
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A comparative computational study of lithium and sodium insertion into van der Waals and covalent tetracyanoethylene (TCNE)-based crystals as promising materials for organic lithium and sodium ion batteries
Y.
Chen
and
S.
Manzhos
*
Department of Mechanical Engineering, National University of Singapore, Block EA #07-08, 9 Engineering Drive 1, Singapore 117575, Singapore. E-mail: mpemanzh@nus.edu.sg
First published on 3rd March 2016
Abstract
We present a comparative ab initio study of Li and Na insertion into molecular (van der Waals) crystals of TCNE (tetracyanoethylene) as well as in covalent Li/Na-TCNE crystals. We confirm the structure of the previously synthesized (covalent) Li-TCNE crystal and predict the existence of its Na-TCNE analogue. In the molecular crystals, we compute the maximum voltages to be 3.5 V for Li and 3.3 V for Na, with theoretical capacities of 1247 mA h g−1 for Li and 416 mA h g−1 for Na. In the covalent crystals, the maximum voltages are 2.2 V for Li and 2.7 V for Na, and theoretical capacities are 394 mA h g−1 for Li and 176 mA h g−1 for Na. Significantly, up to a capacity of 416 mA h g−1 for both Li and Na in the molecular crystal and 197 mA h g−1 for Li and 176 mA h g−1 for Na in the covalent crystal, the insertion of Li and Na would not lead to reactions with common electrolytes. We show that volumetric capacities of organic electrodes need not be low compared to their inorganic counterparts, contrary to popular belief: the molecular TCNE crystal has been computed to achieve the values of 1845 mA h cm−3 for Li and 615 mA h cm−3 for Na, respectively. Tetracyanoethylene-based molecular and covalent crystals could therefore become efficient organic cathode and anode materials for Li and Na ion batteries.
Introduction
The development of novel electrochemical storage technologies is important for a range of applications, not least for grid storage including peak shifting and price arbitrage.1,2 This is critical to achieve the widespread use of renewable but intermittent sources of electricity (such as wind and solar) as well as a larger share of (all-)electric vehicles which are expected to have a longer range and a better efficiency in the future.3 Lithium ion batteries provide today the highest energy density, cycle rate, and cycle life among commercial batteries.4 However, further improvements in performance as well as in sustainability are required.5 In particular, for efficient grid storage, rapid charge–discharge (within minutes) is needed,1,6 which is beyond that of present commercial Li ion batteries.5 In addition, some expensive or poisonous components like Co7 are used in present electrode materials of Li ion batteries. Furthermore, lithium deposits are geographically concentrated and may be insufficient for their use in Li ion batteries on a large scale;8–10 Li might become too costly with the growing battery market. On the other hand, sodium is abundant and cheap, relatively light, and has a qualitatively similar valence shell chemistry to lithium. Sodium ion batteries are a promising candidate technology for bulk electrochemical storage. However, compared to materials for Li storage, the design of inorganic electrode materials with suitable thermodynamics and kinetics for Na storage seems to be more difficult;11–14 this is due, in particular, to the larger ionic radius of Na+.Organic electrodes for rechargeable batteries have been receiving increasing attention in recent years, as they can be used to achieve simultaneously high rate (high power) and environment-friendly batteries. They can, for example, be made from common feedstock such as biomass.15–17 In recent years, significant progress in the development of organic batteries has been made.15 Capacities of up to 900 mA h g−1 (i.e. competitive with inorganic electrode materials) and rates of up to 1000 C (unprecedented for inorganic electrodes) have been reported.15 Moreover, organic electrodes are also promising for post-Li storage,18,19 which will have to be developed to make massive deployment of electrochemical batteries feasible.20
A number of experimental studies of different classes of potential organic electrode materials for Li ion batteries15,18,21–24 have been conducted in recent years. Computational studies are, on the other hand, very few.25,26 Modeling is, however, important to screen for potential new electrode materials, to explain the mechanism of operation of known electrode materials, and ultimately to guide the experimental design towards better performance materials. For Na storage, several organic materials have been proposed, including carboxylate and terephthalate based materials.18,22 One promising class of organic electrode materials is tetracyanides. Cathodes for Li ion batteries made of crystalline tetracyanoquinodimethane (TCNQ)21 were reported to achieve a relatively high capacity exceeding 200 mA h g−1 with an excellent cyclability. The reported lithiation mechanism involves the coordination of Li to CN moieties; that is to say, the aromatic ring would not contribute to the capacity. Furthermore, the voltages reported for this material are in the range 2.5–3.2 V which is not optimal for either cathodic (where voltages ≳4 V are desired) or anodic (where voltages closer to 0 are desired) operation. Therefore, here, we consider a smaller tetracyanide molecule without the (potentially dead-weight) aromatic ring, tetracyanoethylene (TCNE)27 as a potential new organic electrode material which is expected to have a high specific capacity and voltages potentially more suited for anodic operation.26,28,29
TCNE is a lighter molecule which is expected to attach up to 4 Li/Na atoms,27 which would result in a theoretical specific capacity of about 840 mA h g(TCNE)−1. In ref. 26, we studied ab initio lithium and sodium attachment to TCNE molecules, both free and attached to (doped) graphene. We have also studied the mechanism of, and the electronic structure resulting from, Li and Na attachment to isolated TCNE molecules.30 We predicted that up to four (five) Li and Na atoms can be stored on free (adsorbed) TCNE with binding energies stronger than cohesive energies of the Li and Na metals. Interestingly, there was no significant difference either in the specific capacity (per unit mass of material excluding Li/Na) or in the predicted voltage between Li and Na storage. In contrast, for many inorganic electrode materials, Na insertion is thermodynamically inhibited compared to Li insertion.11–14 This makes organic molecules very promising for post-Li storage in general. TCNE, therefore, is a promising candidate molecule for organic Li and Na ion batteries. However, TCNE is stable and easily available under normal conditions in a crystalline form. It is known to form two types of molecular (vdW-bound) crystals: a cubic and a monoclinic phase.31 In contrast to the monoclinic phase, the cubic phase is not stable and transforms into the monoclinic when the temperature is higher than 320 K, which then remains monoclinic upon cooling. Therefore, to understand practical potential of TCNE as the organic electrode, we here study Li and Na interaction with crystalline TCNE, specifically, the monoclinic phase (left panel in Fig. 1). Recently, TCNE has been reported to form covalent crystals with Li of stoichiometry Li-TCNE27 which presents well-defined channels that could be suitable for Li storage and transport (top left panel in Fig. 4). The potential of this material as an organic electrode has not been studied. Also, the existence and potential for Na storage of its putative Na-TCNE analogue remain unknown.
