
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon monoxide protonation in condensed phases and bonding to surface superacidic Brønsted centers†
Evgenii S.
Stoyanov
*ab and
Sergei E.
Malykhin
bc
aVorozhtsov Institute of Organic Chemistry, Siberian Branch of Russian Academy of Sciences (SB RAS), Novosibirsk 630090, Russia. E-mail: evgenii@nioch.nsc.ru
bDepartment of Natural Science, National Research University - Novosibirsk State University, Novosibirsk 630090, Russia
cBoreskov Institute of Catalysis SB RAS, Novosibirsk 630090, Russia
First published on 14th January 2016
Abstract
Using infrared (IR) spectroscopy and density functional theory (DFT) calculations, interaction of CO with the strongest known pure Brønsted carborane superacids, H(CHB11Hal11) (Hal = F, Cl), was studied. CO readily interacted at room temperature with H(CHB11F11) acid, forming a mixture of bulk salts of formyl and isoformyl cations, which were in equilibrium An−⋯H+CO  COH+⋯An−. The bonding of CO to the surface Brønsted centers of the weaker acid, H(CHB11Cl11), resulted in breaking of the bridged H-bonds of the acid polymers without proton transfer (PT) to CO. The binding occurred via the C atom (blue shift ΔνCO up to +155–167 cm−1, without PT) or via O atom (red shift ΔνCO up to −110 cm−1, without PT) always simultaneously, regardless of whether H+ is transferred to CO. IR spectra of all species were interpreted by B3LYP/cc-pVQZ calculations of the simple models, which adequately mimic the ability of carborane acids to form L⋯H+CO, LH+⋯CO, COH+⋯L, and CO⋯H+L compounds (L = bases). The CO bond in all compounds was triple. Acidic strength of the Brønsted centers of commonly used acid catalysts, even so-called superacidic catalysts, is not sufficient for the formation of the compounds studied.
COH+⋯An−. The bonding of CO to the surface Brønsted centers of the weaker acid, H(CHB11Cl11), resulted in breaking of the bridged H-bonds of the acid polymers without proton transfer (PT) to CO. The binding occurred via the C atom (blue shift ΔνCO up to +155–167 cm−1, without PT) or via O atom (red shift ΔνCO up to −110 cm−1, without PT) always simultaneously, regardless of whether H+ is transferred to CO. IR spectra of all species were interpreted by B3LYP/cc-pVQZ calculations of the simple models, which adequately mimic the ability of carborane acids to form L⋯H+CO, LH+⋯CO, COH+⋯L, and CO⋯H+L compounds (L = bases). The CO bond in all compounds was triple. Acidic strength of the Brønsted centers of commonly used acid catalysts, even so-called superacidic catalysts, is not sufficient for the formation of the compounds studied.
Introduction
The formyl cation is an important intermediate in the chemistry of carbon monoxide in an acidic environment.1,2 HCO+ is recognized as an abundant species in interstellar molecular clouds3,4 and has been studied extensively by spectroscopic methods in the gas phase and interstellar space.5 It can be easily generated in the gas phase by a variety of methods.6 Gaseous HCO+ shows superacidic properties: it is solvated with such weak bases as H2,7 He, Ne and Ar.8–10 Solvation decreases the C–H and C–O stretches of the cation. The higher the proton affinity of the rare gas, the greater this decrease is. For Ar⋯HCO+, the red shift of νCO is significant: 48 cm−1 relative to free HCO+.9 The calculated (QCISD(T)/6-311+G(3df,2p) level) energy difference between the formyl and isoformyl cations is large (∼163 kJ mol−1),11 suggesting that the formation of COH+ is unlikely.12 COH+, however, has been detected in interstellar space,13–15 where it is 300-fold less abundant than HCO+, and in laboratories on the ground,16,17 in a 6% mixture with formyl cation.17Direct observation of both HCO+ and COH+ in the condensed phase has been elusive. Attempts to synthesize the formyl cation via direct protonation of CO in liquid superacids based on SbF5 failed: CO remains unprotonated.18,19 De Rege et al.20 first reported spectroscopic observation of the HCO+ formation in the liquid HF-SbF5 superacid under CO pressure of 28–85 atm. They provided some plausible 13C nuclear magnetic resonance (NMR) evidence of the HCO+ existence. Nevertheless, Raugei and Klein12 criticized their empirical findings and proposed another explanation, which excluded the formation of stable HCO+. One of the arguments against the HCO+ formation is significant red shifting of the observed νCO (2110 cm−1) in comparison with that of gaseous CO (by −31 cm−1); this phenomenon requires an explanation. No evidence has been found for the presence of COH+ in solutions of CO in liquid HF-SbF5.12,20
Exploring the characteristics of HCO+ is important for understanding the nature of CO bonding with Brønsted acids because CO is widely used as a test molecule in studies on the acidity strength of Lewis and Brønsted acidic centers of oxide surfaces and acidic catalysts.21–23 The CO bonding via the C atom to Lewis centers as a σ-donor (without a π back donation contribution) increases the CO stretch vibration with respect to the gaseous νCO (blue shift, ΔνCO) by +50 to +100 cm−1.23 ΔνCO is the function of the cation charge density. The greater it is, the greater is the ΔνCO shift.24 One can expect that the CO bonding to H+, which has the highest charge density, will result in the greatest ΔνCO shift. Nonetheless, this does not occur: for the CO bonding even to the superacidic Brønsted centers (sulfate-doped ZrO2 systems), ΔνCO does not exceed +10 cm−1.23 It is surprising that joining with the less basic hydrated Brønsted centers of these systems increases ΔνCO more than twofold (+24 cm−1).23 Even for free HCO+ (2184 cm−1),25 ΔνCO is only +43 cm−1. These peculiarities have not been explained so far.
Thus, the available empirical data indicate that our understanding of the CO protonation or CO bonding with H+ in Brønsted acid centers is incomplete.
In the present work, using infrared (IR) spectroscopy and quantum-chemical methods, we studied how CO interacts with the strongest known solid carborane superacids, H(CHB11Cl11) and H(CHB11F11), and what compounds are formed when the proton is transferred or not transferred to the CO molecule.
Experimental
The carborane acids, H(CHB11Cl11) and H(CHB11F11), (hereafter abbreviated as H{Cl11} and H{F11}, respectively, or H{Hal11} for both) were prepared as described previously.26,27 IR spectroscopic analysis of interaction of CO with the carborane acids was performed as follows. In a specially designed IR cell-reactor, the carborane acids were sublimed at 150–160 °C under pressure 10−5 Torr on cold Si windows as a very thin translucent layer.28 The spectrum of the sublimed acid showed no traces of the H3O+ cation.27,29 Dry gaseous CO was injected anaerobically into the IR cell. Its partial pressure was measured by the intensity of νCO relative to the standard CO spectrum recorded in the same cell filled with 100% CO at atmospheric pressure.The CO interaction with the acid occurred in the IR cell-reactor at room temperature while we recorded the IR spectra at certain time intervals. Weighable quantities of the HCO+{F11−} salt were obtained by aging a portion of H{F11} for 1–3 days in a Schleng tube filled with CO.
All procedures were performed in a Vacuum Atmospheres Corp. glovebox in the atmosphere of N2 (O2 and H2O < 0.5 ppm). The IR spectra were recorded on an ABB MB3000 spectrometer inside a dry box in either transmission or attenuated total reflectance (ATR) mode (525–4000 cm−1). The IR data were processed in the GRAMMS/A1 (7.00) software from Thermo Fisher Scientific.
