
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flexibility and conformation of the cocaine aptamer studied by PELDOR†
C. M.
Grytz
a,
A.
Marko
a,
P.
Cekan
b,
S. Th.
Sigurdsson
*b and
T. F.
Prisner
*a
aInstitute of Physical and Theoretical Chemistry and Center of Biomolecular Magnetic Resonance, Goethe University, 60438 Frankfurt am Main, Max-von-Laue-Str. 9, Hessen, Germany. E-mail: prisner@chemie.uni-frankfurt.de
bUniversity of Iceland, Department of Chemistry, Science Institute, Dunhaga 3, 107 Reykjavik, Iceland. E-mail: snorrisi@hi.is
First published on 18th December 2015
Abstract
The cocaine aptamer is a DNA three-way junction that binds cocaine at its helical junction. We studied the global conformation and overall flexibility of the aptamer in the absence and presence of cocaine by pulsed electron–electron double resonance (PELDOR) spectroscopy, also called double electron–electron resonance (DEER). The rigid nitroxide spin label Ç was incorporated pairwise into two helices of the aptamer. Multi-frequency 2D PELDOR experiments allow the determination of the mutual orientation and the distances between two Çs. Since Ç is rigidly attached to double-stranded DNA, it directly reports on the aptamer dynamics. The cocaine-bound and the non-bound states could be differentiated by their conformational flexibility, which decreases upon binding to cocaine. We observed a small change in the width and mean value of the distance distribution between the two spin labels upon cocaine binding. Further structural insights were obtained by investigating the relative orientation between the two spin-labeled stems of the aptamer. We determined the bend angle between this two stems. By combining the orientation information with a priori knowledge about the secondary structure of the aptamer, we obtained a molecular model describing the global folding and flexibility of the cocaine aptamer.
Introduction
Nucleic acid aptamers have been widely used in the rapidly emerging field of biochemical biosensors due to their ability to bind to specific ligands.1–3 DNA and RNA aptamers are typically selected and identified in vitro using the systematic evolution of ligands by exponential enrichment (SELEX) technology.4–6 In recent years, naturally occurring RNAs, termed riboswitches, which specifically bind to small metabolites and thus regulate their gene expression, have been discovered.7,8 Studies of the structure and dynamics of aptamers provide insights into both ligand-induced conformational changes of nucleic acid architectures and principles of their molecular recognition, which in turn may offer opportunities for structure-based drug design strategies for therapeutics.9The DNA-based cocaine aptamer was discovered and subsequently engineered as an aptamer-based fluorescent sensor for cocaine by Stojanovic and coworkers (Fig. 1B).10 This particular construct was designed to be a single strand in the absence of the ligand. The aptamer folds up into a three-way junction, as detected by fluorescence spectroscopy, upon binding to cocaine11 with the dissociation constant in the micromolar range.11–13 We have previously incorporated the rigid cytidine–analogue, spin-labeled nitroxide Ç (Fig. 1A), into single sites of this cocaine aptamer construct and studied its folding upon cocaine binding.12 Since Ç becomes fluorescent upon reduction,14–16 it offered a unique opportunity to study the folding of this aptamer by two spectroscopic techniques, continuous-wave (CW) electron paramagnetic resonance (EPR) and fluorescence spectroscopies. The interpretation of our data was consistent with a model in which the aptamer was partially folded up into two co-axially stacked helices in the absence of cocaine (helices II and III, Fig. 1B). Our data indicated that the co-axially stacked helices would tilt, relative to each other, upon concomitant binding to cocaine and formation of helix I. More recently, Johnson and coworkers described a stem-length dependent binding mechanism of cocaine to the aptamer using a combination of nuclear magnetic resonance (NMR) spectroscopy and isothermal titration calorimetry.13 In contrast to our previous results,12 they did not observe a pre-formed helix III with the construct having a short helix I (Fig. 1B), while all three helices were formed when helix I was extended to seven base-pairs. For all the constructs that they investigated, binding to cocaine led to a well-defined bound-state structure with all three helices formed. However, these NMR studies were performed in the absence of salts, which affects single-strand to duplex equilibria.
