
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA detailed assignment of NEXAFS resonances of imidazolium based ionic liquids†
Christopher
Ehlert
ab,
Markus
Holzweber
a,
Andreas
Lippitz
a,
Wolfgang E. S.
Unger
a and
Peter
Saalfrank
*b
aBAM Bundesanstalt für Materialforschung und –prüfung, D-12203 Berlin, Germany
bUniversität Potsdam, Institut für Chemie, Karl-Liebknecht-Str. 24-25, 14476 Potsdam-Golm, Germany. E-mail: peter.saalfrank@uni-potsdam.de
First published on 5th February 2016
Abstract
In Near Edge X-Ray Absorption Fine Structure (NEXAFS) spectroscopy X-Ray photons are used to excite tightly bound core electrons to low-lying unoccupied orbitals of the system. This technique offers insight into the electronic structure of the system as well as useful structural information. In this work, we apply NEXAFS to two kinds of imidazolium based ionic liquids ([CnC1im]+[NTf2]− and [C4C1im]+[I]−). A combination of measurements and quantum chemical calculations of C K and N K NEXAFS resonances is presented. The simulations, based on the transition potential density functional theory method (TP-DFT), reproduce all characteristic features observed by the experiment. Furthermore, a detailed assignment of resonance features to excitation centers (carbon or nitrogen atoms) leads to a consistent interpretation of the spectra.
1 Introduction
Ionic liquids (ILs) can loosely be defined as “molten salts” with a melting point lower than about 100 °C. They consist of ion pairs with an organic cation, and an anion which is inorganic or organic. The interesting and very promising chemical and physical properties of ILs have extensively been reviewed elsewhere1–7 and need not be reiterated here.Being liquids with low vapor pressure, high vacuum methods are ideal tools to probe the specific properties of the ILs. Here in particular, we consider Near Edge X-Ray Absorption Fine Structure spectroscopy (NEXAFS) as an analytical method. In NEXAFS, the core electrons of certain types of atoms, in this work the 1s (K shell) electrons of carbon and nitrogen, are X-ray excited to unoccupied molecular orbitals of the system. From the emerging C K and N K NEXAFS spectral information about the orientation, the oxidation state, the electronic structure and the chemical environment of the core-excited atoms can be extracted.8 In fact, also for ILs electron yield NEXAFS is in use since sometime to extract surface-sensitive information on molecular entities – see, e.g., ref. 9–13.
In our own work, we investigated various imidazolium based ionic liquids using NEXAFS measurements as well as other surface-sensitive methods such as X-ray photoelectron spectroscopy and Low Energy Ion Scattering (LEIS) [M. Holzweber et al., to be published]. As in earlier NEXAFS studies on aminothiolate monolayers,14 azide, 1,2,3-triazole and terpyridyl terminated SAMs15 and 2,5-diphenyl-1,3,4-oxadiazole films16 the interpretation of spectra was hindered due to a lack of appropriate reference spectra or materials. The solution in such cases is always a simulation of spectra which substantially reduces the speculative character of assignments for the different resonances occurring in the spectra.
Characteristic signatures were found for NEXAFS spectra arising from C 1s → π* and N 1s → π* as well as higher transitions (notably to σ*). To interprete the rich structure found in these spectra, theoretical simulations are most valuable. In the present paper for two particular systems with very distinct donor–acceptor properties,17 namely an organic/organic and an organic/inorganic IL, simulations based on quantum chemical methods will be performed to interprete experimentally measured C K and N K NEXAFS spectra. Further in experiment, for the organic/organic IL, the size of the cation is systematically increased (via an alkyl chain length, see below), in order to elucidate size-dependent trends in C K NEXAFS.
This paper is structured as follows: In Section 2, some details of the NEXAFS measurements are presented. Section 3 explains the underlying models and theoretical methods used for simulating NEXAFS spectra. In Section 4, we compare the selected experimental results with the information provided by the simulations. Finally, we summarize our findings in Section 5.