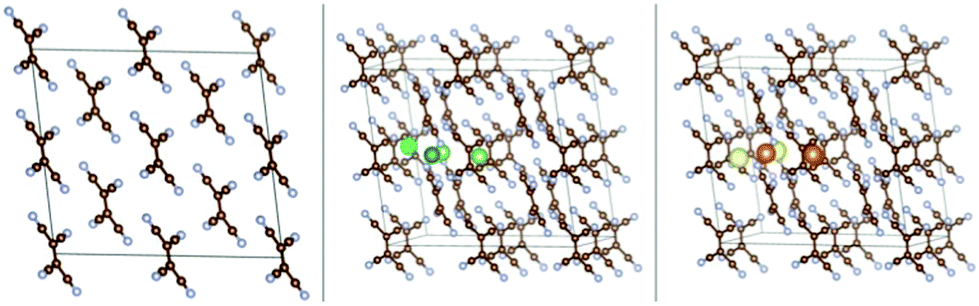 | ||
| Fig. 1 The crystal structure of monoclinic TCNE (left) and the insertion sites of Li (middle) and Na (right) atoms in the crystal. Atom colour scheme here and elsewhere: C-brown, N-grey, Li-green and Na-yellow. Different shades of green and yellow are used for different insertion sites. Visualization here and elsewhere is by VESTA.43 | ||
Here, we present a comparative dispersion-corrected density functional theory (DFT) computational study of the possibilities of Li and Na storage in tetracyanoethylene-based molecular (vdW) and covalent crystals. Specifically, we confirm the previously reported XRD structure of Li-TCNE27 and predict the existence of a covalent Na-TCNE crystal. We identify Li and Na insertion sites and compare the energetics and voltages as well as the theoretical capacities of Li vs. Na storage in vdW vs. covalent crystals.
Methods
Crystalline structures were optimized via DFT32 using the SIESTA code.33 The PBE exchange–correlation functional34 and a DZP (double-ζ polarized) basis set were used. The basis set was optimized to reproduce the cohesive energies of C, Li, Na and N.35–37 Specifically, the cohesive energy of the Li metal ELicoh = −1.67 eV and ENacoh = −1.14 eV computed using these basis sets is very accurate35–37 and can be relied upon to compute the voltages. Geometries were optimized until forces on all atoms were below 0.02 eV Å−1. Simulation cell vectors were optimized until stresses were below 0.1 GPa. A cutoff of 200 Ry was used for the Fourier expansion of the density, and a bcc type oversampling of the Fourier grid was used to minimize the eggbox effect. Smearing equivalent to an electronic temperature of 500 K was used to speed up convergence. To find Li/Na insertion sites, a periodic supercell of size ∼11 × 11 × 12 Å (corresponding to 2 × 2 × 1 unit cells) was used for the covalent crystals, with 8 TCNE-Li/Na molecules per supercell. The Brillouin zone was sampled with a 2 × 2 × 2 grid of Monkhorst–Pack points.38 For molecular (vdW) crystals, insertion sites were computed in a periodic supercell of size 14 × 12 × 14 Å (corresponding to 2 × 2 × 2 unit cells) with 16 TCNE molecules per supercell. The Brillouin zone was sampled at the Γ point. Spin polarization was used in all calculations. The stability of the crystal structures and of insertion sites was confirmed by quenched molecular dynamics (MD) calculations performed following geometry optimization, whereby the structures were relaxed until forces on all atoms were below 0.015 eV Å−1. No appreciable geometry or energy changes were detected.To model the insertion of multiple Li/Na atoms into the covalent crystals, a unit cell (2 TCNE-Li/Na units) was used with a 9 × 9 × 5 Monkhorst–Pack point grid. For vdW crystals, a unit cell (2 TCNE molecules) was used to model the insertion of multiple Li/Na atoms per supercell with a 4 × 4 × 4 Monkhorst–Pack point grid. Calculations for insertion of 0.5 Li/Na atoms per TCNE were also computed in the supercell described above (i.e. corresponding to 2 × 2 × 1 unit cells). For each specific capacity (no. of inserted Li/Na atoms), the configuration with the strongest binding was also rechecked by quenched MD to confirm its stability, as described above.
Charge transfer between Li/Na atoms and TCNE-based crystals was analyzed using Mulliken charges as well as Voronoi charges39 (which are defined in a basis-set independent way). Dispersion forces between TCNE units, which are important for the modeling of vdW-bound crystals, were modelled with the scheme of Grimme40 (DFT-D2) with parameters taken from ref. 40.