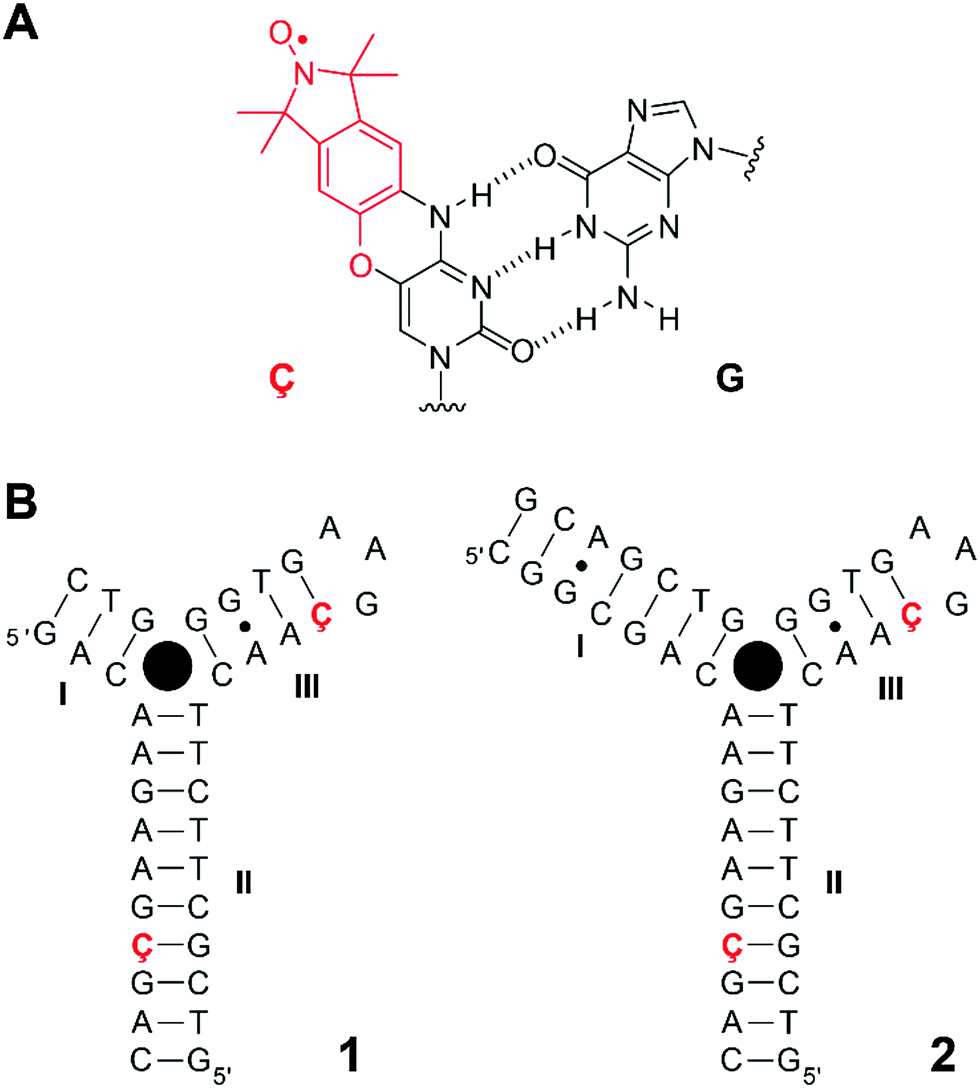 | ||
| Fig. 1 (A) Spin-labeled nucleoside Ç, an analogue of 2′-deoxycytidine (modification in red), base-paired to guanine. (B) Secondary structures of cocaine aptamer constructs 1 and 2 used in this study. Both aptamers have the same sequence and labeling position with the exception of a longer helix I for aptamer 2. The filled black circle represents the ligand cocaine. The positions that were independently labeled with nucleoside Ç are shown in red and bold. | ||
PELDOR (pulsed electron–electron double resonance),17–19 also called DEER (double electron–electron resonance), is an EPR technique that has successfully been used to study the structure and conformational changes of biopolymers.20,21 By recording the PELDOR signal, which oscillates with the frequency νdip of the dipole–dipole interaction between two electron spin centers, one can determine their distance R
R = 3.736·νdip−1/3|3·cos2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Θ − 1|1/3, Θ − 1|1/3, | (1) |
![[R with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/b_i_char_0052_20d1.gif) and the external magnetic field
and the external magnetic field ![[B with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/b_i_char_0042_20d1.gif) . Distances between two spin centers can be determined in the range 2–8 nm with a precision of about 0.3 nm for flexible spin labels.21,22
. Distances between two spin centers can be determined in the range 2–8 nm with a precision of about 0.3 nm for flexible spin labels.21,22
The spin label Ç is an ideal probe for PELDOR studies since it contains a nitroxide fused to the nucleobase14,16 and, therefore, fixes the location and orientation of Ç relative to the framework of the nucleic acid helix14 unlike other PELDOR studies of DNA using flexible spin lables.22–25 Consequentially, a well-defined distance between such a pair of spin labels and their mutual orientations can be extracted from orientation-selective 2D PELDOR experiments (evolution time t vs. pump–probe frequency offset Δν = νA − νB), as we have recently shown for double-stranded DNA molecules.26 The frequency offset averaged PELDOR data reflect only the conformational flexibility of the DNA molecule itself without the need to consider any conformational flexibility of the spin label which is negligibly small in comparison to the DNA mobility.27 Therefore, the orientation between two spin-labeled double-stranded DNA helices can also be extracted from such 2D PELDOR data sets, allowing determination of the global folding and tertiary structure of nucleic acid molecules containing secondary structure elements, such as stems, bulges and hairpin loops.
In this work, we utilize the spin label Ç and 2D PELDOR to investigate conformational changes of the cocaine aptamer. Both constructs (with a short or elongated helix I) undergo a cocaine-induced conformational change, which presumably originates at the junction where cocaine is known to bind.10,11 This is directly visible in the PELDOR time traces by more pronounced oscillations and a small distance change, from the offset-averaged PELDOR time trace applied Tikhonov regularization. A detailed simulation of the 2D PELDOR time traces revealed that there is considerable conformational flexibility in the helical junction, even after binding to cocaine. In most cases so far, PELDOR spectroscopy has been used as a supporting technique to obtain long-range distance restraints for structural studies of large biomolecules. This study underlines the potential of using rigid spin labels to determine directly the flexibility, conformational changes of oligonucleotide molecules and the relative orientations of secondary structural elements which is useful for structure determination.