The calculation procedure
A number of solvated (CO)H+ cations protonated via the C or O atoms were analyzed by quantum-chemical methods. The main objects of this study were the C–O and C–H (or O–H) vibrations and the response of these vibrations when anions or molecules with different proton affinities were binding to the cations. To address the anharmonic nature of the molecular vibrations, an empirical scaling factor was used:30 the ratio of the empirical CO stretch frequency to the calculated harmonic frequency equal to 0.97.Density functional theory (DFT) was applied to the search for optimal geometry and to subsequent vibrational analysis. The method was B3LYP31,32 DFT functional, with correlation-consistent polarized valence quadruple-zeta basis set (cc-pVQZ).33 The GAMESS US quantum-chemical software was used for these tasks.34 Visualization of the results was performed in the MOLDEN software.35
Results
The CO interaction with a very thin (almost transparent) film of the acids covering the Si windows after sublimation, differs from the interaction with powdered acids precipitated from liquid HCl after completion of their synthesis. This difference can be explained as follows: IR spectra of the two samples show significant differences in the frequencies of acidic protons and anions (Fig. S1 and S2, ESI†). In the case of the H{Cl11} acid, the spectrum of the powdered sample coincided with that of the crystalline acid, whose H-bond network forms linear polymers with bridged protons.36 The spectrum of the sample of the thin film of H{Cl11} showed increased frequencies of bridged protons. The same was true for the spectra of the samples of H{F11} acid. This means that the H-bond network structure of the thin-film samples is disordered with bridged H-bonds that are more asymmetrical (than is the case for the crystalline sample) and therefore has greater acidic strength. In the text below, we will refer to the thin-film acidic samples obtained by sublimation as “amorphous” and to the powdered acid precipitated from a solution of liquid HCl as “crystalline.”CO interaction with the amorphous H{Cl11} acid
It was studied at partial CO pressure of 0.4 atm. IR spectra were recorded at certain intervals, and after 2 h 30 min, the reaction was stopped by pumping gaseous CO out. IR spectra showed some νCO bands (Fig. 1, blue). Such a band at 2144 cm−1 actually coincides with νCO of gaseous CO and may belong to physically absorbed CO that was not removed by vacuum treatment. Two bands, at 2298 and 2260 cm−1, were significantly blue shifted (>100 cm−1), suggesting that they belong to CO attached to H atoms of the H{Cl11} acid via the C atom. We will denote the resulting compounds OC·H{Cl11} as “type I.” The conjugated bands of X–H stretches (where X is a basic atom) were observed at 2951 and 2872 cm−1 (Fig. 1, inset). The last weak band of the CO stretch at 2133 cm−1 was red shifted by −10 cm−1. This means that it belongs to another type of compounds, which we denoted as “type II.” Later, we will describe the more detailed spectra of this compound.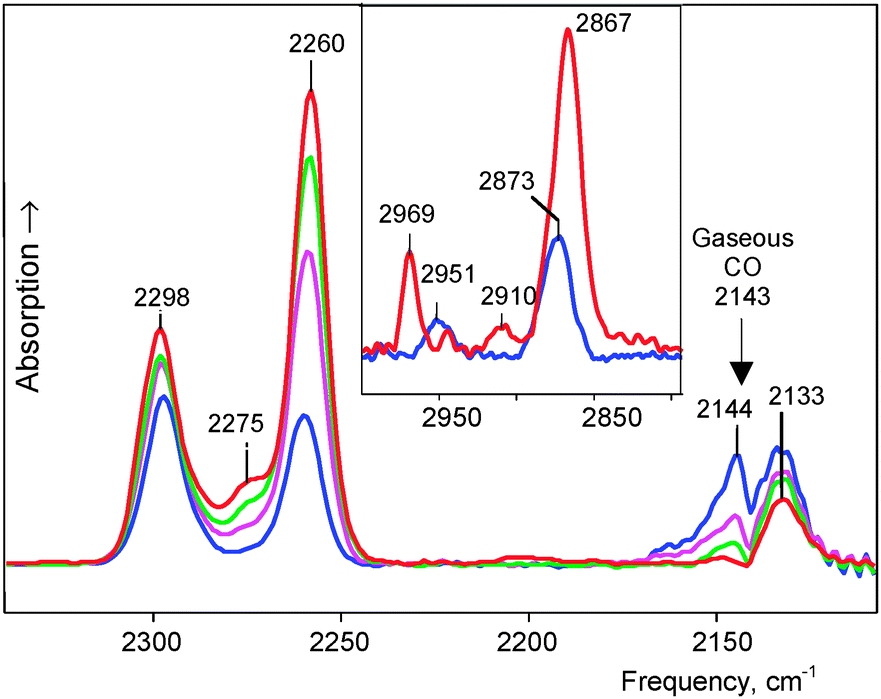 | ||
| Fig. 1 IR spectra of the surface compounds formed during the adsorption of CO by amorphous H{Cl11}. The blue spectrum was recorded within 2 h 30 min of the input of CO and subsequent evacuation. Then, the sample was kept in a vacuum, and spectra were recorded after 1 (violet), 2 (green), and 5 days (red). Absorption corresponding to unreacted H{Cl11} was subtracted. Inset shows the frequency region of X-H stretches. | ||
This sample was kept in vacuum, and IR spectra were recorded after 1, 2, and 5 days (Fig. 1). The spectra showed that the intensity of the νCO bands at 2298 and 2260 cm−1 (and conjugated νCH bands) continued to increase, while the intensity of the band at 2144 cm−1, which corresponds to physically adsorbed CO, decreased and eventually disappeared (Fig. 1 red). Therefore, the kinetics of the formation of compounds I from the surface-absorbed CO was slow:
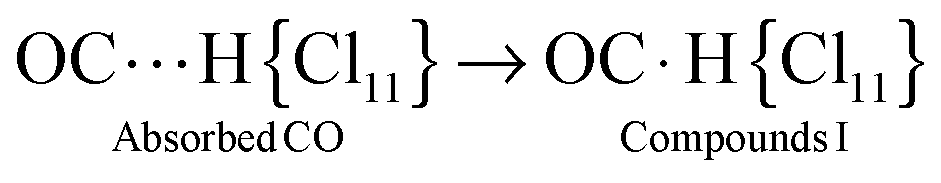
The IR spectrum of the sample with fully exhausted adsorbed CO, shown in Fig. 1 (red), was obtained by subtracting the spectrum of the unreacted acid via multiplication by the adjustment factor f = 0.983. This means that ∼2% of the acid was consumed with CO, and the formed compounds I were mostly the surface compounds.
This experiment was repeated at twofold higher partial pressure of CO (0.8 atm). After 24 h, the experiment was stopped by pumping CO out. The last spectrum of the formed products was obtained by subtracting absorption from the unreacted H{Cl11} acid via multiplication by the adjustment factor f = 0.907 (Fig. 2). That is, approximately 9–10% of the acid reacted with CO, and the resulting products still could be regarded as mostly superficial. The spectrum signals were much stronger and showed more bands from CO vibrations. A pair of known νXH and νCO bands from type I compounds was accompanied by a third one at 2911 and 2275 cm−1, respectively. Thus, three subtypes of the OC·H{Cl11} compounds were formed: Ia, Ib and Ic (Fig. 2). As compared to the first experiment (Fig. 1), the intensity and frequencies of these bands were slightly changed indicating that compounds Ia–Ic are sensitive to the nearest surrounding. The low-frequency band at 1321 cm−1 (Fig. 2) can be attributed to the bend X–H–C(O) vibrations of these compounds because its intensity increased proportionally with the sum of intensity levels of CO stretch absorption phenomena of Ia–Ic.
 | ||
| Fig. 2 An IR spectrum of the surface compounds formed during the adsorption of CO at 0.8 atm by amorphous H{Cl11} for 24 h. Absorption by unreacted H{Cl11} was subtracted. The bands marked with an asterisk belong to the {Cl11−} anion. The insets show some of the frequency ranges in more detail. | ||
IR spectra also showed a band at 2133 cm−1 of a type II compound and three weak but definitively identified νCO bands at 2096–2034 cm−1 (Fig. 2, right inset). Their significant red shifting relative to the 2143 cm−1 band of gaseous CO suggested that they may be CO molecules attached to the H{Cl11} via the O atom. Hereafter, we will denote them as OIa, OIb and OIc.