2 Experimental setup
NEXAFS spectroscopy was carried out at the HE-SGM monochromator dipole magnet beam line at the synchrotron radiation source BESSY II (Berlin, Germany). Spectra were acquired at the C K-edge in the total energy electron yield (TEY) mode.8 The resolution E/ΔE of grid 1 at the carbonyl π* resonance of CO (hν = 287.4 eV) was found to be in the order of 2500. The slit-width used was 200 μm. Raw spectra were divided by the monochromator transmission function which was obtained with a freshly sputtered Au sample.8 Spectra were recorded at an angle of 55° measured between the surface plane of the sample and the direction vector of the light beam. Energy alignment of the energy scale was achieved by using an I0 feature referenced to a C 1s → π* resonance measured on a fresh surface of a HOPG (Highly Ordered Pyrolytic Graphite, Advanced Ceramic Corp., Cleveland, USA) at 285.4 eV.18 Experimental spectra are shown with the pre-edge count rate subtracted and after normalization in units of the absorption edge jump.8Ionic liquids were either purchased from IoLiTec Ionic Liquids Technologies GmbH (Heilbronn, Germany) in the highest available grade or were a donation from AC2T research GmbH (Wiener Neustadt, Austria). Small amounts of ionic liquid were bottled into small vials, dried and stored in a vacuum at 10−6 to 10−7 mbar. Samples were prepared to exhibit a flat surface by filling up a cavity in a customized copper or steel sample holder and immediately transferred into the vacuum. XPS survey and high resolution (for C, N, O, F, and I) spectra confirmed the ionic liquid surface purity.
3 Computational models and methods: simulation of NEXAFS spectra
3.1 Models
From a theoretical point of view, the simulation of liquids is a challenging task. To account for the motion of the liquid's constituents, an (ab initio) molecular dynamics simulation would be necessary to probe snapshots along a trajectory in order to evaluate thermally averaged spectra. This has been done for example in ref. 19. Here we avoid this costly strategy by considering experimentally reported crystal structures as a starting point instead, neglecting molecular motion altogether. Furthermore, we adopt minimal cluster models rather than periodic models20–22 for calculating NEXAFS spectra. Cluster models, well-established also for NEXAFS,23,24 not only simplify the theoretical treatment but may also be physically more sensible for a liquid than periodic models. Also, the cluster ansatz allows for the treatment of charged systems, and can easily be extended to include vibronic effects and nuclear motion if needed. It should also be noted, though, that the minimal, static cluster models used below will only allow for an approximate treatment of NEXAFS spectra of ILs. It will turn out, however, that already this minimal model captures and explains the essential features of experimental spectra.In this paper, two different ILs are considered, both based on the use of alkylimidazolium+ ions as organic cations, and either organic or inorganic anions. Specifically, we consider as an example for an IL with an organic anion 1,3-dimethylimidazolium bis(trifluoromethylsulfonyl) imide, abbreviated as [C1C1im]+[NTf2]− in what follows. Here, “im” stands for imidazolium, and “C1C1” indicates that two C1-alkyl groups (methyl, –CH3) are attached to the N atoms of imidazolium. Furthermore, [NTf2]− denotes the bis(trifluoromethylsulfonyl)imide counter ion. While the experiments have been done with the IL, the theoretical modeling is based on a structural model derived from the experimental crystal structure of Rogers et al.25 Accordingly, at low temperatures [C1C1im]+[NTf2]− forms a solid with a triclinic cell containing four cation–anion pairs, as shown in the ESI.† For quantum chemical investigation, we take a single such cation–anion pair out of the crystal without reoptimizing any coordinates. The model ion pair is shown in Fig. 1(a).
 | ||
| Fig. 1 Cluster models used for the spectral simulation, as obtained from crystal structure data (see the text). (a) 1,3-Dimethylimidazolium bis(trifluoromethylsulfonyl) imide, [C1C1im]+[NTf2]−. (b) 1-Butyl-3-methylimidazolium iodide, [C4C1im]+[I]−. Color coding for atoms: red for oxygen, blue for nitrogen, light blue for fluorine, white for hydrogen, yellow for sulfur, purple for iodide, and grey for carbon. | ||
Similarly, as a second system we consider 1-butyl-3-methylimidazolium iodide as an example for an IL with an inorganic anion, abbreviated as [C4C1im]+[I]− in the following. Here, a methyl (–CH3, “C1”) and a butyl group (–C4H9, “C4”) are attached to imidazolium with the counter ion now being iodide ([I]−). A crystal structure for the solid was published by Nakakoshi et al.,26 showing an orthorhombic unit cell containing again four ion pairs (see ESI†). Again, a single ion pair was cut out of this structure to establish a minimal model for simulation, which is shown in Fig. 1(b).