Results and discussion
PELDOR experiments
Orientation-selective PELDOR experiments at X-band frequencies were performed on the aptamers in absence and presence of cocaine in order to obtain information regarding both the distances and the mutual orientations between two Ç spin labels (Fig. 2 and Fig. S10, ESI†). In a typical PELDOR time trace, the echo signal decays to a non-zero value (1 − λ), where λ is the modulation depth. If angular correlations between both spin labels exist, the modulation depth as well as the oscillation frequency is a function of the frequency offset between pump and probe pulses, reflecting the specific geometry.27 In the 2D PELDOR experiments on the aptamer samples, the varied modulation depth as a function of the frequency offset is easily visible in 3 out of the 4 samples (Fig. 2B–D).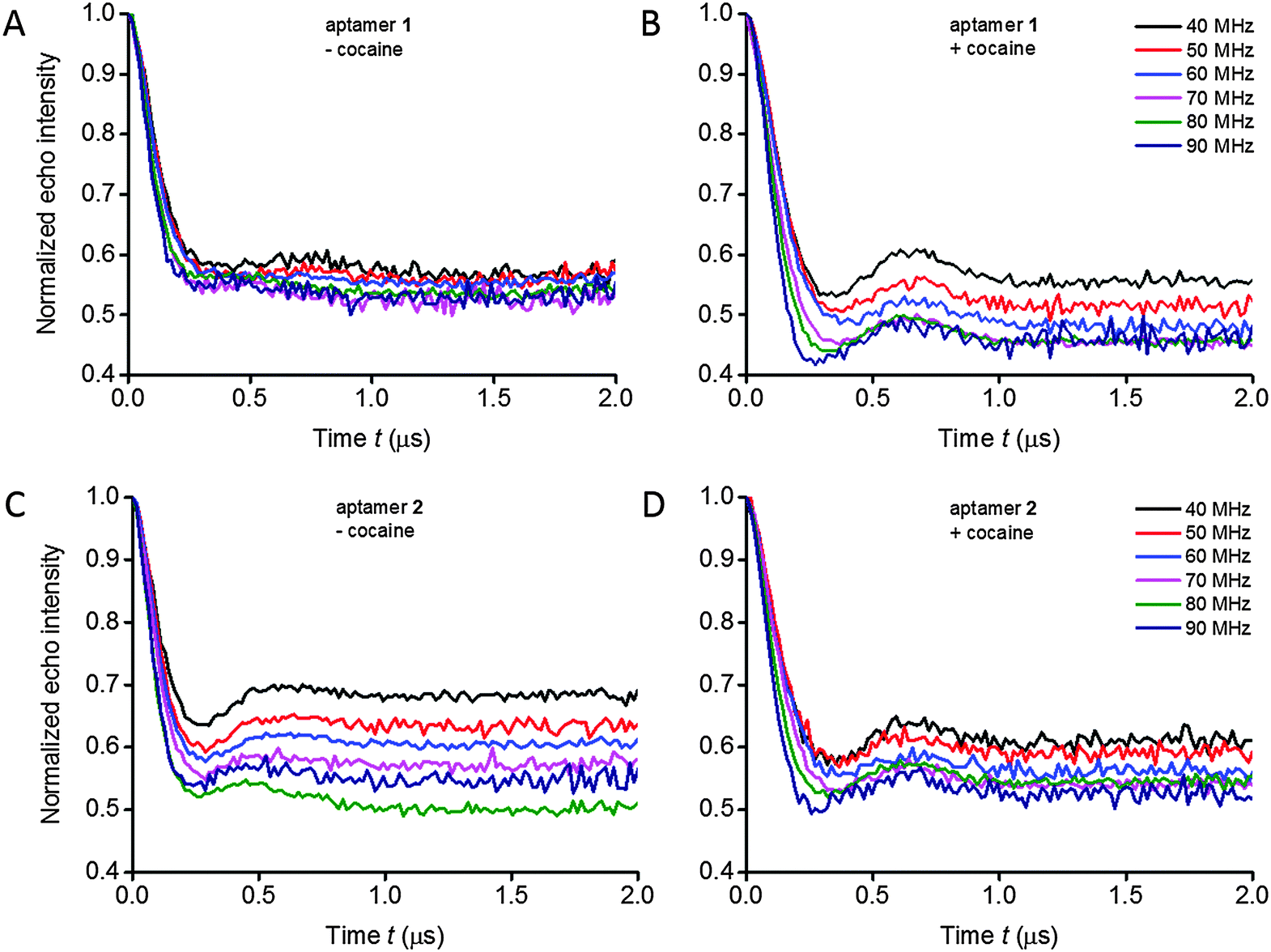 | ||
| Fig. 2 Experimental background-divided PELDOR time traces measured at frequency offsets Δν from 40 to 90 MHz at the X-band for spin-labeled aptamers 1 (A, B) and 2 (C, D) in the absence and presence of cocaine, respectively. | ||
Determination of distances and distance distributions
For distance determination, the X-band PELDOR time traces (Fig. 3) were summed up over the measured frequency offsets Δν. Summing up the time traces from a set of experimental time traces measured at different frequency offsets is a good approximation to average out the orientation information and obtain accurate distances as suggested by Jeschke28 and supported by simulations.27,29 The averaged time traces and the corresponding distance distributions, obtained by Tikhonov regularization are illustrated in Fig. 3. The extracted mean distances are summarized in Table S1 (ESI†).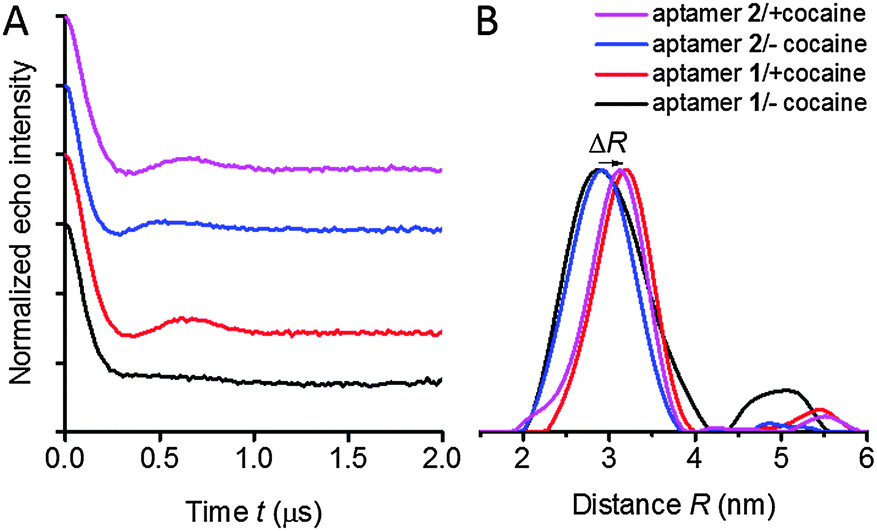 | ||
| Fig. 3 (A) Averaged and background-divided time traces of spin-labeled cocaine aptamers 1 (red, black) and 2 (magenta, blue) in the presence and absence of cocaine at X-band frequencies. The upper three time traces have been shifted in the vertical direction for better display. (B) Distance distributions obtained by Tikhonov regularization for aptamers 1 and 2. The shift of the mean distance ΔR induced by the binding of cocaine is depictured with an arrow. | ||
Two peaks can be observed in the distance distributions obtained by Tikhonov regularization of aptamer 1 in the absence of cocaine (Fig. 3B, black curve), whereas all other samples show a single peak. These two peaks might correspond to two different conformations: the major peak is at 2.9 nm, while another broad and weak peak appears at around 4.8 nm. The observation of two peaks in the distance distributions would be consistent with a previously proposed two-state model for folding of the cocaine aptamer consisting of T- and Y-shaped junctions,12 that are expected to give different distances. However, the reliability of distance distributions depends strongly on the maximum dipolar evolution time tmax.30 Due to this reason, we recorded time traces with an extended dipolar evolution time up to 5 μs with samples of aptamer 1 in the presence and the absence of cocaine in a deuterated solvent. These measurements were performed at the Q-band to avoid artifacts induced by a poor signal-to-noise ratio. Measurements at Q-band frequencies have higher sensitivity compared to X-band and a somewhat reduced orientation selectivity. Fig. S2 in the ESI,† shows that we again observe a second peak in the distance distribution of aptamer 1 without cocaine. The PELDOR data for the cocaine-bound state display also a second peak, but with considerably reduced intensity. A model distribution consisting of one and two Gaussian peaks were applied to fit the averaged PELDOR time traces. Two Gaussians have to be used to fit the averaged PELDOR time trace of aptamer 1 in the absence of cocaine which also supports a second conformer with a larger distance (data not shown). Nevertheless because of the low intensity of the second broad peak the assignment to the proposed two-state model remains somewhat speculative.