CO did not interact with the powder of crystalline H{Cl11} acid even during several days of storage in a sealed flask.
CO interaction with the crystalline H{F11} acid
The powder of H{F11} was kept for two days in the sealed flask filled with CO, and then the ATR IR spectrum of the solid was recorded. It showed three weak νCO bands at 2311–2260 cm−1 from the Ia–Ic compounds and strong absorption in the frequency region of CO stretches (at 2152–2134 cm−1), which belong to the compounds denoted above as type II (Fig. 3). The broad strong band of H+ vibration at 2920 cm−1 is obviously conjugated with the strong band at 2152 cm−1. They both belong to one basic compound IIa. The weak νCO band at 2133 cm-1 points to the existence of the second compound IIb, whose conjugated stretch vibration from the H atom overlapped with the strong absorption from IIa. | ||
| Fig. 3 An IR spectrum of the products formed in the reaction of CO with crystalline powder H{F11}. Absorption by the unreacted H{F11} acid was subtracted. | ||
The IR spectrum also showed absorption of the parent H{F11} acid, whose intensity was 27% of that of the starting acid. Therefore, the formed salts represented the bulk product.
CO interaction with the amorphous H{F11} acid
This interaction proceeded much more rapidly than in the case of powdered H{F11}. At partial pressure of 0.53 atm, all the acid was fully utilized within ca. 2 h, and the reaction was complete.The spectrum of the formed products, HCO{F11}, showed three weak νCO bands of Ia−Ic compounds at 2309–2262 cm−1 (Fig. 4, inset). Their conjugated weak bands from the CH stretches were also identified (Table 1).
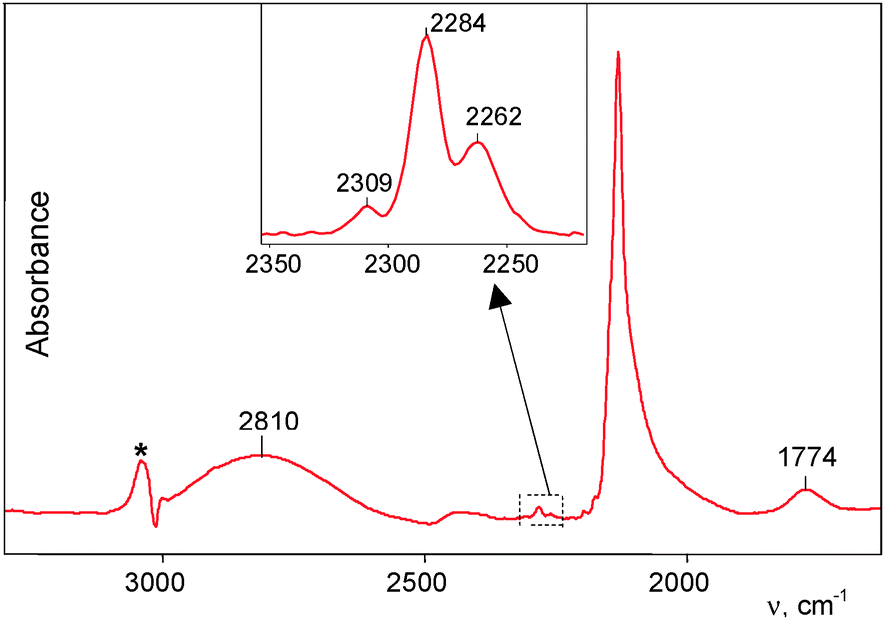 | ||
| Fig. 4 An IR spectrum of the products formed after complete interaction of CO with amorphous H{F11}, followed by removal of gaseous CO. Absorption of the {F11−} anion was subtracted by means of the spectrum of the Cs{F11} salt. The remnant from the νCH band of the {F11−} anion after subtraction is marked with an asterisk. | ||
| Compound | Anion | ν(Hal-H+) | νCO | δ(Hal-H-C) | Cation | Anion | ν asHCO | ν sHCO | δHCO |
|---|---|---|---|---|---|---|---|---|---|
| a Not determined. b In parentheses: ν(Hal-H+) variation for different samples. | |||||||||
| CO(gas) | 2143 | HCO+(gas) | 308941 | 218425,42 | 83037 | ||||
| Ar⋯ HCO+(gas) | 281510 | 21369 | |||||||
| Ia | {F11−} | 2970 | 2310 | IIa | {F11−} | 2920 | 2152 | 867 | |
| Ib | 2910 | 2284 | IIb | 2810 | 2133 | 867 | |||
| Ic | 2873 | 2260 | IIc | 2700 | 2117 | 867 | |||
| Ia | {Cl11−} | 2951 (2969)b | 2298 | 1321 | II | {Cl11−} | 2133 | ||
| Ib | 2910 | 2275 | 1321 | ||||||
| Ic | 2873 (2867)b | 2260 | 1321 | ||||||
The main features of the spectrum are three strong bands: broad complex νCH at 2810 cm−1, unsymmetrical νCO at 2133 cm−1 (Fig. 4), and low-frequency absorption at 867 cm−1 (Fig. S3 in the ESI†). The intensity of these three bands increased proportionally in the course of the reaction, thus confirming that they correspond to the compounds of the same type II with the characteristic νCO frequencies near 2133 cm−1.
The complex νCH band can be subdivided into three components ca. 2920, 2810, and 2700 cm−1 (Fig. 5a). Similarly, the asymmetrical νCO band can be separated into four components (Fig. 5b). One pair of the bands, νCH = 2920 cm−1 and νCO = 2150 cm−1, coincides with the bands of a IIa compound (Fig. 3). The second, mostly strong pair of signals νCH = 2810 cm−1 and νCO = 2133 cm−1, likely belongs to the basic compound IIb of this sample. We conventionally attributed the third pair, νCH = 2700 cm−1 and νCO = 2117 cm−1 to compound IIc. The fourth νCO at 2097 cm−1 and a low-frequency band at 1774 cm−1 (Fig. 4) belong, as we will prove below, to the isoformyl cation, COH+.