While these two systems were the focus of our combined experimental/theoretical study, the organic/organic IL of above was investigated experimentally also for other n in the series [CnC1im]+[NTf2]−. In particular, the effect of the alkyl chain length (–CnH2n+1, “Cn”) on experimental C K NEXAFS spectra was studied also for n = 2, 3, 4, 6, in addition to n = 1.
3.2 Calculation of NEXAFS spectra
Our calculation procedure for simulation of NEXAFS spectra is based on density functional theory in the Kohn–Sham scheme. All results presented in this work were obtained using the CP2K27 program package. Herein, we use the GAPW (Gaussian and augmented plane wave)28 method. Specifically, a 6-311G** basis29,30 for all atoms was used, and a plane-wave cutoff of 300 eV. The PBE functional31 was adopted as an exchange–correlation energy functional.The theoretical method used here for X-ray absorption is the so-called transition potential method (TP-DFT).32–34 The method, well established also for NEXAFS spectra, needs not to be described in much detail here, and also no details of implementational specifics for CP2K are given; the reader is referred to ref. 34. Briefly, in order to calculate NEXAFS spectra with many possible electronic excitations from an initial, atomic core orbital ψi, to final states, ψf, the TP-DFT method introduces a transition state for one excitation center (for example a carbon atom for a C K NEXAFS), which represents all excitations to unoccupied orbitals. This transition state is characterized by a core orbital (C 1s in the case of C K NEXAFS) with a fractional occupation number of 0.5, and is calculated self-consistently. This is “half way” between the initial (electron occupation 1) and the final occupation number (electron occupation 0), of ψi. As shown elsewhere, this procedure accounts for a large portion of the relaxation energy of the core hole, and for a balanced yet economic, description of the final states.
Within the dipole approximation, valid for soft X-ray spectroscopy as considered here, the absorption cross-section for excitation center/core orbital i is then expressed by the Golden Rule type expression
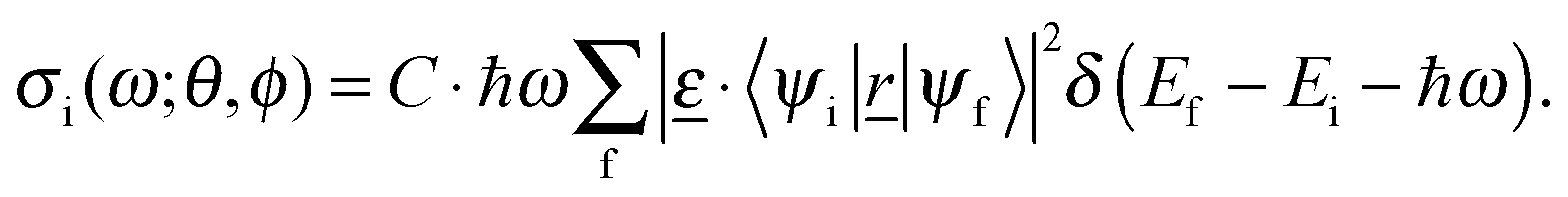 | (1) |
![[r with combining low line]](https://www.rsc.org/images/entities/i_char_0072_0332.gif) |ψf 〉 are transition dipole matrix elements, with = (x, y, z). Furthermore,
|ψf 〉 are transition dipole matrix elements, with = (x, y, z). Furthermore,  the polarization vector of the excitation light with electric field strength
the polarization vector of the excitation light with electric field strength  (with E0 being the field amplitude), which can be expressed as
(with E0 being the field amplitude), which can be expressed as  . In order to account for all possible orientations of the molecular system with respect to the X-ray field (as realized in an un-polarized NEXAFS experiment on an IL), we average over both angles θ and ϕ, such that an angle-integrated spectrum corresponding to center i is obtained as
. In order to account for all possible orientations of the molecular system with respect to the X-ray field (as realized in an un-polarized NEXAFS experiment on an IL), we average over both angles θ and ϕ, such that an angle-integrated spectrum corresponding to center i is obtained as | (2) |
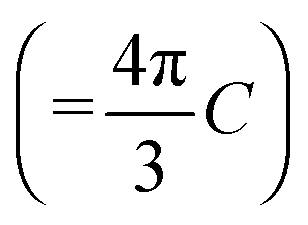 , and sif2 =|〈ψi|s|ψf〉|2 (where s = x, y, z). The total cross-section is obtained by summing over all excitation centers of the same chemical element,
, and sif2 =|〈ψi|s|ψf〉|2 (where s = x, y, z). The total cross-section is obtained by summing over all excitation centers of the same chemical element, | (3) |
Furthermore, we introduced empirical shifts, which align the experimental and theoretical spectra, as is common practice.32 We used a shift of −0.9 eV for C K and −1.6 eV for N K NEXAFS spectra.