Another small but significant effect can be observed upon binding of cocaine for both constructs 1 and 2, namely a shift ΔR of the mean distance of about 0.3 nm, indicating a conformational change upon binding to cocaine. This shift of the maximum peak of the distance distribution is clearly visible for both aptamer samples and for the PELDOR experiments performed at X- and Q-band frequencies in non-deuterated as well as deuterated solvent (Fig. S2 and S11, ESI†). For flexible spin labels such a shift could also be induced by changes of the rotameric state of the spin label itself but that can be excluded for the rigid spin label Ç, again demonstrating the ideal properties of this spin label with respect to quantitative distance determination. However, the change in distance can also be due to orientation selection.
Extracting orientational information from 2D PELDOR data
In the case of a strong offset frequency dependence of the PELDOR time traces, the mutual orientation between the spin labels can be determined in addition to the distance.26,29 Changes in oscillation frequency, modulation depth and damping of the PELDOR signals as a function of the probe frequency offset (Δν varied between 40 and 90 MHz) at X-band frequencies give rise to information about the relative orientation between the two spin labels. For the non-bound as well as for the cocaine-bound state for both aptamer constructs, the oscillation frequency and damping of the PELDOR signals change only weakly as a function of the frequency offset, indicating a high flexibility of the aptamer at the junction between helices II and III. However, all 2D PELDOR data sets (except for aptamer 1 in the absence of cocaine which has the most flexible structure) show a variation of the modulation depth as a function of the offset frequency. A change in the modulation depth by spectral overlap between pump and probe pulses31 accounts for less than 10% under our experimental conditions, which is less than the experimental accuracy. This is consistent with the experimental PELDOR time traces of aptamer 1 without cocaine (Fig. 2A). Therefore, the modulation depth variation of all other samples is attributed to non-random orientations between the two spin labels. Interestingly, the modulation depth variation and dipolar oscillation frequency of the longer aptamer construct 2 in the absence of cocaine already resemble the data set of the cocaine-bound aptamer 1 very much.The dependence of the oscillation frequency on the angle Θ can easily be visualized by the dipolar oscillation formula, whereas the dependence of the modulation depth on the specific geometry and offset frequency requires a quantitative modelling of pumped and probed (A and B) spins for a given spin pair geometry. The modulation depth parameter is also sensitive to experimental parameters such as the MW power, resonator bandwidth, pulse length and B1 homogeneity over the sample length. We have shown that the modulation depth is reproducible within 5–10% error from experiment to experiment, if the experiments are performed under identical conditions.32 Within a 2D PELDOR data set, where each offset measurement is performed immediately after the other, this error will be even lower. Thus, the different variations of the modulation depth as a function of the offset frequency in the experiments on aptamers 1 and 2, with and without cocaine, (Fig. 2) reflect different conformational distributions of relative orientations between the two spin labels of the different samples, rather than experimental errors.
Fitting of the experimental 2D PELDOR data set of the cocaine-bound aptamer 1, the spin label conformers of the PELDOR database library reproduced all PELDOR time traces very well including the modulation depth (Fig. 4A and Fig. S5, ESI†). In total, 100 iterative fitting steps where performed yielding 53 non-redundant spin-label conformers for the cocaine-bound state of this aptamer (Fig. 4B). A clustering of all spin label conformers in the β angle range 60–90° is obvious (as mentioned above β1 and β2 can be interchanged). The same procedure has been applied to the 2D PELDOR data set of aptamer 1 in the non-cocaine bound state. This data show no clear offset dependence and indeed, as can be expected, a much broader orientation distribution results from the fit procedure (see the ESI† for more details about the fit procedure especially with respect to the modulation depth). This confirms that aptamer 1 shows high conformational flexibility in the absence of the ligand. In contrast, the cocaine-bound state of the aptamer is more ordered.
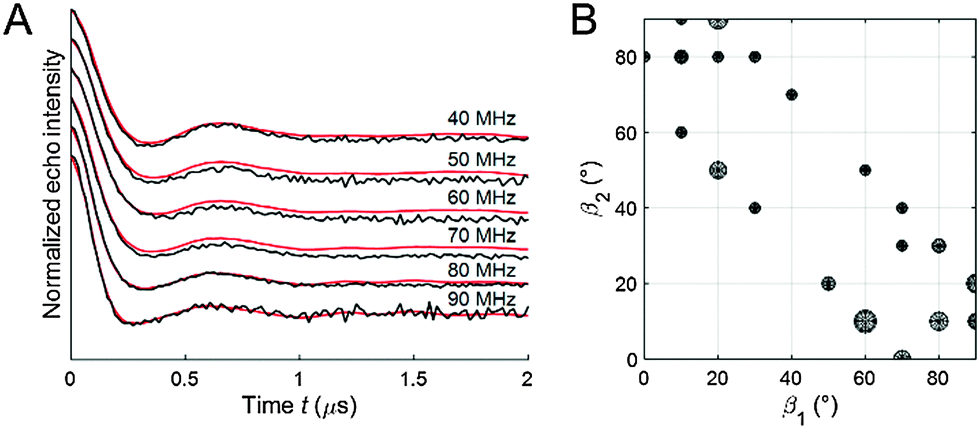 | ||
| Fig. 4 (A) Experimental X-band 2D PELDOR data set for aptamer 1 in the cocaine-bound state (black) and fit by an ensemble of spin label conformers (red). (B) Angles β1 and β2 of the 53 spin label conformers used to fit the experimental 2D PELDOR data set. Each circle represents a spin label conformer and the radius of the circle corresponds to the weight of this conformer in the fit. | ||
As explained above, a priori knowledge about the secondary structure of the three-way junction was subsequently used to construct physically possible solutions for the relative orientation between helix II and III from this angular information. The procedure yielded an ensemble of 1125 possible structural models from 549504 possible helix orientations that are in agreement with the experimental PELDOR data (Fig. S7, ESI†).