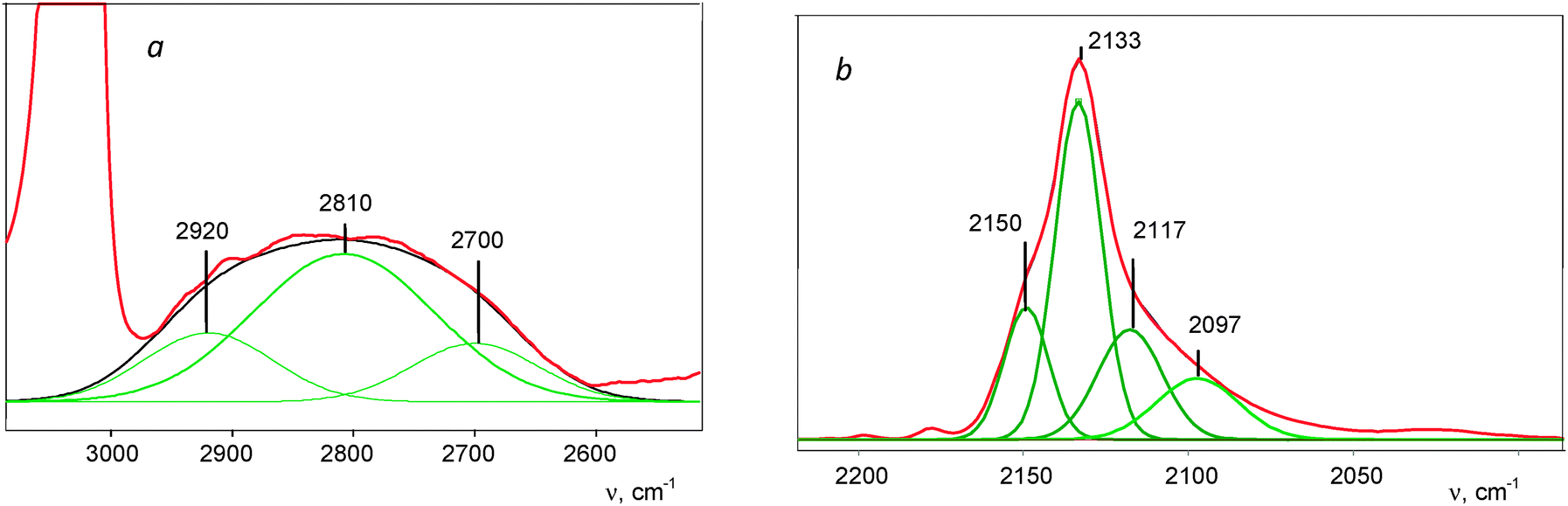 | ||
| Fig. 5 A deconvoluted IR spectrum of the formyl cations, shown in Fig. 4, in the regions of νasHCO (a) and νsHCO (b) frequencies. | ||
The low-frequency absorption at 867 cm−1 (Fig. S3, ESI†) is very close to the empirical bend vibration of the HCO+ cation in a vacuum.37 Its intensity showed linear dependence on the sum of intensity values of CH stretches of IIa–IIc compounds during their formation. Therefore, this effect can be attributed to the bend HCO vibrations of the HCO+ cations in all three compounds IIa–IIc.
Results of calculations
Carborane acids are quite large molecules for ab initio simulation of their vibrational spectra by such a reliable but very demanding method as coupled cluster theory.38 Nevertheless, they are tractable at the DFT level. We also performed calculations for their analogs, the simpler (CO)·H+L compounds, were L = He, Ne, Ar, H2O, or C2H2. A wide range of L basicity, which includes basicity of {Hal11−} anions, allows us to get broader and deeper insights into the features of CO bonding with superacidic molecules.The L⋯H+CO and LH+⋯CO compounds
The calculated frequencies for optimized structures of the formyl cation and its solvates, L⋯H+CO (L = He, Ne, or Ar), are shown in Table S1 (ESI†). The forms of their vibrational modes are shown in Fig. 6. One can see that the two CH and CO stretching vibrations of the bare H+CO ion are significantly mixed. Consequently, they can hardly be referred to as νCH and νCO. When a cation is solvated by molecule L the mixing of the and vibrations of L⋯H+CO increases to a greater extent with the higher basicity of L. A major contribution to the higher frequency νasHCO is made by the CH stretch and to the lower frequency νsHCO by the CO stretch. With the increasing basicity of L, both frequencies νasHCO and νsHCO decrease (Table S1, ESI†). | ||
| Fig. 6 The form and amplitudes of the normal vibrational modes for compounds with CO bonded to the proton via C-atom (a–c) or via O-atom (d–f) with and without proton transfer to CO. The length of the arrows indicates the amplitude of the atoms deviated from the equilibrium state for the normal vibrations. | ||
If L was the {F11−} ion, the ab initio simulation showed more a complex situation. The {F11−} anion has three sites of F atoms with different basicity: “a,” “b,” and “c” (Fig. 7). When CO was attached to the “a” site of H{F11}, the H+ was transferred to the C atom forming a salt of the H+CO cation. Its stretch frequencies are typical for L⋯H+CO type compounds (Table S1, ESI†). The CO attaching to the “b” and “c” sites of H{F11} caused the formation of compounds with a rather bridged proton (Fig. S4, ESI†), with the frequencies corresponding to bridged-proton oscillation (Table S1, ESI†).
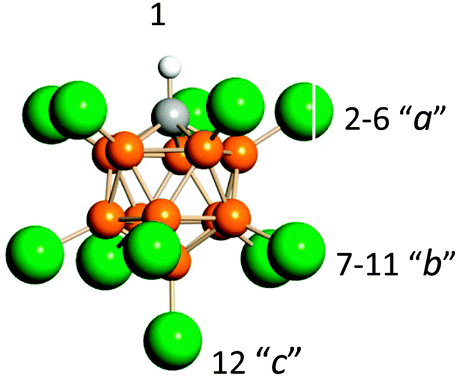 | ||
| Fig. 7 Icosahedral carborane anions, CHB11Hal11−, (Hal = F, Cl) with the numbering of three types of Hal atoms differing in basicity. | ||
With a further increase in L basicity (L = H2O, SO2), the proton is transferred to L, and stretch frequencies are sharply changed (Table S2, ESI†). The proton oscillations are now localized to the L–H+ bond and hardly affect oscillations of the CO bond. That is, νCO becomes highly characteristic (Fig. 6c).
Different forms of vibrational modes of the L⋯H+CO and LH+⋯CO species did not allow us to obtain information on the subtle differences in the nature of their CO bonds. To resolve this issue, the normal νasHCO and νsHCO modes of the H+CO cation are presented as a sum of contributions from their localized counterparts, “intrinsic” frequencies νiCH and νiCO39 (Table S1, ESI†). They yielded a single vibrational frequency for each internal coordinate and represent the force constant and bond length. Later, they will be used to trace the variation of CO bond strength (and thus its length), when basicity of L increased and the proton was transferred from H+CO to the ligand L.
The isoformyl cation and its solvates
The calculated IR spectra for the optimized structure of neat COH+ showed that its normal vibrations νOH = 3401 and νCO = 1965 cm−1 are quite characteristic (Fig. 6d).Solvation with Ar led to the transition of a proton to a somewhat bridged state (Table S3, ESI†) with stronger mixing of the CO and OH stretch vibrations (Fig. 6e). As a result, νasCOH (2100 cm−1) became the highest frequency with a predominant contribution from the stretch. The lower frequency νsCOH (1777 cm−1) is mainly determined by the contribution from the bridging-proton oscillation.
When solvated molecule L was more basic, such as H2O, the proton was transferred to L. The νCO frequency of the formed H3O+⋯OC becomes characteristic (Fig. 6f) with red shifting of −83 cm−1 compared to the frequency of free (Table S3, ESI†).
Discussion
The CO adsorption on the surface of H{Hal11} acids results in the formation of two major types of compounds, I and II, which vary greatly in IR spectra (Table 1). They belong to the {Hal11}H+CO family, with CO binding to the acidic center via the C atom; this binding energetically is much more favorable than binding via the O atom.40 Calculation of optimized structures of their analogs, LH+CO, shows that depending on the basicity of the L, two types of species can be formed: the L⋯H+CO with proton transfer to the CO molecule, and LH+⋯CO with the proton transfer to the base L. The CO stretches of LH+⋯CO correspond to those empirically determined for the type I compounds, and the stretch vibrations for L⋯H+CO match those empirically determined for type II compounds (Table 1 and Tables S1, S2, and S3, ESI†). Therefore, compounds I are the {Hal11−}H+⋯CO salts with proton transfer onto the anion, and compounds II are salts of the formyl cation, {Hal11−}⋯ H+CO.IR spectra also show the weak bands of minor products, which will be discussed below.