4 Results
4.1 C K NEXAFS resonances of [CnC1im]+[NTf2]− and [C4C1im]+[I]− ionic liquids
We now apply the methodology to C K and later, N K NEXAFS spectra of the two ILs mentioned above, [C1C1im]+[NTf2]− and [C4C1im]+[I]−. In Fig. 2, C K NEXAFS spectra are presented and analyzed. A summary of all resonance positions and assignments can be found in Table 1. Measured C K NEXAFS spectra of other species of the type [CnC1im]+[NTf2]−, with different alkyl chain lengths n = 2, 3, 4, 6, can be found in Fig. 3. | ||
| Fig. 2 C K NEXAFS spectra of [C1C1im]+[NTf2]− (a) and [C4C1im]+[I]− (b) and their simulation. The upper panels show the experimental NEXAFS spectra (black), with resonances indicated, as well as the theoretical spectra (blue). The middle panels analyze atomic contributions to theoretical NEXAFS spectra, with a color coding and numbering of carbon atoms as indicated in the lowest panels. | ||
| Resonance | Character | Excitation energy (eV) | Excitation center |
|---|---|---|---|
| [C1C1im]+[NTf2]− | |||
| I | C1s → π* | 287.0 | C5, C4 |
| 286.7 | C2 | ||
| II | C1s → σ* | 289.3 | C6, C7 |
| III | C1s → σ* | 290.6 | C6, C7 |
| IV | C1s → σ* | 294.7 | C8, C9 |
| V | C1s → σ* | 298.2 | C4, C5 & C8, C9 |
| [C4C1im]+[I]− | |||
| I | C1s → π* | 286.5 | C4, C5 |
| 286.9 | C2 | ||
| II | C1s → σ* | 290.4 | C6, C7, C8, C9, C10 |
| III | C1s → σ* | 292.5 | C6, C7, C8, C9, C10 |
 | ||
| Fig. 3 C K NEXAFS spectra of [CnC1im]+[NTf2]−. n represents the length of the alkyl chain (see the text). | ||
A first observation from the upper panels of Fig. 2 is that the experimental spectra of both ILs differ significantly. The [C1C1im]+[NTf2]− spectrum (a) consists of five (roman numbers I–V) characteristic, well-separated resonance features. The [C4C1im]+[I]− IL (b), on the other hand, shows only three resonances (roman numbers I–III), and at least resonances II and III are broader and more overlapping than the high-energy resonances of [C1C1im]+[NTf2]−. The lowest-energy peak I corresponds to a C 1s → π* (see below). It appears for both ILs, being similar in the energetic position, the width and its double-resonance nature. In previous measurements and interpretations,35 a double resonance feature was expected, but not as clearly resolved as in this work.
The simulated spectra are in good overall agreement with experiment. In particular for [C1C1im]+[NTf2]− the main features I–V are well reproduced. It should be noted, though, that the theoretical spectra are too structured, as a consequence of neglected molecular motion which would lead to additional broadening. (Of course that could be mimicked by choosing larger broadening factors. However, then one would also loose details of the double-resonance nature of resonance I, which is why we do not follow this route here.)