For visualization of the extracted relative conformational flexibility between aptamer helices II and III, their geometry was described by the model sketched in Fig. 5A, defined by the bend angle φ (angle between both helix axes) and the two twist angles ψ and θ (rotation of the two nitroxide N–O vectors ![[A with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/b_i_char_0041_20d1.gif) xx,II and xx,III of the respective helix axis). The bend angle can be directly extracted from the angles β1 and β2, whereas the twist angles ψ and θ are only restricted by the geometrical restraints imposed by the a priori knowledge from the secondary structure of the aptamer.
xx,II and xx,III of the respective helix axis). The bend angle can be directly extracted from the angles β1 and β2, whereas the twist angles ψ and θ are only restricted by the geometrical restraints imposed by the a priori knowledge from the secondary structure of the aptamer.
 | ||
| Fig. 5 (A) Definition of bend φ and twist angles (θ and ψ). Both helices are represented as cylinders. More details can be found in the ESI.† (B) A density plot of twist angle θ against bend angle φ for the conformers found from the simulation of the 2D PELDOR data set for aptamer 1 in the cocaine-bound state. (C) A density plot of twist angle ψ against twist angle θ. Color bar specifies the density of the conformers to the solution. The data points are interpolated. | ||
As can be seen for the cocaine-bound aptamer 1 the bend angle is very well conserved within the conformational ensemble. The analysis gives a weighted mean bend angle of 86 ± 19°. The twist angles θ and ψ scatter much more but with some correlation between them. The molecular models using the helices as rigid bodies can be found in the ESI† (Fig. S9).
The geometrical parameters resulting from the algorithm fit to the PELDOR data might not be unique, especially for rather flexible systems, where different ensembles might lead to almost identical good fits.33 To test the robustness of our fit we removed the area of the spin label conformational space where the solutions were concentrated (Fig. 6B, blue squared region) from the library and tried to fit the experimental PELDOR data set by such a restricted library. As can be seen in Fig. 6A, the fit gets significantly worse. Interestingly, the new conformers found from this fit border on the edges of the cut-region (Fig. 6B) approaching the missing structures as much as possible. These results confirm that our found spin label conformations from the PELDOR database fit are essential and not a solution replaceable by another ensemble of spin label conformations.
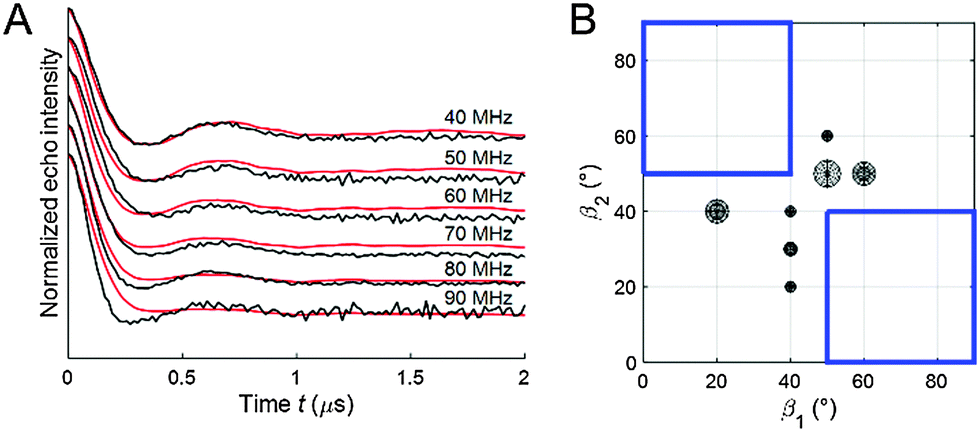 | ||
| Fig. 6 (A) Experimental X-band 2D PELDOR data set for the cocaine bound aptamer 1 (black) and fit by ensemble of spin label conformers (red) from the restricted conformational space library. (B) Angles β1 and β2 of the new spin label conformers found to fit the experimental 2D PELDOR data set. Each circle represents a spin label conformer and the radius of the circle corresponds to the weight of this spin label conformer in the fit. The blue-squared region represents the spin label orientations that were removed from the PELDOR database library. | ||
The cocaine aptamer is a flexible three-way junction
Three-way DNA junctions can take on different shapes, depending on the sequence at the junction.34 When the bases at such junctions are perfectly paired (i.e. when there are no additional unpaired bases), a three-way junction is a Y-shaped molecule without any helix–helix stacking, as originally shown by gel electrophoretic studies35 and later confirmed by Förster resonance energy transfer (FRET) experiments36 and atomic force microscopy.37 Since helices at junctions prefer to be stacked, the Y-shape of junctions appears to break this general folding principle.34 However, additional conformational flexibility that is introduced at the junction, such as unpaired nucleotides, enables coaxial stacking of two helices to form a T-shaped molecule, sometimes along with a Y-shaped conformation, as determined by NMR spectroscopy.34,38,39The rigid spin label Ç, used in this EPR study, contains a nitroxide rigidly fused to the nucleobase (Fig. 1A). Therefore, Ç is sensitive to the motion of the base and thereby to helical motions when the spin label is paired with guanine in a duplex region.14 Thus, in addition to accurate distance measurements between two Ç-labels, the damping of the dipolar oscillations in the PELDOR time traces give detailed information on the conformational flexibility of the DNA molecule.27 We have previously shown that PELDOR signals from duplex DNA that contains two Çs, show well resolved dipolar oscillations (depending on the labelling position and the mutual orientation of the two spin labels).26 Furthermore, the oscillation frequency, modulation depth and damping of the PELDOR signals vary strongly as a function of the probe frequency offset Δν.29 In comparison, the oscillations in the PELDOR signals for both aptamers 1 and 2 are more strongly damped (Fig. 2 and 3) and show much less variation in the oscillation frequency as a function of the frequency offset (Fig. 2), indicating that both have considerable conformational flexibility, albeit less so in the cocaine-bound state. In summary, these data show that the cocaine aptamer can be considered as a fairly flexible junction, especially in the absence of cocaine.
Aptamer 1 shows much higher flexibility in the absence of cocaine compared to aptamer 2
Aptamer 1 is a construct of the cocaine aptamer that was designed by Stojanovic and coworkers to have helices I and III unfolded in the absence of cocaine.10 In aptamer 2, helix I has been extended to stabilize the formation of a three-way junction.11 In the absence of cocaine, construct 2, which contains the elongated helix I, shows a clearly visible oscillation in the averaged PELDOR signal (Fig. 3) and dependence of the modulation depth as a function of the probe frequency offset (Fig. 2), in contrast to the mutant with the short helix (Fig. 2 and 3). Thus, extension of helix I yields a stiffer structure, despite the fact that a rather similar mean inter-helical distance is observed for both aptamers in the absence of cocaine (Fig. 3B). The main difference in the distance distribution functions of the two aptamers without cocaine is therefore the broad peak at large distances which is necessary to simulate the fast damping of the dipolar oscillations of aptamer 1.Our previous studies of the cocaine aptamer using fluorescence and CW-EPR spectroscopy indicated that helices II and III are already formed in the absence of cocaine.12 Furthermore, upon addition of cocaine an increase in fluorescence of Çf, located in helix II immediately at the junction (Fig. 1B), was taken as evidence for helical stacking of aptamer 1 before addition of cocaine.12 A NMR study has also been performed by Johnson and coworkers on a cocaine aptamer that was constructed similar to aptamer 1 of this study, in which helix I contained three base pairs and helix III was a four base-pair stem loop. According to their data, “only a small amount of secondary structure” was formed in the absence of cocaine.13 However, the NMR measurements were performed in the absence of salt, which reduces duplex stability, and may explain the lack of secondary structure. The PELDOR studies of aptamers 1 and 2, performed in the presence of salt, show a well-defined distance at 2.9 nm without cocaine (Fig. 3B), confirming that helix III already forms in the absence of cocaine. The additional peak at higher distances could reflect the presence of two conformations; speculatively T- and Y-shaped junctions12 with a difference in the spin–spin distance between the two spin labels of around 1.9 nm.