CO interaction with H+via the C atom
The H+ oscillations of {Hal11−}H+⋯CO characterize vibrations of the Cl–H+ or F–H+ bonds, which are not mixed with those of the CO bond. That is, both νHalH+ and νCO are highly characteristic. The dependence of νHalH+ on νCO shows two separate functions for each counterion (Fig. 8, red and green data points). This means that the three compounds Ia, Ib and Ic are formed by {F11−} anions, and the other three Ia, Ib and Ic are formed by {Cl11−} anions. In accordance with our calculations and empirical data,37 these phenomena are caused by features of undeca-halogen anions, CHB11Hal11−: the basicity of their Hal atoms at positions 2–6 (a), 7–11 (b), and 12 (c) (Fig. 7) slightly increases in the order of a, b, and c. | ||
| Fig. 8 Frequency dependences of νHalH on νCO for type I compounds, and frequency dependences of νasHCO on νsHCO for type II compounds. Empirical data points for H+CO, H2⋯H+CO, and Ar⋯H+CO were taken from other studies.7–9,10,25,41,42 Blue points show results of DFT calculations scaled by 0.97 to the experimental CO stretch of gaseous CO. | ||
This effect determines the formation of the three compounds Ia, Ib, and Ic, which can be schematically depicted as is shown on the Scheme 1.
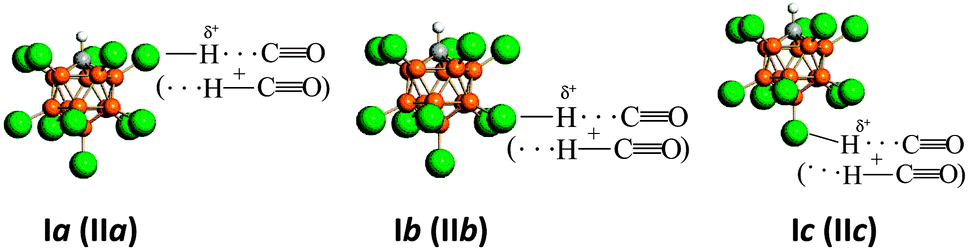 | ||
| Scheme 1 Schematic representation of compounds Ia, Ib, and Ic and compounds IIa, IIb, and IIc (in parentheses). | ||
The empirical valence vibrations of formyl cations in IIa–IIc and those of neat H+CO and its solvates L⋯H+CO (L = H2 and Ar) in the gas phase show concordant dependence (Fig. 8, black). This result proves that all these cations belong to one family, and that the influence of the environment on the formyl cations is insignificant.
The reason for the existence of three compounds, IIa, IIb and IIc, with the identical {F11−} anion obviously is the same as the reason for the compounds Ia –Ic and is shown in Scheme 1.
CO interaction with H+via the O atom
IR spectra of the surface (CO)·H{Cl11} compounds show weak νCO bands at 2096, 2072, and 2034 cm−1 with a red shift ΔνCO of 47–110 cm−1 relative to gaseous CO (Fig. 2, right inset). These frequencies may belong to CO molecules, which are bonded to the three sites “a”, “b” and “c” of the H{Cl11} acid via the O atom, thus forming compounds denoted as “OIa”, “OIb”, and “OIc”. Because they are formed simultaneously with Ia, Ib, and Ic, all these compounds can be in equilibrium. The calculated frequencies of the optimized structure of CO⋯HOH2+, which is an analog of OIa–OIc compounds, show that their CO stretches are also highly characteristic, as was determined for the Ia–Ic compounds. In this case, the correlation between νCO of OIa–OIc and corresponding Ia–Ic compounds should be observed. Such a correlation is indeed present (Fig. S5, ESI†), which proves the existence of equilibria Ia ⇌ OIa, Ib ⇌OIb, and Ic ⇌OIc. As an example, the equilibrium Ia ⇌OIa is shown in Scheme 2.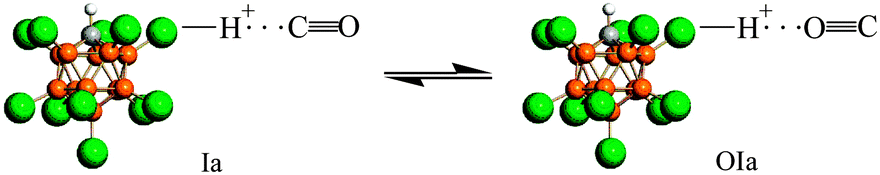 | ||
Scheme 2 Representation of the equation Ia ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) OIa. OIa. | ||
The weak νClH bands from OIa–OIc compounds cannot be reliably identified because of the overlap with strong νClH absorption from compounds Ia–Ic.
The calculated spectrum of the naked isoformyl cation shows that both its CO and CH stretches interact only slightly and are mostly characteristic (Fig. 6d). Solvation with an Ar atom converts the cation to a rather asymmetric disolvate CO–H+⋯Ar with specific νasCOH and νsCOH frequencies (Table S3, ESI†) because of mixing of the CO stretch with bridging-proton oscillation (Fig. 6e). The major contribution to the higher frequency νasCOH (2198 cm−1) is now caused by the CO stretch, and bridging-proton oscillation makes a major contribution to the lowering of frequency νsCOH, which decreases to 1758 cm−1. Both frequencies have their counterparts in the empirical IR spectrum of the OIIb compound: 2097 cm−1 (Fig. 5b) and 1774 cm−1 (Fig. 4), respectively. Thus, the bridged type of cation OIIb coexists with the cation IIb:

In the IR spectrum of the type II compounds with dominant IIa, the νsCOH band is split into two components (Fig. 3); this finding implies that IIa is in equilibrium with OIIa.
CO protonation in the liquid superacids
Interpretation of the IR spectra of the products formed during the reaction of CO with the solid superacids gives us a key to the interpretation of the spectrum of the CO solution in liquid “magic” superacid SbF5 + HF (comparable in strength with H{F11}). This spectrum, published elsewhere,20 remains unexplained.It shows two bands, a broad one at 2110 cm−1 and a sharp one at 1671 cm−1 (Fig. 2 of ref. 20). When 12CO was replaced with 13CO, the broad band was red-shifted by ca. 30 cm-1 that allowed attributing it to valence HCO+ vibrations. The red shift of the sharp band was much smaller. It is noteworthy that the broad band clearly consists of two Gaussian components, at 2110 and ca. 2065 cm−1, to which the authors20 did not pay attention. From the results of the present work, it follows that the sharp band at 1671 cm−1 corresponds to the vibration of the bridged proton in the solvated isoformyl cation, CO–H+–An−. Contribution of the CO stretch to this vibration is low, and the isotopic 12C/13C red shift is small. It seems to be reasonable to attribute the conjugated frequency of this vibration, νasCOH, to the band at 2065 cm−1, just as with CO−H+⋯{F11−}. Because the isoformyl cation can exist only in equilibrium with the formyl cation, the second band, at 2110 cm−1, can be attributed to νsHCO of the formyl cation.
Because the contribution of the CO stretch to both vibrations –νasCOH (isoformyl cation) and νsHCO (formyl cation)–is significant, their isotopic 12C/13C red shifts are large and comparable. The conjugated νasHCO frequency of the formyl cation is expected at ca. 2660 cm−1, according to extrapolation of the dependence of νasHCO on νsHCO (Fig. 8) to the value νsHCO = 2110 cm−1. The authors of ref.12 could not detect the νasHCO band because of its broadening as well as overall weakness of signals in the spectrum of the compounds under study.