The transition potential method allows us to identify those excitation centers (carbon atoms) which contribute to a given, selected resonance. A C-atomwise analysis of spectra is shown in the middle panels of Fig. 2, with color coding and atom numbering as indicated in the lowest panels of the figure.
Considering Fig. 2(b), [C4C1im]+[I]− first, we note that the first resonance (I, around 287 eV) is caused by core excitation of the three C atoms in the imidazolium ring. The double-resonance nature arises from the fact that one C atom (labeled C2 in the figure, red) is chemically somewhat different from the two other C atoms (labeled C4 and C5, green). In particular, C2 has two N atoms as nearest neighbours, while C atoms C4 and C5 have only one. This difference in electrostatic environments leads to a small shift of excitation energies, and hence to two sub-signals. The signals contributing to resonance I are C 1s → π* transitions, and the sub-peak arising from C atom C2 should appear at slightly higher photon energy than the signals arising from C4 and C5, because of the two electronegative N neighbours (atom labels 1 and 3), rather than only one. Our assignment C 1s → π* of signal I arises from the fact that a π* orbital localized at imidazolium is the lowest unoccupied molecular orbital (see ESI†) and that the transition dipole moment is perpendicular to the imidazolium ring.
We see further from Fig. 2(b) that resonances II (at 290.4 eV) and III (292.5 eV) are almost exclusively due to the alkyl side chain carbon atoms (indicated by the orange color, C atoms 6–10). Closer, alkyl C-atom resolved inspection shows that their signals are widely distributed over most of the energy range shown, i.e., many final states ψf are accessible. Their character is not further analyzed in detail here. Furthermore, every alkyl C atom has a slightly different chemical environment, and different dipole matrix elements to different final states are expected. As a consequence, the overall width of signals II and III is larger than the width of resonance I. The intensity of resonance II is about equal to the one for excitation I, in agreement with measurements. However, the intensity of the NEXAFS signal at higher energies is at some variance with experiment: there, resonance III is more intense than II, while in the simulation the spectrum looses intensity after resonance II at energies around 290 eV, and later rises again. These shortcomings may be due to the (too) simple model which neglects dynamical flexibility in particular of the longer alkyl (butyl) chain, and also the presence of neighbouring ions in a real IL.
Apart from this, as a summary the experimentally observed feature I can clearly be assigned to 1s → π* transitions of the ring C atoms, while the two other features II and III are due to side chain carbon atoms.
In Fig. 2(a), lower two panels, an analogous analysis is performed for the [C1C1im]+[NTf2]− system. Again, the double-resonance at around 287 eV (feature I in Fig. 2(a)) can be assigned to the three carbon atoms in the imidazolium ring (here, C atoms 2, 4, and 5, indicated in red and green), and is due to C 1s → π* transitions. The argument for the splitting is the same as before. The resonances marked as II (289.3 eV) and III (290.6 eV) are dominated by the methyl carbons of the side chains, labeled C atoms 6 and 7, respectively, and indicated in orange. There is a small shift of the signal of atom 6 relative to the one of atom 7 (not shown), due to a slightly unsymmetric neighbourhood in our ion pair model. Compared to [C4C1im]+[I]−, resonance II of [C1C1im]+[NTf2]− is also slightly shifted to lower excitation energies (from 290.4 eV to 289.3 eV). This shift is also observed in experiment. We see further that the intensity of resonance II is much lower than the intensity of I in (a), in contrast to (b). This is due to a much shorter side chain in [C1C1im]+[NTf2]− compared to [C4C1im]+[I]−, methyl rather than butyl, with less C atoms contributing to the signal intensity. Such an interpretation is further supported by the fact that experimental C K NEXAFS spectra of [CnC1im]+[NTf2]− show increasing intensity of the “alkyl features” II–V relative to signal I, with increasing n (see Fig. 3). Returning to our two model systems of above, we note that the resonances IV (294.7 eV) and V (298.2 eV) are only found for [C1C1im]+[NTf2]−, but not for [C4C1im]+[I]−. A tempting explanation is that carbon atoms of the organic anion [NTf2]− are responsible for the difference, since the latter are absent in the IL containing [I]−. The simulations indeed support this hypothesis: we observe an intense resonance at 295 eV for [C1C1im]+[NTf2]−, with contributions exclusively from the carbon atoms 8 and 9 (pink) of the anion. The other high-energy resonance, V, however, has also contributions from carbons located in the imidazolium ring. Again, a deeper analysis was not attempted due to the simplicity of the applied model.