The cocaine aptamer undergoes a conformational change and stiffens upon binding to cocaine
The PELDOR data explicitly show that addition of cocaine to both constructs of the cocaine aptamer changes the structure and flexibility of the aptamer. Upon addition of cocaine, a clear visible oscillation appeared in the time traces for aptamer 1 and became more pronounced for aptamer 2 (Fig. 2 and 3). Thus, the conformational flexibility of the cocaine aptamer is reduced and the Y-shaped structure is stabilized by the cocaine upon its binding. Johnson and coworkers also concluded, based on NMR data, that the aptamer rearranges to a “more rigid structure” upon binding to cocaine.13The averaged time traces and distance distributions are almost identical for aptamers 1 and 2 in the presence of cocaine, which shows that elongation of helix I in aptamer 2 does not influence the structure of the cocaine-bound state (Fig. 3 and Fig. S1, ESI†). This is consistent with the previous results that have shown that cocaine stabilizes the formation of helix I and the aptamer.10,12,13
The mean distance between the spin labels in both aptamer constructs increased by about 0.3 nm upon addition of cocaine. This small change in the distance is at the edge of what is detectable by PELDOR. However, since this shift is observed for both aptamer samples and in all our control PELDOR experiments at X- and Q-band frequencies (Fig. S2, ESI†), we judge it to be significant. It is doubtful that this relatively short change in the distance between the two spin labels could have been determined using flexible spin labels, although small differences in the distance have been reported.40 This underscores the advantage of using rigid spin labels such as Ç. This change in the inter-spin distance indicates a cocaine-induced conformational change, which presumably originates at the junction where cocaine is known to bind.10,11 The approximation to average out the orientation information is qualitatively very good.27 Nevertheless, the small shift observed upon cocaine binding is at the limit of resolution of the PELOR method.
The bend angle between helices II and III of the cocaine-bound aptamer can be determined by 2D PELDOR
For aptamer 1, we observed a pronounced change in the modulation depth of the PELDOR signal as a function of the probe frequency offset Δν after cocaine binding, whereas without cocaine no such dependence could be observed (Fig. 2). We interpreted this as non-random orientations between both spin labels in the cocaine-bound state. Due to the rigidity of the Ç spin label, the estimation of the relative orientation of two Ç spin labels and thereby of the two helices of the aptamer is possible. The three angles (α2, β1, β2) and the distance R describing the relative orientation of the two spin labels Ç1 and Ç2 were determined by an iterative fit procedure of the 2D PELDOR data recorded at X-band frequencies. Thus, only 4 out of 6 parameters necessary to fully describe the relative geometry between both spin labels are known. In addition, because the two spin labels are indistinguishable and the spectra are insensitive to an inversion of the magnetic field B0,41 even more symmetry related solutions are possible. Here, we used additional a priori knowledge about the secondary structure of the aptamer to obtain a consistent description of the conformational flexibility of aptamer 1 in its cocaine-bound state. It could be shown that despite the large flexibility of the aptamer, the bend angle φ between helices II and III is rather well-defined to 86 ± 19°.Experimental
Sample preparation and aptamer constructs
For the EPR experiments, the rigid spin label Ç was incorporated pairwise into helices II and III of the cocaine aptamer DNA (Fig. 1B). Two aptamer constructs were prepared, each containing two Ç-spin labels on the same position, but differing in the lengths of helix I.Aptamer 1 consists of a 13-mer with the sequence 5′-d(GACAAGAAGÇGAC) which is paired with a 24-mer with the sequence 5′-d(GTCGCTTCTTCAAÇGAAGTGGGTC). Aptamer 2 contains a longer helix I (seven base pairs instead of three, Fig. 1B). A GT-mismatch in helix III of the original construct12 was substituted by a Watson–Crick base pair GÇ which results in a tighter binding of cocaine.13 Helix II, which is known to form in the absence of cocaine,12 was elongated to enable spin labels to be incorporated into helix II further away from the junction. A two-strand construct was used to give more flexibility in preparation of spin-labeled oligonucleotides and higher yields.
The spin-labeled oligonucleotides were synthesized as previously described.14,16 10 nmol of spin-labeled DNA strands were annealed in 100 μl of 10 mM phosphate buffer (pH 7), 100 mM NaCl and 0.1 mM EDTA. Aptamer samples with the cocaine ligand (5 mM) were prepared in a similar manner. After evaporation of the solvent, each sample was dissolved in 100 μl of 20% non-deuterated or deuterated ethylene glycol/water (H2O or D2O) mixture. Ethylene glycol was used as a cryoprotectant. Before the PELDOR experiments at X-band frequencies, 25 μl of the spin-labeled samples were filled in EPR tubes (2.8 mm OD) and frozen in liquid nitrogen.