The binding energy (Eb) per Li/Na atom was computed as
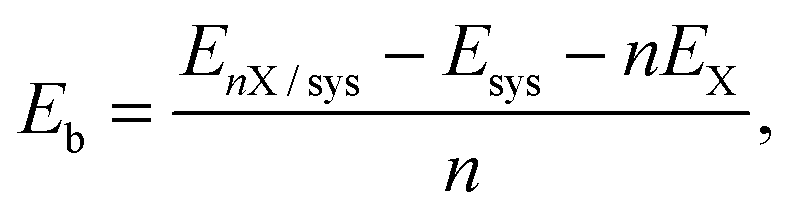 | (1) |
The average voltage between concentrations x and x0 of Li or Na is computed using the following equation:41,42
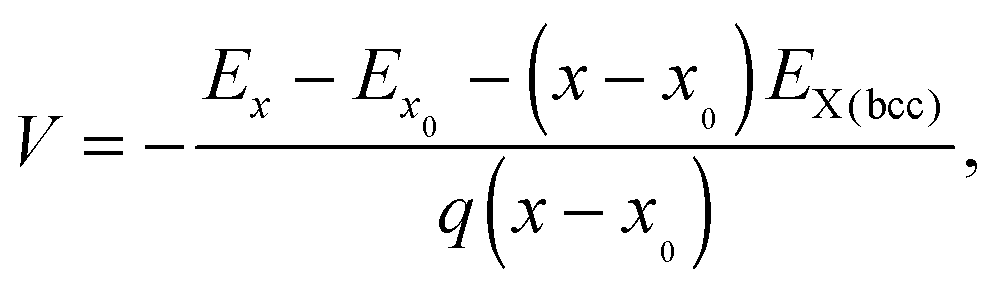 | (2) |
Results and discussion
Li and Na insertion into vdW crystals
The crystal structure of the TCNE molecular crystal was taken from ref. 31 and was optimized with the present setup (the first configuration in Fig. 1). The optimized lattice parameters of this crystal are a = 7.38 Å, b = 5.87 Å and c = 6.70 Å; α = 90.00°, β = 97.58° and γ = 90.00°. These parameters can be compared with experimental results that are in the following range: a = 7.48–7.51 Å, b = 6.20–6.21 Å and c = 6.99–7.00 Å; α = 90.00°, β = 97.10–97.35° and γ = 90.00°.31,44–47 The agreement can be considered to be good for vdW systems.24,37 The cohesive energy of this crystal is −1.39 eV per TCNE molecule.We performed a search for possible insertion sites by inserting Li/Na atoms in many possible locations within the supercell and performing optimization. Four stable non-equivalent insertion sites were found in the molecular crystal for both Li and Na insertions (Fig. 1), with Eb stronger than Ecoh of the Li and Na metals. The lowest energy site for Li/Na has Eb = −4.36/−3.71 eV which is 2.69 and 2.57 eV stronger than the Ecoh of Li and Na, respectively. The binding energies vs. Ecoh of Li and Na are shown in the leftmost red empty squares of Fig. 7, top panels. This means that insertion of dilute concentrations of Li and Na is thermodynamically favored at potentials of about 2.7 and 2.6 V vs. Li/Li+ and Na/Na+, respectively, making the molecular TCNE crystal a potential cathode material. We also note that the binding of alkali atoms to the TCNE crystal is much stronger than to isolated TCNE molecules (see ref. 30 and specifically its ESI), highlighting the important effect of long-range electrostatic interactions on the electrode potential.48
We then studied insertion of multiple Li and Na atoms into the molecular TCNE crystal. Multiple configurations were tried. We found that up to 6/3 Li/Na atoms per TCNE unit can be inserted while preserving the crystal structure. This corresponds to a capacity of 1247/623 mA h g−1 for Li/Na. The binding energies vs. Ecoh of Li and Na are shown in Fig. 7, top panels. We note that the top panels of Fig. 7 contain similar information to conventional convex hull plots. For each no. of inserted atoms, the configuration with the strongest Eb is shown in Fig. 2 and 3. For Li insertion, at higher concentrations, there are significant distortions in the TCNE molecular structure, so that a four-membered cyclic structure is observed. These, however, relax back to the original TCNE structure after the removal of Li. A similar effect was observed in ref. 30 for Li attachment to a free TCNE molecule.
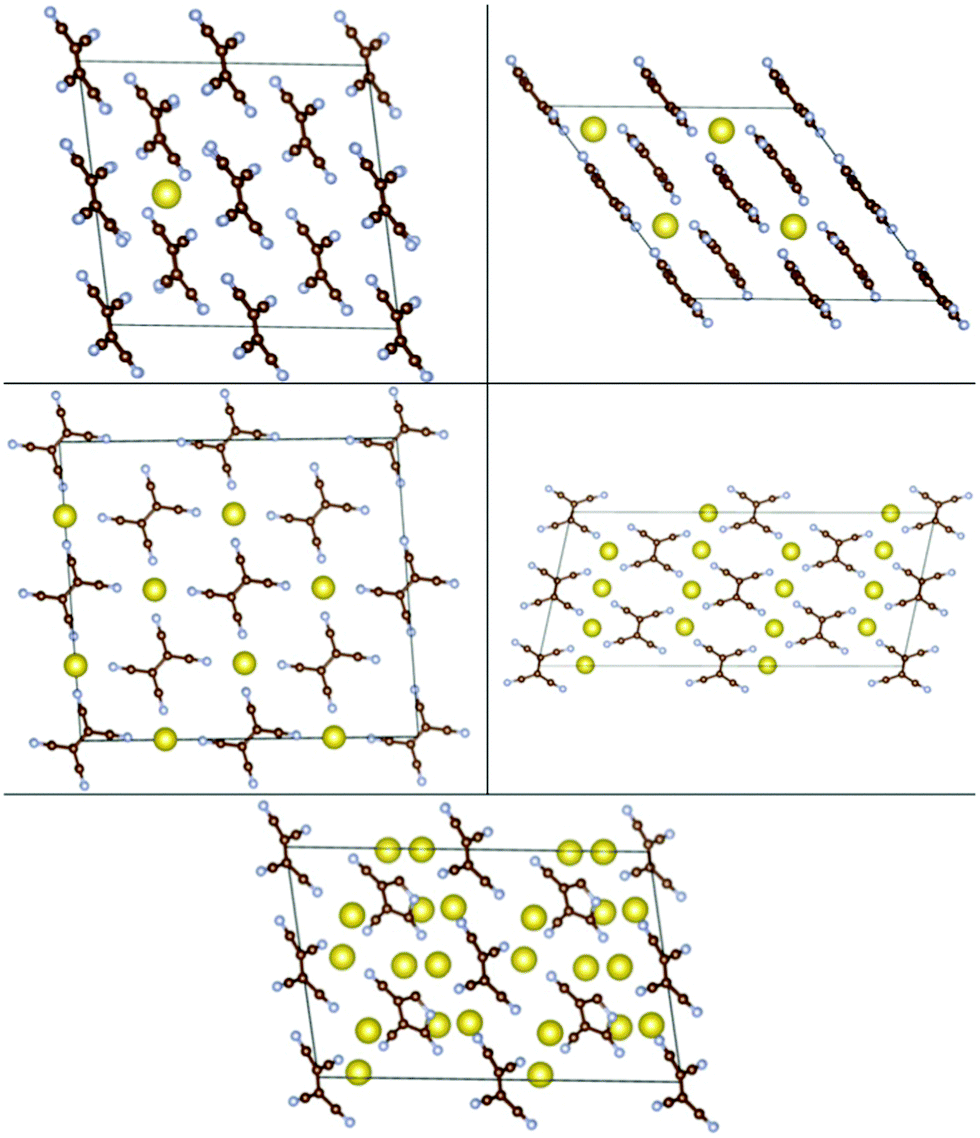 | ||
| Fig. 2 Configurations of Nan-TCNE crystals with the strongest Eb, left to right and top to bottom: n = 0.0625, 0.5, 1, 2, 3. | ||