We may summarize for [C1C1im]+[NTf2]− that experimental resonance I can clearly be assigned to the imidazolium ring system. The methyl groups mainly contribute intensity to resonances II and III, while resonances IV and V are attributed to the anionic counterion.
4.2 N K NEXAFS resonances of [C1C1im]+[NTf2]− and [C4C1im]+[I]− ionic liquids
A similar analysis to that for C K NEXAFS spectra has been carried out for N K NEXAFS spectra of the two ILs in question. According to Fig. 4, the N K resonance spectra of [C4C1im]+[I]− (b) and [C1C1im]+[NTf2]− (a) are very similar to each other, in contrast to what was found for C K NEXAFS. In the figure, five distinct resonance features I–V are marked and their resonances positions and assignments are summarized in Table 2. For [C4C1im]+[I]−, only two nitrogen atoms (per ion pair) can contribute to the spectrum of Fig. 4(b). Both are located in the imidazolium ring and have a very similar chemical environment, apart from slight asymmetries introduced by side chains of different lengths and the anion. As a consequence, the contributions of individual N atoms to NEXAFS spectra are almost identical, and no atomwise analysis is therefore presented in the figure. Specifically, we assign feature I (at 401.9 eV) to a N 1s → π* and feature II (at 403.5 eV) to a N 1s → σ* excitation. Resonance I is much more intense than resonance II, both in experiment and theory. Experimentally, peaks I and II are separated by 1.6 eV, and by 1 eV according to theory. Generally, the agreement between experiment and theory is not as good as for the C K spectra in Fig. 2, in their higher-energy part (resonances III–V). Nevertheless, theory correctly features a broad distribution of signals in the high-energy region above 404 eV, with peaks having intensities between those of I and II. Experimentally, qualitatively the same holds true for signals III–V. | ||
| Fig. 4 N K NEXAFS spectra of [C1C1im]+[NTf2]− (a) and [C4C1im]+[I]− (b) and their simulation. The upper panels show the experimental NEXAFS spectra (black), with resonances indicated, as well as the theoretical spectra (blue). The lower panel of (a) analyzes atomic contributions to theoretical NEXAFS spectra, with a numbering of nitrogen atoms as indicated in the lowest panel of Fig. 2(a). The corresponding atom-wise analysis for (b), [C4C1im]+[I]−, is not shown, because the two N atoms have practically identical spectra, with the same shape as the total spectrum. | ||
| Resonance | Character | Excitation energy (eV) | Excitation center |
|---|---|---|---|
| [C1C1im]+[NTf2]− | |||
| I | N1s → π* | 402.0 | N1, N3 |
| II | N1s → σ* | 403.7 | N1, N3, N10 |
| III | N1s → σ* | 405.5 | — |
| IV | N1s → σ* | 408.4 | — |
| V | N1s → σ* | 414.8 | — |
| [C4C1im]+[I]− | |||
| I | N1s → π* | 401.9 | N1, N3 |
| II | N1s → σ* | 403.5 | N1, N3 |
| III | N1s → σ* | 405.4 | N1, N3 |
| IV | N1s → σ* | 408.5 | N1, N3 |
| V | N1s → σ* | 415.1 | N1, N3 |
While the experimental N K spectra of [C4C1im]+[I]− and [C1C1im]+[NTf2]− are overall rather similar, a difference occurs regarding the intensity in the energy region right above the first N1s → π* excitation (I). For [C4C1im]+[I]−, the intensity decreases to almost zero according to theory, while we observe a significant intensity for [C1C1im]+[NTf2]−. In experiment, the signal intensity of [C1C1im]+[I]− drops also less between features I and II, compared to [C1C1im]+[NTf2]−. Again, one may expect the organic anion [NTf2]− in [C1C1im]+[NTf2]− to be responsible for this effect. This is indeed supported by the simulation: from the lower panel of Fig. 4(a), we see that the N atom of [NTf2]− (atom 10 in Fig. 2(a)) contributes significantly to the NEXAFS signal in the region slightly above signal I (red curve). Similar results can be found in the paper of Rodrigues et al.,12 where N K NEXAFS spectra of [C4C1im]+[Br]− and [C4C1im]+[NTf2]− are presented.