The valence vibrations of the formyl cation in solution of the SbF5 + HF acid have a lower frequency than do the valence vibrations of compounds IIa–IIc. This result means that the SbF5 + HF acid is weaker than H{F11}, and according to the equilibrium I ↔ II, the concentration of the type I compound is increased. Compound I should be unstable in this solution and should easily decompose:
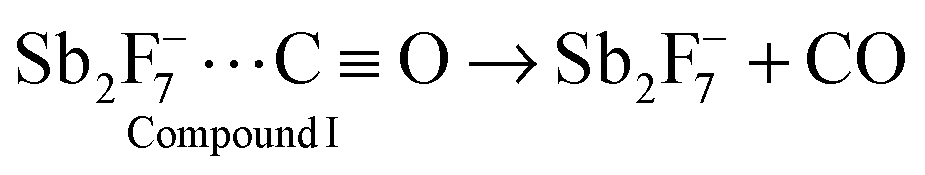
The nature of the CH and CO bonds
The length of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O bond of carbon monoxide is 1.128 Å,43 which is consistent with a triple bond.
O bond of carbon monoxide is 1.128 Å,43 which is consistent with a triple bond.
When CO is attached to the H{Hal11} acid via the C atom without the proton transfer to CO, the Ia–Ic compounds are formed, which have the highly ionic {Hal11}H⋯CO bond, whose oscillation is not mixed with that of the CO bond. Analogs of the Ia–Ic compounds are the H2OH+⋯CO and SO2H+⋯CO ions and Lewis complexes with only the σ-Metal– bond.24,44 The CO stretching frequencies of all these compounds are red shifted as compared to free CO; this phenomenon may be explained in terms of Cation(σ*) ← CO(σ) donation, that is, the e−-donation from the 7σ highest occupied molecular orbital (HOMO) of CO to the “free” σ-orbital of the cation.45 (According to ref. 45 the 7σ HOMO of CO is not antibonding as it is often stated. The increased νCO in H+CO is more likely caused by the effect of the charge on polarization of the bonding orbitals.) Therefore, the Ia–Ic compounds may be associated Lewis-like compounds. The greater the charge density on the cation, the stronger is its interaction with CO, the higher the CO stretch frequency, the shorter the RCO distance. Hence, the strength of the triple CO bond increases in the order Ic, Ib, and Ia reaching a maximum value of νCO = 2310 cm−1 for Ia with the counterion {F11−} (Table 1).
One would expect that the H+ transfer to CO with further solvation with L would increase the strength of the CO bond and its stretch vibration. Nonetheless, the mixing of CH and CO oscillations in L⋯H+CO does not allow for tracing of the changes in the CO bonding strength to changes in the basicity of L. This problem can be overcome if we use calculated “intrinsic” frequencies νiCO and νiCH, which correlate with RCO/CH bond length. With the decreasing basicity of L, νiCH increased (and RCH decreased) significantly, whereas νiCO (and RCO) varied insignificantly (Table S1, ESI†). Thus, the basicity of L in compounds II affects mainly the C–H bond and has almost no effect on the CO bond.
It is a valid experiment to compare the νCO frequencies of type I compounds with “intrinsic” νiCO frequencies of type II compounds scaled by 0.97. For compounds {F11−}H+⋯CO (I), the greatest value of νCO is 2310 cm−1; for Ar⋯H+CO (the closest analogue of {F11−}⋯H+CO [II]), the scaled νiCO is 2319 cm−1. That is, {F11−}H+⋯CO (I) and {F11−}⋯H+CO(II) do not differ greatly in CO strength.
The empirical νCH frequencies of L⋯H+CO cations show linear dependence on proton affinity (PA) of L (He, Ne, or Ar).9 This effect allowed us to evaluate “effective PA” of the {F11−} anion in compounds IIa−IIc according to their νCH. Fig. 9 shows that they are 295 (IIa), 382 (IIb), and 451 kJ mol−1 (IIc). These results allow us to say that basicity of the three sites of the {F11−} anion (“a,” “b,” and “c” in {F11−}⋯H+CO) is close to that of the Ar atom.
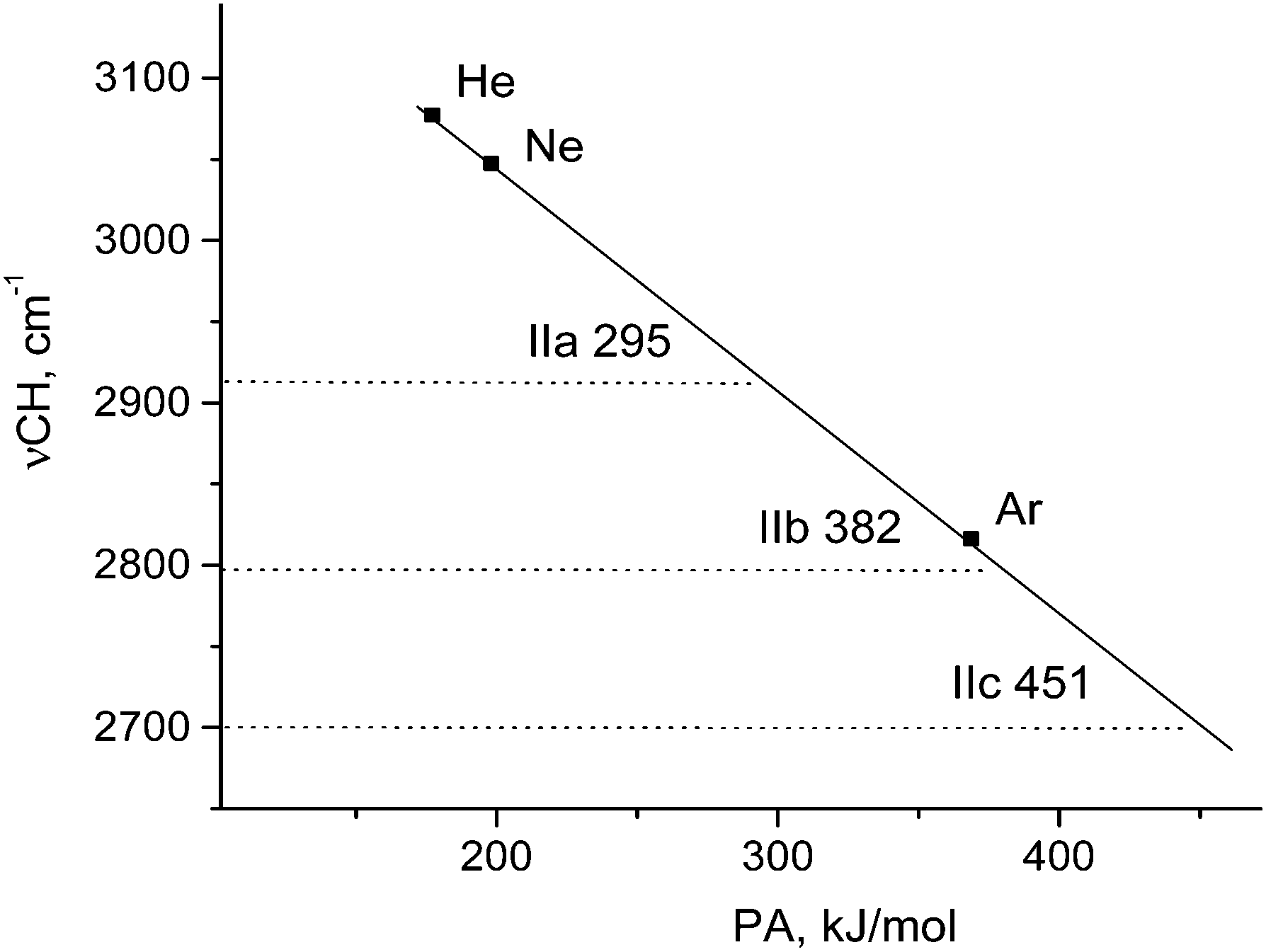 | ||
| Fig. 9 Dependence of the empirical νCH of L⋯H+–CO cations on proton affinity (PA) of L. νCH of compounds IIa–IIc allowed us to evaluate the “effective PA” of sites “a”, “b,” and “c” of the {F11−} anion (presented numerically). | ||
The optimized structure of CO–H+ for vacuum corresponds to a linear cation with the C–O–H angle of 180°.45 It means that the CO bond retains its triple character. The joining of H+ with the O atom of CO obviously takes place through interaction with the nonbonding e− pairs of the O atom. When H+ was transferred to {Hal11−} (OIa–OIc compounds), the O⋯H bond became highly ionic, and its oscillation was not mixed with that of the CO bond (Fig. 6f). The optimized structure of their analog, CO⋯HOH2+, preserves linearity (C–O–H angle is 177.5°) and properties of O⋯H and CO bonds. With the strengthening of the O⋯H bond (in the order OIc, OIb, and OIa), νCO decreases, but even for OIA, νCO = 2034 cm−1 is still much higher than that of the double-bonded C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of aldehydes and ketones (1740–1700 cm−1).