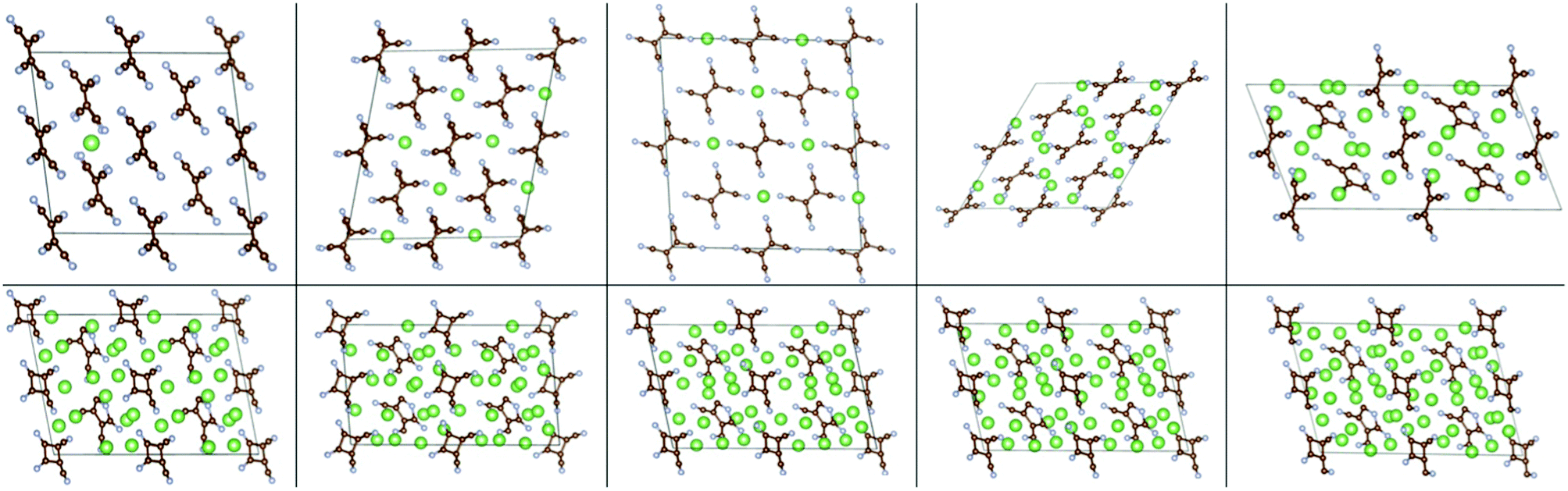 | ||
| Fig. 3 Configurations of Lim-TCNE with the strongest Eb, left to right and top to bottom: m = 0.0625, 0.5, 1, 2, 3, 4, 4.5, 5, 5.5, 6. | ||
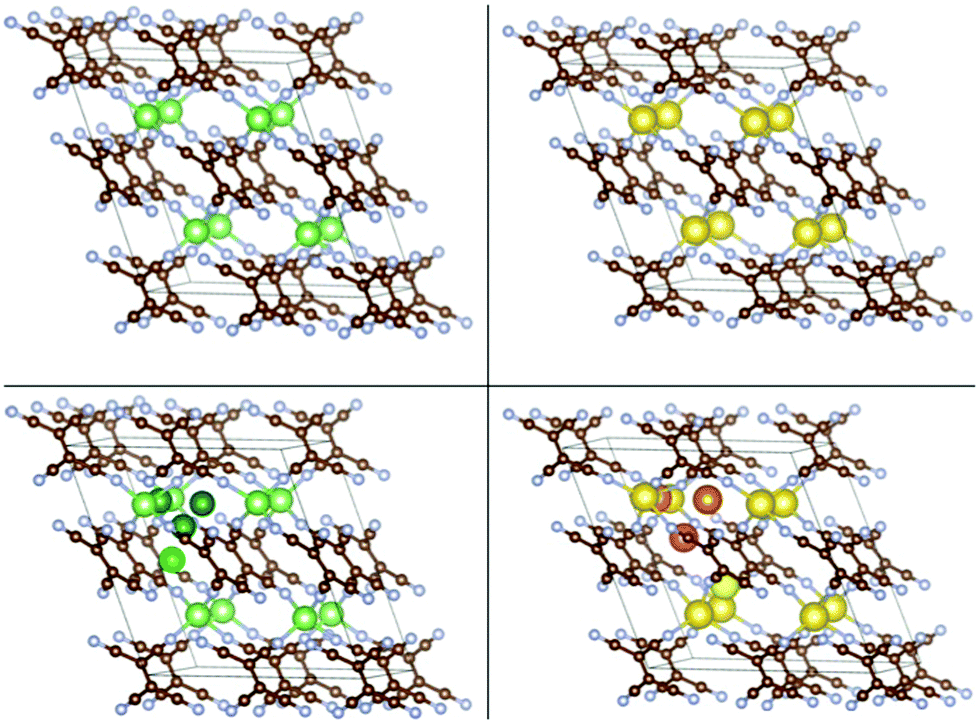 | ||
| Fig. 4 Top panels: The crystal structures of covalent Li-TCNE (left) and Na-TCNE (right) crystals. Bottom panels: Insertion sites of Li (left) and Na (right) atoms in the crystals (different shades of green/yellow are used for different Li/Na sites). | ||
As can be seen in Fig. 7, top panels, the binding of the Li/Na atom in the molecular crystal is strongest when 1/0.5 Li/Na atom per TCNE molecule is inserted into the crystal (corresponding to a specific capacity of 208/104 mA h g−1). Beyond this point, the binding of Li/Na atoms would be weakened. This means that during the insertion of up to 1/0.5 Li/Na atom per TCNE molecule, Li and Na atoms will concentrate into zones with these concentrations, i.e. a separation of lithiated/sodiated and non-lithiated/sodiated phases is expected at a constant voltage of 3.54/3.31 V vs. Li/Na bulk. The expected voltage profiles for Li and Na insertion are shown in Fig. 7, bottom panels, taking into account the expected phase segregation. The profiles confirm that molecular TCNE crystals can be used as organic cathodes of Li and Na ion batteries. Significantly, up to the capacity of 416 mA h g−1 for both Li and Na insertion, the voltage is within the electrochemical stability window for common liquid organic electrolytes, such as LiPF6 in EC:DEC between 1.3 V and 4.5 V49 and NaClO4 in EC:DMC between 1.2 V50 and 4.5 V51 (vs. Li/Li+ and Na/Na+, respectively). The insertion of Li and Na in TCNE up to these capacities would therefore not lead to reactions with the electrolyte and is expected to be safe. The voltage drops to zero at 1247 mA h g−1 for Li and 416 mA h g−1 for Na. These gravimetric capacities correspond to computed volumetric capacities of about 1845 and 615 mA h cm−3 for Li and Na, respectively. To put these numbers into perspective, the common graphite anode used in Li ion batteries has a specific capacity of 372 mA h g−1 and a volumetric capacity of 975 mA h cm−3;52 the common LiCoO2 cathode possesses reversible capacities of about 140 mA h g−1 and 710 mA h cm−3 and LiFePO4, 170 mA h g−1 and 610 mA h cm−3.53 This result shows that organic materials can have volumetric capacities competitive with their inorganic counterparts. The largest variation of volume due to the insertion of Li and Na is +22% and +20%, respectively, achieved at 2 Li and 1 Na inserted per molecule, respectively. The volume change at the maximum capacity (zero voltage) is 12% and 19% for Li and Na, respectively. One can estimate the energy density of this material when used with a Li/Na anode by multiplying the average voltage by the capacity; this gives about 1670 W h kg−1 for Li and 1140 W h kg−1 for Na.