The green line corresponds to the imidazolium nitrogens 3 and 1 (see Fig. 2(a) for atom numbering), which give weaker signals in the energy region in question.
In summary, the N K NEXAFS spectra of [C1C1im]+[NTf2]− and [C1C1im]+[I]− are similar, with a lowest, most intense feature (I) arising from the core (1s) excitation of the two imidazolium N atoms to the lowest imidazolium π* orbital. One observes small differences slightly above resonance I between the two ILs, which are due to contributions of the N atom of [NTf2]−.
5 Summary and outlook
In this study, we investigated NEXAFS spectra of several imidazolium based ionic liquids. Measured C K NEXAFS spectra were presented for [CnC1im]+[NTf2]− for n = 1, 2, 3, 4, 6, as well as [C4C1im]+[I]−. We further presented simulations of C K and N K NEXAFS spectra based on density functional theory and the transition potential method (TP-DFT), for [C1C1im]+[NTf2]− and [C4C1im]+[I]−, and compared them with experiment.The following observations were made:
• Simulations based on static, single-ion pair models seem to capture the essential features found in the experiment.
• In C K NEXAFS spectra of the two ILs, a characteristic splitting occurs for the lowest-energy, C 1s → π* resonance (I), which arises from slightly different signals for two types of C atoms with slightly different neighbourhoods in the imidazolium ring.
• Contributions to the C K NEXAFS spectra at higher energies originate from the core excitation of C atoms in alkyl chains attached to imidazolium, or from C atoms in the organic anion, [NTf2]−, and can be clearly assigned.
• Also N K NEXAFS spectra can be well explained by theoretical simulations. They are quite similar for the two ILs studied in greatest detail.
• As a small detail, an additional N atom in [NTf2]− has some effect on the intensity in the region between low-energy resonances I and II, as found both in theory and experiment.
The route of using experimental, static crystallographic geometries proved to be quite successful, at least for simulation of NEXAFS spectra of the two investigated ILs. However, not for every IL a crystal structure is available. Therefore, it is clearly desirable to go beyond this approach. As we stated before, a methodology which combines spectral calculation with molecular dynamics (either classical or ab initio) is desirable, although quite expensive. Using such an approach, the influence of intermolecular interactions could also be investigated, as well as thermal motion of ions in the IL. The latter will be particularly important at higher temperatures and/or when flexible building units (such as longer alkyl side chains) are used.
Acknowledgements
C. E. thanks BAM for financial support through Project Ideen_2012_59 as part of the BAM MIS Program. The authors acknowledge support from the team at the BESSY II synchrotron radiation facility and Dr A. Nefedov (Karlsruhe Institute of Technology, KIT) from the HE-SGM Collaborate Research Group as well. M. H. is grateful for financial support from the Austrian Science Found (FWF) through the Erwin-Schrödinger fellowship program (project number J 3471-N28).References
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357 CrossRef CAS PubMed.
- H. Weingärtner, Angew. Chem., Int. Ed., 2008, 47, 654 CrossRef PubMed.
- K. R. J. Lovelock, I. J. Villar-Garcia, F. Maier, H.-P. Steinrück and P. Licence, Chem. Rev., 2010, 110, 5158 CrossRef CAS PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS PubMed.
- R. Caminiti, The structure of ionic liquids, Springer, Cham, 2014 Search PubMed.
- N. Plechkova, Ionic liquids uncoiled critical expert overviews, John Wiley & Sons, Hoboken, New Jersey, 2012 Search PubMed.