O stretch of aldehydes and ketones (1740–1700 cm−1).
Formyl cation–isoformyl cation rearrangement
From both experimental and theoretical studies, it follows that the H+CO cation is much less stable than the COH+ cation, with the energy difference being ∼160 kJ mol−1,11,40 and the significant barrier separating the two isomers: ∼150 kJ mol−1.40 Nonetheless, solvation of the cations with L molecules, whose PA lies between the PAs of CO at the O atom (427 kJ mol−1) and at the C atom (594 kJ mol−1) reduces the barrier so much that the H+ migration takes place without an overall barrier.40 This is the case for the {F11−}⋯H+CO and COH+⋯{F11−} compounds: “effective PA” of {F11−} is ca. 300–450 kJ mol−1. Their mixture readily interacts with gaseous CH3Cl with HCl elimination. In the course of the reaction, the IR absorption of both cations, CO+H and H+CO, which significantly differ in acidity, decreases proportionally (Fig. S6, ESI†). This observation confirmed that they are in rapid equilibrium:| {F11−}⋯H+CO ⇌ COH+⋯{F11−} | (1) |
As for the I ⇌ II and OI ⇌ OII transitions, according to the calculations, the COH+ cation may exist as a distinct entity only in vacuum or when solvated with He or Ne. With Ar solvation, it forms a rather asymmetric proton disolvate CO–H+–Ar (Table S3, ESI†), which is smoothly converted to an OI type compound with further increasing basicity of L. This finding is consistent with experimentally observed OI ⇌ OII transition via bridged disolvates. The same is predicted for the II → I transition: it should proceed via intermediate bridged proton species. For example, the calculated “a” isomer of HCO·{F11} is of type IIa, whereas “b” and “c” are species with a bridged proton (Fig. S4, ESI†). Nonetheless, our experiments show that all three isomers are type II, and the II → I transition occurs abruptly. In any case, formation of the bridged proton OC-H+-L species is not detected.
CO interaction with the surface Brønsted centers
Superacids in the solid phase are polymeric and contain bridged H atoms that reduce their acidic strength significantly. Even in the monomeric molecules of H{Cl11} in the gas phase at 180 °C, the H+ atoms are intramolecularly H-bonded.36When CO is adsorbed on the Brønsted centers of the H{Cl11} acid with preserved crystallinity, its attachment is weak, close to physical adsorption:
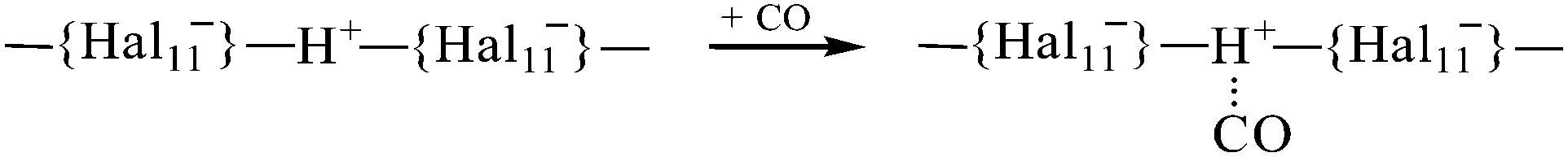
The basicity of CO is not sufficient to break up the bridged H-bond, and the adsorption stops at the stage of physical absorption. The H atoms of the amorphous H{Cl11} formed bridged H-bonds that are more asymmetrical. This effect increases acidic strength of the Brønsted centers so that basicity of CO appears to be sufficient to break up the bridge, with subsequent formation of the surface Lewis-like compounds: Ia–Ic and OIa–OIc. Velocity of their formation decreases with time and reaches a plateau as the surface layer is filling (Fig. S7, ESI†).
In the case of the strongest acid (H{F11}), the CO molecules easily break the bridged H-bonds. The proton is transferred to CO, and the bulk salts of formyl and isoformyl cations are formed.
Currently, in widely used acid catalysts, even in so-called superacidic catalysts (such as sulfate-doped ZrO2), acid strength of the Brønsted centers is much lower than that of H{Cl11}. Therefore, the formation of Lewis-like compounds Ia–Ic and OIa–OIc, and especially the formyl cations, cannot occur. The CO adsorption is stopped at the stage of physical adsorption with a blue shift ΔνCO ∼10 cm−1. Attachment of a water molecule to the Brønsted center leads to the breakage of the bridged H-bond and to the formation of the asymmetric H3O+ cation. The acidity strength of such Brønsted centers increases, and ΔνCO of the attached CO molecules increases more than twofold (+24 cm−1).23
Conclusions
The binding of CO to superacidic Brønsted centers with the bridged H-atoms can occur via three steps:(1) Physical adsorption. CO is a weak base, and the strength of its binding to the acidic bridged H-atom may not be sufficient to break the bridge. This type of adsorption occurs during the use of all modern acidic and superacidic catalysts.
(2) Adsorption with the breakage of the H-bridge and binding of CO to the H atom without the proton transfer to CO. In this case, the Lewis-like compounds are formed, OC⋯H{Hal11} and CO⋯H{Hal11}. Their blue shift Δν(CO) (up to +167 cm−1) or red shift Δν(CO) (up to −110 cm−1), respectively, reaches the limit values for Lewis compounds (in the absence of a π back donation contribution) because the charge density on H+ is maximal for cations. This type of adsorption occurs on the surface of the strongest solid superacids, H{Cl11} and H{F11}, not currently used in chemical practice.
(3) Chemisorption of CO with the proton transfer to the CO can take place when PA of the Brønsted acidic centers drops to the values of PA of the noble gases, krypton and argon, or falls even lower. This condition is satisfied only by the solid H{F11} acid. The formyl and isoformyl cations can also be formed, under certain conditions (high CO pressure), in solutions of the liquid SbF5 + HF superacid.
Solvation of formyl and isoformyl cations with the nearest environment in condensed phases decreases the difference in their energies and the energy barrier separating them, so that the equilibrium L⋯H+CO ⇌ COH+⋯L acquires fast dynamics, with a significant detectable fraction of the COH+⋯L compound. The same is true for the equilibrium LH+⋯CO ⇌ CO⋯H+L, where L is a neutral molecule or anion. Acidic properties of the mixture of +OC–H and H–OC+ are determined by the more acidic isoformyl cation.