The average charge donation of Li/Na in the molecular crystal was also computed. For the configurations shown in Fig. 2 and 3, up to 0.70/0.84 |e| (Mulliken) and 0.28/0.30 |e| (Voronoi) per Li/Na atom are donated to TCNE molecules, even at the largest Li/Na concentrations. This is in contrast to the Li/Na attachment to a single TCNE molecule, where there were clear and matching weakening trends of Eb and of average charge donation with the number of attached Li/Na atoms.26,28,29
Li and Na insertion into covalent Li/Na-TCNE crystals
We also performed ab initio optimization of the covalent Li-TCNE crystal structure (top left panel of Fig. 4) and its Na analogue (top right panel of Fig. 4), both of which have stable configurations. The unit cell of the covalent Li-TCNE crystal has a structure with lattice parameters a = 5.42 Å, b = 5.56 Å and c = 11.93 Å; α = 90.00°, β = 110.12° and γ = 90.00°. The volume is only about 1% larger than that of the crystal structure reported in an experimental study27 (a = 5.43 Å, b = 5.41 Å and c = 11.91 Å; α = 90.00°, β = 107.70° and γ = 90.00°). After replacing Li with Na atoms, we have found a stable structure of the Na-TCNE analogue, with lattice parameters a = 5.92 Å, b = 5.87 Å and c = 12.43 Å; α = 90.00°, β = 111.40° and γ = 90.00°. The formation energy of Li/Na-TCNE crystals is −4.82/−4.38 eV per formula unit (vs. TCNE molecules and bcc Li/Na, negative sign means favorable formation).Similarly to the molecular crystal, multiple possible insertion sites were tried, and four stable non-equivalent insertion sites were found in these covalent crystals (the bottom panels in Fig. 4), with Eb stronger than Ecoh of the Li and Na metals. The lowest energy site for Li/Na has Eb = −3.45/−2.89 eV which is 1.78 and 1.75 eV stronger than the Ecoh of Li and Na, respectively. The binding energies vs. Ecoh of Li and Na are shown in the leftmost black filled squares of Fig. 7, top panels. This means that insertion of dilute concentrations of Li and Na is thermodynamically favored at potentials of about 1.8 V vs. Li/Li+ and Na/Na+, respectively, making Li/Na-TCNE a potential anode material.
The insertion of multiple atoms into the covalent crystals with many possible configurations was then modeled. Up to 2.5/2 Li/Na atoms per TCNE unit can be inserted in the covalent crystal which corresponds to a capacity of 493/353 mA h g−1, respectively, while preserving the crystal structure. The binding energies vs. Ecoh of Li and Na are shown in Fig. 7, top panels. For each no. of inserted atoms, the configuration with the strongest Eb is shown in Fig. 5 and 6. Comparing Fig. 2 and 3 with Fig. 5 and 6, we can find that the structures of covalent crystals are much less distorted by the insertion than that of the molecular crystal, which, as expected, shows that the covalently bound Li-TCNE framework is more stable than the vdW-bound framework of the molecular crystal. Furthermore, by comparing the vacancy formation energy, which is 5.4/4.6 eV for the extraction of a Li/Na atom from the structures shown in the top panel of Fig. 4 into vacuum to the insertion energy Eb, which peaks at −3.83/−3.81 eV for Li/Na insertion, we also confirmed that Li/Na-TCNE crystals are stable under Li/Na insertion/extraction.