- B. Kirchner, Ionic liquids, Springer, Heidelberg New York, 2009 Search PubMed.
- J. Stöhr, NEXAFS Spectroscopy, Springer Berlin Heidelberg, Berlin, Heidelberg, 1992 Search PubMed.
- C. Hardacre, Annu. Rev. Mater. Res., 2005, 35, 29 CrossRef CAS.
- M. Mezger, B. M. Ocko, H. Reichert and M. Deutsch, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3733 CrossRef CAS PubMed.
- T. Nishi, T. Iwahashi, H. Yamane, Y. Ouchi, K. Kanai and K. Seki, Chem. Phys. Lett., 2008, 455, 213 CrossRef CAS.
- F. Rodrigues, D. Galante, G. M. do Nascimento and P. S. Santos, J. Phys. Chem. B, 2012, 116, 1491 CrossRef CAS PubMed.
- F. Rodrigues, G. M. do Nascimento and P. S. Santos, J. Electron Spectrosc. Relat. Phenom., 2007, 155, 148 CrossRef CAS.
- X. Song, Y. Ma, C. Wang, P. M. Dietrich, W. E. S. Unger and Y. Luo, J. Phys. Chem. C, 2012, 116, 12649 CAS.
- E. Darlatt, A. Nefedov, C. H.-H. Traulsen, J. Poppenberg, S. Richter, P. M. Dietrich, A. Lippitz, R. Illgen, J. Kühn, C. A. Schalley, C. Wöll and W. E. Unger, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 621 CrossRef CAS.
- R. Giebler, B. Schulz, J. Reiche, L. Brehmer, M. Wühn, C. Wöll, A. P. Smith, S. G. Urquhart, H. W. Ade and W. E. S. Unger, Langmuir, 1999, 15, 1291 CrossRef CAS.
- M. Holzweber, R. Lungwitz, D. Doerfler, S. Spange, M. Koel, H. Hutter and W. Linert, Chem. – Eur. J., 2013, 19, 288 CrossRef CAS PubMed.
- P. E. Batson, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 2608 CrossRef CAS.
- C. Ehlert, D. Kröner and P. Saalfrank, J. Electron Spectrosc. Relat. Phenom., 2015, 199, 38 CrossRef CAS.
- C. Ehlert, W. E. S. Unger and P. Saalfrank, Phys. Chem. Chem. Phys., 2014, 16, 14083 RSC.
- M. Taillefumier, D. Cabaret, A.-M. Flank and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 195107 CrossRef.
- C. Gougoussis, M. Calandra, A. P. Seitsonen and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 075102 CrossRef.
- C. Kolczewski and K. Hermann, Surf. Sci., 2004, 552, 98 CrossRef CAS.
- W. Zhang, A. Nefedov, M. Naboka, L. Cao and C. Woll, Phys. Chem. Chem. Phys., 2012, 14, 10125 RSC.
- J. D. Holbrey, W. M. Reichert and R. D. Rogers, Dalton Trans., 2004, 2267 RSC.
- M. Nakakoshi, M. Shiro, T. Fujimoto, T. Machinami, H. Seki, M. Tashiro and K. Nishikawa, Chem. Lett., 2006, 35, 1400 CrossRef CAS.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15 CrossRef CAS.
- G. Lippert, J. Hutter and M. Parrinello, Theor. Chem. Acc., 1999, 103, 124 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- M. N. Glukhovtsev, A. Pross, M. P. McGrath and L. Radom, J. Chem. Phys., 1995, 103, 1878 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- L. Triguero, L. Pettersson and H. Ågren, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 8097 CrossRef CAS.
- M. Leetmaa, M. Ljungberg, A. Lyubartsev, A. Nilsson and L. Pettersson, J. Electron Spectrosc. Relat. Phenom., 2010, 177, 135 CrossRef CAS.
- M. Iannuzzi and J. Hutter, Phys. Chem. Chem. Phys., 2007, 9, 1599 RSC.
- E. Apen, A. P. Hitchcock and J. L. Gland, J. Phys. Chem., 1993, 97(26), 6859 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp07434g |
| This journal is © the Owner Societies 2016 |