2D PELDOR experiments at X-band
Orientation-selective 2D PELDOR experiments at X-band frequencies (9 GHz/0.3 T) were performed at 50 K on a Bruker ELEXSYS E580 pulsed X-band EPR spectrometer. The spectrometer contains a standard flex line probehead, housing a splitring resonator (ER 4118X-MS3), equipped with a continuous flow helium cryostat (CF935) and a temperature control system (ITC 502), the latter two from Oxford Instruments. A commercially available setup (E580-400U) from Bruker was used to couple the second microwave frequency into the microwave bridge. All pulses were amplified via a gated travelling wave tube amplifier (TWT) from Applied Systems Engineering (117X). The over-coupled resonator had a quality factor, Q, of about 100. The dead-time- free four-pulse DEER sequence42 was used:| π/2(νA) − τ1 − π(νA) − (τ1 + t) − π(νB) − (τ2 − t) − π(νA) − τ2 − echo signal |
For the samples with non-deuterated solvent, the detection pulses (νA) were set to 32 ns for both the π and the π/2 pulses. The pump pulse (νB) had a length of 12 ns and was located at the maximum of the anisotropic powder spectrum selecting the nitrogen spin mI = 0 sublevel with random orientations of the nitroxide radical with respect to the external magnetic field (Fig. 7c). This frequency was set as the central resonator frequency. The amplitude of the pump pulse was optimized by the inversion of a Hahn-echo at the frequency νB. The frequency offset Δν = νA − νB was varied between 40 and 90 MHz in steps of 10 MHz, selecting different orientations of the nitroxide radicals with respect to the external magnetic field through the anisotropy of the nitrogen hyperfine tensor A. Therefore, the probe pulses (νA) were slightly off-resonant, but still within the bandwidth of the microwave resonator. Proton modulation was suppressed by averaging of 8 spectra of variable τ1 ranging from τ1 = 132 to 196 ns in steps Δτ1 of 8 ns. The time delay between the second and the third detection pulse (τ1 + τ2) was set to 2.5 μs. In order to eliminate receiver offsets, the π/2 pulse was phase-cycled (0°, 180°). A video amplifier bandwidth of 25 MHz was chosen. The spectra were recorded with a repetition time of the pulse sequence of 4000 μs or 5000 μs. Depending on the frequency offset Δν, the approximate accumulation time for each time trace was usually 2–3 h (for Δν = 40 − 70 MHz) and about 8–15 h (for Δν = 80 and 90 MHz).
Q-band PELDOR
PELDOR experiments at Q-band (33.6 GHz/1.2 T) were performed on an ELEXSYS E580 spectrometer equipped with a Bruker ELEXSYS SuperQ-FT. The DNA was solved in deuterated solvent, which allows elongating the dipolar evolution time to 5 μs21 for a better background-division. The temperature was 50 K, which was controlled by an Oxford continuous flow helium cryostat. A Bruker EN5107D2 resonator was used and the pulsed microwave (mw) power was 10 W, delivered from a solid state mw-amplifier (HA8019). The detection pulses νA were set to 32 ns for the π and π/2 flip angles. The frequency νA was about 50 MHz lower than the cavity resonance. Lengths of 20 and 26 ns were chosen for the pump pulse with frequency νB, which was set on-resonant with the cavity. The time delay τ1 between the first and the second probe pulses was set to an initial value of 124 ns and incremented in 8 steps of 2 ns each to reduce deuterium modulation. The pulse separation between the second and the third probe pulses (τ1 + τ2) was adjusted to 5 μs.Data analysis with respect to distance distribution
For distance determination, a single time trace was generated from the 2D PELDOR data set by summing up all time traces measured with different frequency offsets Δν. Prior to summing up, an exponential background was fitted to the last 2/3 of the PELDOR time traces; the PELDOR trace was then divided by this background function. Data points at the end of the time trace were cut off to avoid artefacts at tmax caused by the overlap between the pump pulse and the second π-pulse of the probe-channel. Tikhonov regularization of the averaged PELDOR signal was performed using the software ‘DeerAnalysis2013’27,43 to obtain the distance information. The regularization parameter α was set to 100.Data analysis with respect to orientation dependence
For the nitroxide molecular frames, the x-axis is parallel to the NO-bond and the z-axis is oriented perpendicular to the nitroxide plane. The y-axis is the cross product of x and z. These axes coincide with the principle axes of the g and the hyperfine interaction tensors, A (Fig. 7A). The relative orientation of two nitroxides 1 and 2 with respect to the common interconnecting dipolar vector is defined by two consecutive Euler rotations: first between the g-frame of nitroxide 2 into the principal axis of the dipolar tensor which is aligned with the interspin axis, D2(α2, β2, γ2) and second from the latter into the g-frame of nitroxide 1, D1(α1, β1, γ1).33 Therefore, the relative orientation of two nitroxide spin labels with respect to the dipolar tensor is characterized by the Euler angles α1, β1, γ1 and α2, β2, γ2 (Fig. S3, ESI†). It should be noted that the relative orientation of the two paramagnetic centers with respect to each other can be characterized using only 5 angles due to the axial symmetry of the dipolar interaction tensor without losing generality. Due to this reason, α1 is set to zero. To completely describe the relative positions of the two spin labels with respect to each other the inter-spin distance, R, must also be known. Further details can be found in the ESI† and ref. 27, 33, 44 and 45.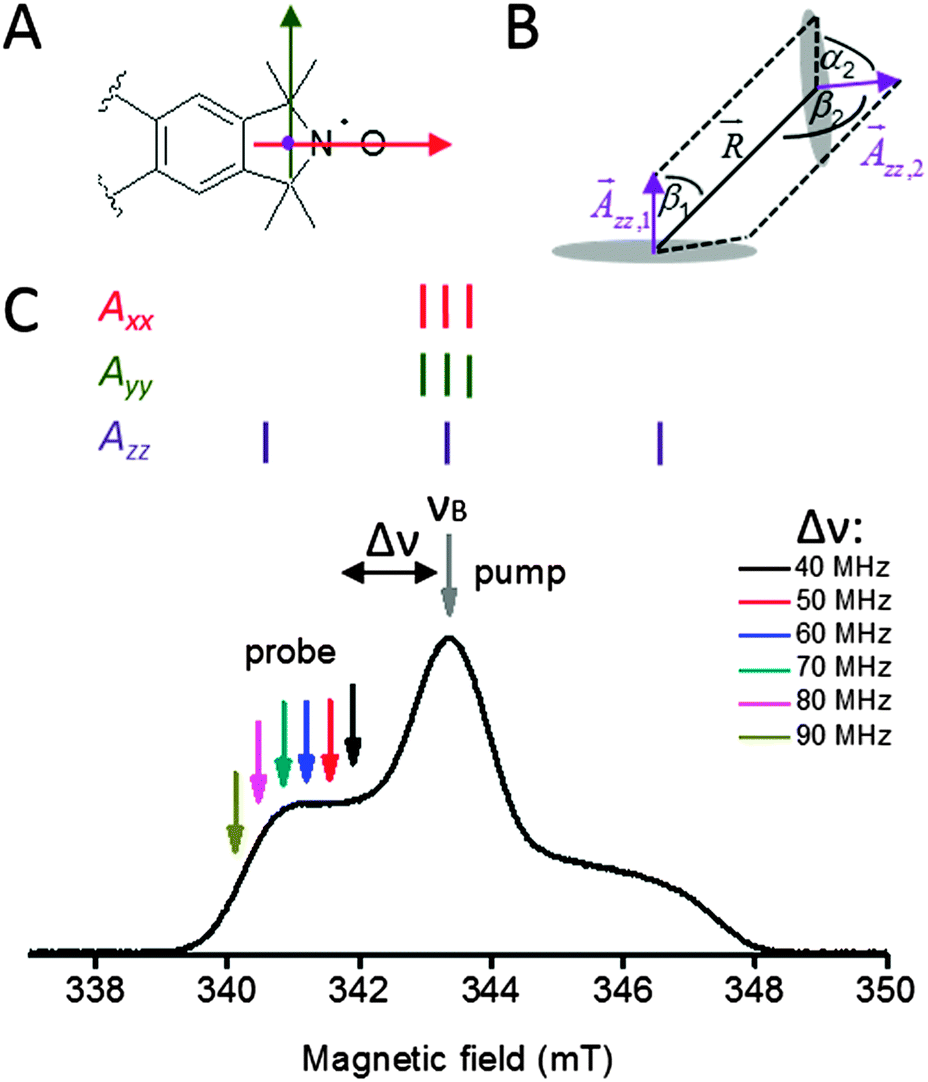 | ||