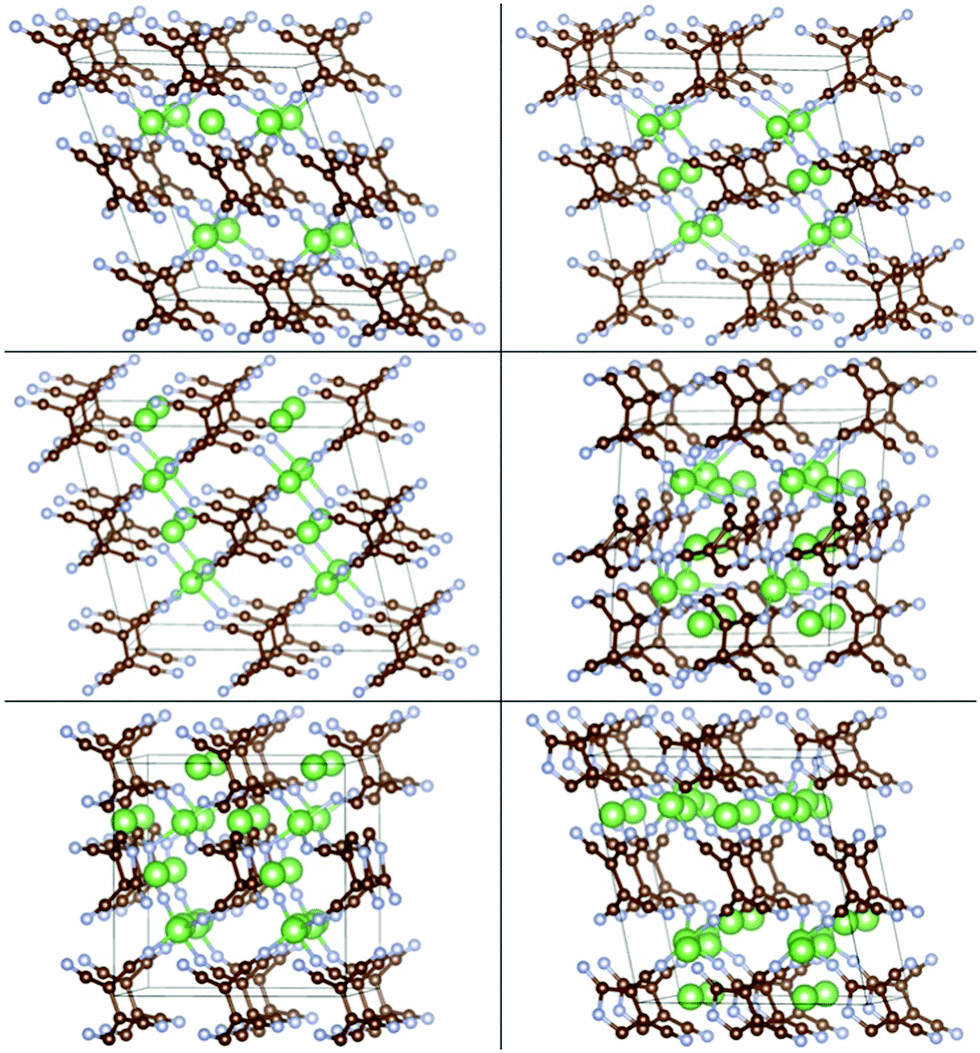 | ||
| Fig. 5 Configurations of Lim-(Li-TCNE) crystals with the strongest Eb, left to right and top to bottom: m = 0.125, 0.5, 1, 1.5, 2, 2.5. | ||
 | ||
| Fig. 6 Configurations of Nan-(Na-TCNE) crystals with the strongest Eb, left to right and top to bottom n = 0.125, 0.5, 1, 1.5, 2. | ||
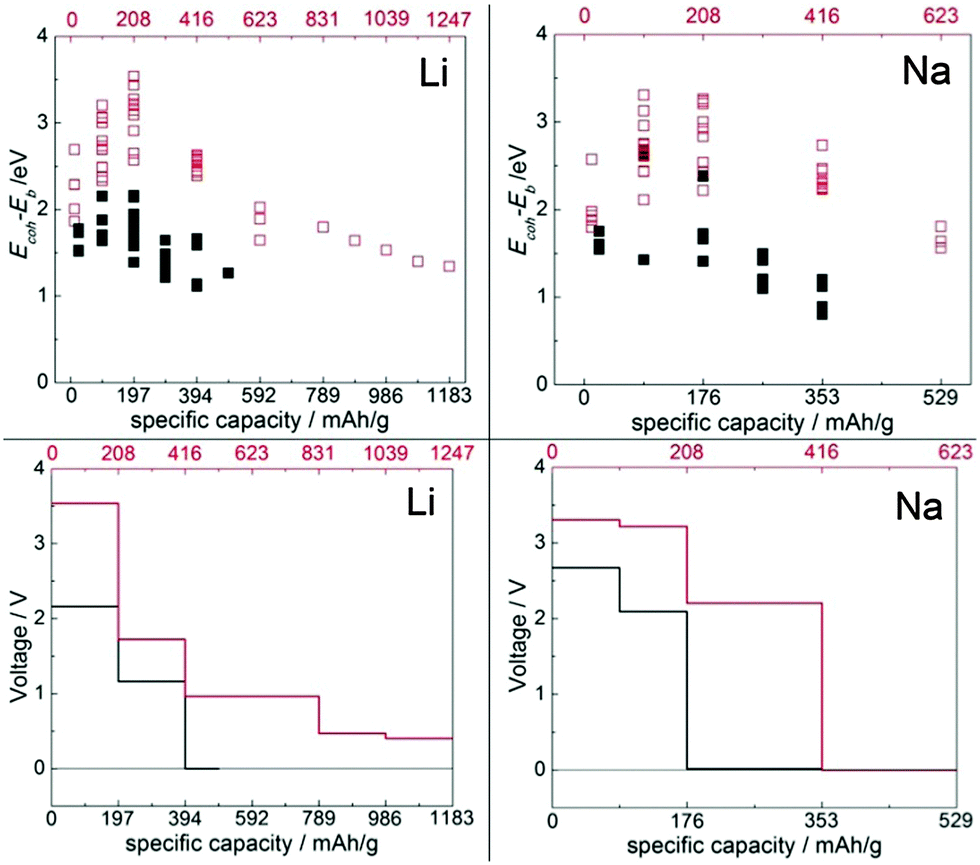 | ||
| Fig. 7 Top: Binding strength per alkali atom on the “voltage” scale (EcohLi/Na − Eb) vs. specific capacity corresponding to the no. of inserted Li/Na atoms in the monoclinic TCNE crystal (red empty squares and top axes) and covalent Li/Na-TCNE crystal (black filled squares and bottom axes). Bottom: Computed voltage profiles vs. specific capacity in the monoclinic TCNE crystal (red lines and top axes) and covalent Li/Na-TCNE crystal (black lines and bottom axes). | ||
The strongest binding energy per Li/Na atom peaks at the concentration of 1/0.5 Li/Na atoms per Li/Na-TCNE unit, which would correspond to a specific capacity of 197/88 mA h g−1 for Li/Na. Up to this capacity, therefore, a separation of lithiated/sodiated and non-lithiated/sodiated regions is expected to occur at a constant voltage of 2.16/2.67 V vs. Li/Na bulk. The computed voltage profile is shown in Fig. 7, bottom panels, taking into account the expected phase segregation. The voltage drops to zero after 394/176 mA h g−1 for Li/Na insertion, which corresponds to computed volumetric capacities of 523 and 220 mA h cm−3, respectively. The energy density of this material when used with a Li/Na electrode is estimated to be about 660 W h kg−1 for Li and 420 W h kg−1 for Na. The largest variation of volume due to the insertion of Li and Na is −16% and −17%, respectively, achieved at 1 Li or Na atom inserted per molecule. The volume change at the maximum capacity (zero voltage) is −3% and −15%, respectively. In contrast to the vdW crystals, therefore, the Li and Na insertion into the covalent Li/Na crystals may lead to a contraction rather than expansion. This may be rationalized by considering that in the covalent crystal, the Li/Na atoms already present in the crystal structure position the TCNE units further apart than in the vdW crystal (indeed, the volume per formula unit is about 17% larger in Li-TCNE and 40% larger in Na-TCNE). Because of the attractive nature of Li/Na-TCNE interactions, the inserted Li/Na pull the structure together. The voltage remains above the electrolyte reduction potential up to 197/176 mA h g−1 for Li/Na. Therefore, up to these capacities the covalent Li/Na-TCNE crystalline electrodes will not promote reactions with the electrolyte and are promising candidates as anode materials.