Spectroscopic properties of protonated CO confirmed that the triple character of the CO bond does not change when CO interacts with H+. Binding of H+ to the C atom of CO without proton transfer strengthens and shortens the CO bond, which reaches the limit value of ca. 1.110 Å. The H+ transfer to the C atom has a weak additional impact on the CO bond; this finding proves that the H+ influence is caused by the effect of its charge on the polarization of the CO bonding orbitals. When CO interacts with H+via the O atom without the proton transfer, the CO bond is weakened and elongated. The subsequent proton transfer to the O atom results in further weakening and elongation of the CO bond (up to ca. 1.153 Å in COH+) confirming that the O atom is an e− donor from non-bonding and bonding orbitals. Furthermore, the CO bond preserves its triple nature.
Interpretation of the IR spectra of protonated CO entities allowed us to explain such a finding as the decrease in the lowest stretch vibration of the solvated OC–H+⋯Ar cation below νCO of gaseous CO. In addition, this analysis made it possible to interpret the published spectrum of a CO solution in liquid SbF5 + HF20 and to prove that together with the OC − H+ cation, a significant portion of H+−OC is formed, whose existence in the condensed phase was not recognized previously.12,20
Acknowledgements
This work was supported by the Russian Foundation for Basic Research Grant 16-03-00357 and the Ministry of Education and Science of the Russian Federation within the Project of the joint Laboratories of the Siberian Branch of the Russian Academy of Sciences and National Research Universities. Calculations were performed at the Siberian Supercomputer Centre SB RAS. The authors thank Irina S. Stoyanova for providing the carborane acids and technical support.References
- G. A. Olah, G. K. Prakash and J. Sommer, Superacids, John Wiley and Sons, New York, 1985 Search PubMed.
- G. A. Olah, Angew. Chem., Int. Ed., 1993, 32, 767 CrossRef.
- D. Buhl and L. E. Snyder, Nature, 1970, 227, 267 CrossRef PubMed.
- W. Klemperer, Nature, 1970, 227, 1230 CrossRef CAS.
- C. F. Neese, P. S. Kreynin and T. Oka, J. Phys. Chem. A, 2013, 117, 9899 CrossRef CAS PubMed and references therein.
- P. W. Harland, N. D. Kim and H. S. A. Pertie, Aust. J. Chem., 1989, 2, 9 CrossRef.
- E. J. Bieske, S. A. Nizkorodov, F. R. Bennett and J. P. Maier, J. Chem. Phys., 1995, 102, 5152 CrossRef CAS.
- S. A. Nizkorodov, O. Dopfer, M. Meuwly, J. P. Maier and E. J. Bieske, J. Chem. Phys., 1996, 105, 1770 CrossRef CAS.
- H. Linnartz, T. Speck and J. P. Maier, Chem. Phys. Lett., 1998, 288, 504 CrossRef CAS.
- S. A. Nizkorodov, O. Dopfer, T. Ruchti, M. Meuwly, J. P. Maier and E. J. Bieske, J. Phys. Chem., 1995, 99, 17118 CrossRef CAS.
- N. L. Ma, B. J. Smith and L. Radom, Chem. Phys. Lett., 1992, 197, 573 CrossRef CAS.
- S. Raugei and M. L. Klein, J. Phys. Chem. B, 2001, 105, 8212 CrossRef CAS.
- P. Caselli, P. C. Myers and P. Thaddeus, Astrophys. J., 1995, 455, L77 CAS.
- L. M. Ziurys and A. J. Apponi, Astrophys. J., 1995, 445, L73 CrossRef.
- W. Irvine, Chem. Eng. News, 1982, 14, 19 Search PubMed.
- T. Amano, J. Mol. Spectrosc., 1990, 139, 457 CrossRef CAS.
- A. J. Illies, M. F. Jarrold and M. T. Bowers, J. Chem. Phys., 1982, 77, 5847 CrossRef CAS.
- G. A. Olah, K. Laali and O. Farooq, J. Org. Chem., 1985, 50, 1483 CrossRef CAS.
- G. A. Olah, K. Dunne, Y. K. Mo and P. Szilagyi, J. Am. Chem. Soc., 1972, 94, 4200 CrossRef CAS.
- P. J. F. Rege, J. A. Gladysz and I. T. Horvath, Science, 1997, 276, 776 CrossRef PubMed.
- M. I. Zaki and H. Knözinger, Mater. Chem. Phys., 1987, 17, 201 CrossRef CAS.
- K. I. Hadjiivanov and G. N. Vayssilov, Adv. Catal., 2002, 47, 307 CAS.
- C. Morterra, G. Cerrato and F. Pinna, Spectrochim. Acta, Part A, 1999, 55, 95 CrossRef.
- H. Knözinger, in Acid–Base Catalysis, Proc. Int. Symp., ed. K. Tanabe, H. Hattori, T. Yamaguchi and T. Tanaka, Sapporo, 1988 Search PubMed.
- S. C. Foster and A. R. W. McKellar, J. Chem. Phys., 1984, 81, 3424 CrossRef CAS.
- M. Juhasz, S. Hoffmann, E. S. Stoyanov, K. Kim and C. A. Reed, Angew. Chem., Int. Ed., 2004, 43, 5352 CrossRef CAS PubMed.
- M. Nava, I. V. Stoyanova, S. Cummings, E. S. Stoyanov and C. A. Reed, Angew. Chem., Int. Ed., 2014, 53, 1131 CrossRef CAS PubMed.
- E. S. Stoyanov, I. V. Stoyanova and C. A. Reed, J. Am. Chem. Soc., 2011, 133, 8452 CrossRef CAS PubMed.
- E. S. Stoyanov, K.-C. Kim and C. A. Reed, J. Am. Chem. Soc., 2006, 128, 1948 CrossRef CAS PubMed.
- A. P. Scott and L. Radom, J. Phys. Chem. A, 1996, 100, 16502 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- R. A. Kendall Jr, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796 CrossRef.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- G. Schaftenaar and J. H. Noordik, J. Comput.-Aided Mol. Des., 2000, 14, 123 CrossRef CAS PubMed.
- E. S. Stoyanov, S. P. Hoffmann, M. Juhasz and C. A. Reed, J. Am. Chem. Soc., 2006, 128, 3160 CrossRef CAS PubMed.
- P. B. Davies and W. J. Rothwell, J. Chem. Phys., 1984, 81, 5239 CrossRef CAS.
- I. Shavitt and R. J. Bartlett, Many-Body Methods in Chemistry and Physics: MBPT and Coupled-Cluster Theory, Cambridge University Press, 2009 Search PubMed.
- J. A. Boatz and M. S. Gordon, J. Phys. Chem., 1989, 93, 1819 CrossRef CAS.
- A. J. Chalk and L. Radom, J. Am. Chem. Soc., 1997, 119, 7573 CrossRef CAS.
- C. S. Gudeman, M. H. Begemann, J. Pfaff and R. Saykally, Phys. Rev. Lett., 1983, 50, 727 CrossRef CAS.
- S. C. Foster, A. R. W. McKellar and T. J. Sears, J. Chem. Phys., 1984, 81, 578 CrossRef CAS.
- W. M. Haynes, Handbook of Chemistry and Physics, CRC Press, Boca Raton, Florida, 2010, 91 edn, pp. 9–33, ISBN 978-1439820773 Search PubMed.
- A. J. Lupinetty, S. H. Strauss and G. Frenking, in Progress in Inorganic Chemistry, ed. K. D. Karlin, Wiley, New York, Vol. 49, 2001 Search PubMed.
- G. Frenking, C. Loschen, A. Krapp, S. Fau and S. H. Strauss, J. Comput. Chem., 2006, 28, 117 CrossRef PubMed.
- S. G. Lias, J. E. Bartmess, J. F. Liebman, J. L. Holmes, R. D. Levin and W. G. Mallard, J. Phys. Chem. Ref. Data, Suppl., 1988, 17 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp07441j |
| This journal is © the Owner Societies 2016 |