spectral analysis and time series, Academic Press, 1981, ch. 9, vol. 2 Search PubMed.
- S.-T. Lin, M. Blanco and W. A. Goddard, J. Chem. Phys., 2003, 119, 11792–11805 CrossRef CAS.
- M. Heyden and D. J. Tobias, Phys. Rev. Lett., 2013, 111, 218101 CrossRef PubMed.
- E. C. Dykeman and O. F. Sankey, J. Phys.: Condens. Matter, 2010, 22, 423202 CrossRef PubMed.
- Z. Yang, B. Shi, H. Lu, P. Xiu and R. Zhou, J. Phys. Chem. B, 2011, 115, 11137–11144 CrossRef CAS PubMed.
- A. Kitao, S. Hayward and N. Go, Proteins, 1998, 33, 496–517 CrossRef CAS.
- A. Amadei, A. B. Linssen and H. J. Berendsen, Proteins, 1993, 17, 412–425 CrossRef CAS PubMed.
- A. Kitao and N. Go, Curr. Opin. Struct. Biol., 1999, 9, 164–169 CrossRef CAS PubMed.
- I. J. Finkelstein, A. M. Massari and M. D. Fayer, Biophys. J., 2007, 92, 3652–3662 CrossRef CAS PubMed.
- A. Kuffel and J. Zielkiewicz, Phys. Chem. Chem. Phys., 2012, 14, 5561–5569 RSC.
- J. M. Zanotti, G. Gibrat and M. C. Bellissent-Funel, Phys. Chem. Chem. Phys., 2008, 10, 4865–4870 RSC.
- R. C. Tolman, Phys. Rev., 1918, 11, 261–275 CrossRef CAS.
- S. H. Crandall and W. D. Mark, Random vibration in mechanical systems, Academic Press, New York, London, 1963 Search PubMed.
- S. Kazemirad, H. K. Heris and L. Mongeau, J. Acoust. Soc. Am., 2013, 133, 3186–3197 CrossRef PubMed.
- F. Monroy, F. Ortega, R. G. Rubio and M. G. Verlade, Adv. Colloid Interface Sci., 2007, 134–135, 175–189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp07628e |
| This journal is © the Owner Societies 2016 |