We also calculated the average charge donation of Li/Na in the covalent crystals. For the configurations shown in Fig. 5 and 6, up to 0.69/0.81 |e| (Mulliken) and 0.32/0.33 |e| (Voronoi) per Li/Na atom is donated to TCNE units.
During the insertion of Li/Na, similar (Li/Na)x-TCNE stoichiometries may be formed with both vdW and covalent crystals. For example, the insertion of one alkali atom per TCNE unit into the vdW crystal results in the same stoichiometry as that of the pristine covalent Li/Na-TCNE crystal. The total energy (of the lowest energy configuration) is about 0.1/0.3 eV (per TCNE unit) lower than the energy of the covalent Li/Na-TCNE crystal. At higher Li concentrations, structures obtained with the vdW crystal have higher energy (by 0.3 eV at Li2-TNE and by about 0.7 eV at Li3-TCNE) than those obtained with the covalent crystal at the same stoichiometry. At higher Na concentrations, structures obtained with the vdW crystal remain lower in energy than those obtained from the covalent crystal at the same stoichiometry (by 0.1 eV at Na2-TCNE and by 0.03 eV at Na3-TCNE). However, the geometries of the two types of crystals remain significantly different. This, together with the facts that (i) the Li-TCNE covalent structure was stable in ref. 27 even though it is computed to be slightly higher in energy and (ii) a high energy cost is required to remove Li/Na from their positions in the pristine covalent crystals, suggests that the two types of crystals might not interconvert, although such conversion is a possibility and a definitive answer should be given by an experiment.
Conclusions
We carried out a comparative dispersion-corrected density functional theory study of the possibilities of Li and Na storage in tetracyanoethylene(TCNE)-based van der Waals (molecular) and covalent crystals. Firstly, we confirmed the structure of the previously synthesized (covalent) Li-TCNE crystal and predicted the existence of its Na-TCNE analogue. For both kinds of crystals, we identified Li and Na insertion sites, including insertion of multiple atoms. We found that up to 6/3 and 2.5/2 Li/Na atoms per TCNE unit can be inserted into the molecular and covalent crystals, respectively, while preserving the structure. The computed voltage can reach 3.54/2.16 V vs. Li/Li+ for Li insertion and 3.31/2.67 V vs. Na/Na+ for Na insertion into the molecular/covalent crystals, respectively. The molecular crystal could therefore be used as a cathode. Significantly, up to capacity of 416 mA h g−1 for both Li and Na in the molecular crystal and 197 mA h g−1 for Li and 176 mA h g−1 for Na in the covalent crystal, the insertion of Li and Na would not lead to reactions with the electrolyte. This is a desirable property for both conventional liquid carbonate based electrolytes as well as for some of the promising solid state electrolytes (SSE) such LGPS (Li10GeP2S12) and other sulfide SSE that do not form a self-arresting solid electrolyte interface.54With the vdW TCNE crystal, the computed voltages reach zero at the specific capacities of 1247 mA h g−1 for Li and 416 mA h g−1 for Na, corresponding to volumetric capacities of 1845 and 615 mA h cm−3 for Li and Na, respectively. This shows that volumetric capacities of organic electrodes need not be low compared to their inorganic counterparts, contrary to popular belief. With the covalent Li/Na-TCNE crystals, the voltages are computed to fall to zero at the specific capacities of 394 mA h g−1 for Li and 176 mA h g−1 for Na, corresponding to volumetric capacities of 523 and 220 mA h cm−3 for Li and Na, respectively. The computed volume changes along the charge–discharge curve for all cases remained under about 20%.
Therefore, we conclude that tetracyanoethylene (TCNE) – based molecular and covalent crystals could become efficient organic cathode and anode materials, respectively, for both Li and Na ion batteries.
Acknowledgements
This work was supported by the Tier 2 AcRF grant MOE2014-T2-2-006 by the Ministry of Education of Singapore.References
- E. Barbour, I. A. G. Wilson, I. G. Bryden, P. G. McGregor, P. A. Mulheran and P. J. Hall, Energy Environ. Sci., 2012, 5, 5425–5436 Search PubMed.
- D. Yue, P. Khatav, F. You and S. B. Darling, Energy Environ. Sci., 2012, 5, 9163–9172 CAS.
- T. Kousksou, P. Bruel, A. Jamil, T. El Rhafiki and Y. Zeraouli, Sol. Energy Mater. Sol. Cells, 2014, 120(part A), 59–80 CrossRef CAS.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 CAS.
- J. B. Goodenough, Energy Environ. Sci., 2014, 7, 14–18 CAS.
- C. J. Barnhart, M. Dale, A. R. Brandt and S. M. Benson, Energy Environ. Sci., 2013, 6, 2804–2810 Search PubMed.
- C. Lupi and M. Pasquali, Miner. Eng., 2003, 16, 537–542 CrossRef CAS.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-Gonzalez and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 CAS.
- J. M. Tarascon, Nat. Chem., 2010, 2, 510 CrossRef CAS PubMed.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- I. M. Oleksandr, L. T. Teck and M. Sergei, Appl. Phys. Express, 2013, 6, 027301 CrossRef.
- O. I. Malyi, T. L. Tan and S. Manzhos, J. Power Sources, 2013, 233, 341–345 CrossRef CAS.
- O. Malyi, V. V. Kulish, T. L. Tan and S. Manzhos, Nano Energy, 2013, 2, 1149–1157 CrossRef CAS.
- F. Legrain, O. I. Malyi and S. Manzhos, Solid State Ionics, 2013, 253, 157–163 CrossRef CAS.
- Y. L. Liang, Z. L. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 CAS.
- H. Chen, M. Armand, G. Demailly, F. Dolhem, P. Poizot and J.-M. Tarascon, ChemSusChem, 2008, 1, 348–355 CrossRef CAS PubMed.
- A. Abouimrane, W. Weng, H. Eltayeb, Y. Cui, J. Niklas, O. Poluektov and K. Amine, Energy Environ. Sci., 2012, 5, 9632–9638 CAS.
- Y. NuLi, Z. Guo, H. Liu and J. Yang, Electrochem. Commun., 2007, 9, 1913–1917 CrossRef CAS.
- A. K. Shukla and T. P. Kumar, J. Phys. Chem. Lett., 2013, 4, 551–555 CrossRef CAS PubMed.
- Y. Hanyu and I. Honma, Sci. Rep., 2012, 2, 453–458 Search PubMed.
- Y. Park, D. S. Shin, S. H. Woo, N. S. Choi, K. H. Shin, S. M. Oh, K. T. Lee and S. Y. Hong, Adv. Mater., 2012, 24, 3562–3567 CrossRef CAS PubMed.
- T. Yasuda and N. Ogihara, Chem. Commun., 2014, 50, 11565–11567 RSC.
- N. Ogihara, T. Yasuda, Y. Kishida, T. Ohsuna, K. Miyamoto and N. Ohba, Angew. Chem., Int. Ed., 2014, 53, 11467–11472 CrossRef CAS PubMed.
- N. Dardenne, X. Blase, G. Hautier, J.-C. Charlier and G.-M. Rignanese, J. Phys. Chem. C, 2015, 119, 23373–23378 CAS.
- Y. Chen and S. Manzhos, Mater. Chem. Phys., 2015, 156, 180–187 CrossRef CAS.
- J. H. Her, P. W. Stephens, R. A. Davidson, K. S. Min, J. D. Bagnato, K. van Schooten, C. Boehme and J. S. Miller, J. Am. Chem. Soc., 2013, 135, 18060–18063 CrossRef CAS PubMed.
- Y. Chen and S. Manzhos, Proceedings of the 14th Asian Conference on Solid State Ionics (ACSSI-2014), Research Publishing Services, Singapore, pp. 374–380.
- Y. Chen and S. Manzhos, MRS Online Proc. Libr., 2014, 1679 DOI:10.1557/opl.2014.1849.
- Y. Chen and S. Manzhos, Phys. Chem. Chem. Phys., 2016, 18, 1470–1477 RSC.
- M. Khazaei, M. Arai, T. Sasaki and Y. Kawazoe, J. Phys. Chem. C, 2013, 117, 712–720 CAS.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- M. S. José, A. Emilio, D. G. Julian, G. Alberto, J. Javier, O. Pablo and S.-P. Daniel, J. Phys.: Condens. Matter, 2002, 14, 2745 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- C. Kittel, Introduction to Solid State Physics, Wiley, Hoboken, NJ, 2005 Search PubMed.
- E. Kaxiras, Atomic and Electronic Structure of Solids, Cambridge University Press, Cambridge, 2003 Search PubMed.
- N. Capel, D. Bharania and S. Manzhos, Computation, 2015, 3, 574 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188–5192 CrossRef.
- C. Fonseca Guerra, J.-W. Handgraaf, E. J. Baerends and F. M. Bickelhaupt, J. Comput. Chem., 2004, 25, 189–210 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- W. Luo, J. Wan, B. Ozdemir, W. Bao, Y. Chen, J. Dai, H. Lin, Y. Xu, F. Gu, V. Barone and L. Hu, Nano Lett., 2015, 15, 7671–7677 CrossRef CAS PubMed.
- G. Ceder, G. Hautier, A. Jain and S. P. Ong, MRS Bull., 2011, 36, 185–191 CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- R. Mukhopadhyay and S. L. Chaplot, J. Phys.: Condens. Matter, 2002, 14, 759 CrossRef CAS.
- D. A. Bekoe and K. N. Trueblood, Z. Kristallogr., 1960, 113, 1–22 CrossRef CAS.
- H. Yamawaki, K. Aoki, Y. Kakudate, M. Yoshida, S. Usuba and S. Fujiwara, Chem. Phys. Lett., 1992, 198, 183–187 CrossRef CAS.
- R. G. Little, D. Pautler and P. Coppens, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 1493–1499 CrossRef CAS.
- M. Saubanère, M. B. Yahia, S. Lebègue and M. L. Doublet, Nat. Commun., 2014, 5, 5559 CrossRef PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2009, 22, 587–603 CrossRef.
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS.
- A. Bhide, J. Hofmann, A. Katharina Durr, J. Janek and P. Adelhelm, Phys. Chem. Chem. Phys., 2014, 16, 1987–1998 RSC.
- L. Croguennec and M. R. Palacin, J. Am. Chem. Soc., 2015, 137, 3140–3156 CrossRef CAS PubMed.
- R. J. Brodd, Batteries for Sustainability: Selected Entries from the Encyclopedia of Sustainability Science and Technology, Springer, New York, 2012 Search PubMed.
- A. C. Luntz, J. Voss and K. Reuter, J. Phys. Chem. Lett., 2015, 6, 4599–4604 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2016 |