| Fig. 7 (A) Hyperfine tensor-frame of the nitroxide Ç spin label (14N) A with the principal components x, y and z (orthonormal to the nitroxide plane) marked in red, green and purple, respectively. (B) Three angle restraints and one distance restraint are accessible in an orientation selective PELDOR experiment at X-band frequencies from one spin label pair. β1 and β2 are the angles between the distance vector and the, zz,1 and zz,2 vectors, respectively. α2 is the angle between the plane containing the vectors and zz,1 and the plane containing vectors and zz,2. (C) The nitroxide EPR spectrum with the corresponding 14N hyperfine stick diagram for X-band frequencies. The arrows indicate the fixed pump (νB) frequency and the variable probe (νA) frequencies with offsets ranging from Δν = 40 to 90 MHz. | ||
Quantitative analysis of the orientation dependency of the PELDOR time traces recorded at X-band frequencies was performed using a numerical algorithm to globally fit the 2D PELDOR data set; the algorithm was developed by our group for the application on rigid spin labels.27 In the first step, a library of all 2D PELDOR data sets that can occur at X-band frequencies for nitroxide radical pairs with arbitrary orientations and inter-spin distances in the experimentally accessible range was created. In the second step, an iterative fitting was applied to find an optimal combination of these pre-simulated PELDOR time traces to reproduce the experimental data set. The fitting algorithm searches for an ensemble of spin label conformers that minimize the mean squared deviation between the experimental and set of library data sets:
| ∥n−1(Sr,1 + … + Sr,n−1 + Sr,n) − Sexp∥2 → min | (2) |
At X-band frequencies, only the z-component of the nitrogen hyperfine tensor is resolved as shown in Fig. 7C. Due this reason, only the angles β1, α2, β2 and the distance R (Fig. 7B) have to be varied to create the library of data sets needed for the fitting procedure.
The γ1 and γ2 angles cannot be determined from X-band PELDOR measurements, because of the similar values of Axx and Ayy such that the asymmetry in x and y directions cannot be determined (Fig. 7C). It should be noted however that γ1 and γ2 are required for unique description of the mutual orientation between both Ç labels. This means that while it is possible to obtain the bend angle, φ, between both helices for the X-band 2D PELDOR datasets, the twist angle, θ, of one helix relative to the other cannot be determined. Therefore, it is not possible to define a unique solution from our experimental data. This problem could in principle be solved by performing orientation dependent PELDOR measurements at higher magnetic fields, where the x and y components of the anisotropic g-tensor are spectrally resolved. To achieve this, a frequency of 95 GHz or above would be needed. However, unfortunately the inherent flexibility of the aptamer would lead to a very small modulation depth at this frequency due to variations in the Lamor frequency of the spin pairs. This made these experiments unfeasible. Instead, we used geometrical knowledge about the helices to restrict the conformational space and provide further refinement of the simulation.
Two B-DNA helices were constructed using the web interface software w3dna.46 For helix II the sequence 5′d-(AAGAAGCGAC) paired with 5′d-(GTCGCTTCTT) and for helix III the sequence 5′-d(GTTG) was paired with 5′-d(CAAC). The GA mismatch in helix III was not considered. The rigid nitroxide spin label Ç was modeled into both DNA helices. The coordinates of all atoms were loaded into a custom written Matlab program, which orients the DNA according to the nitroxide spin label orientation found by the fitting algorithm (for nitroxide 1 the Euler angle β1 and for nitroxide 2 the Euler angles α2, β2) and the varied γ1 and γ2 angles. The second Ç labeled helix was additionally translated by the distance R between the two electron spins each located in the middle of the NO-bond. In addition, because the two nitroxide spin labels cannot be distinguished, we considered all possible solutions for spin label 1 in helix II and spin label 2 in helix II and vice versa. Similar, we considered that the nitroxide radical normal (zz) of each spin can point up or down, which turns the helix upside down. Within the analysis, the Ç spin labeled DNA helices were treated as rigid bodies. With the iterative fit algorithm, the non-redundant parameter sets (α2, β1, β2, R), corresponding to spin label conformers were found to best fit the experimental 2D PELDOR data set. From this data set all possible helix orientations were calculated, which yielded all possible helix orientations.
Fortunately, not all of these structure models for the cocaine-bound state are compatible with the basic geometrical knowledge of the aptamer structure and can therefore be discarded. All structures were filtered by three conditions, which are based on the secondary structure of the cocaine aptamer. The criterion for choosing the threshold conditions was that the experimental PELDOR data could still be satisfactorily simulated with the remaining bundle of conformers. From the secondary structure of the aptamer it is known that helices II and III have to be covalently bound. One can imagine that the junction might stretch the pitch height. Therefore, we chose a threshold value that allows stretching of the helix by maximum 0.8 nm. Structural models with a separation distance between the P-atom at the 5′ end of Cytosin24 and 3′OH at Thymin23 longer than 0.8 nm were filtered out. Consideration of the maximum and the minimum space required by the non-labeled helix I (diameter of a B-DNA helix 2 nm) completed the geometry of the three-way junction. All structures with a separation between the 3′OH at the 3′ end of helix III and the P-atom at the 5′ end of helix II (deoxyadenosine 14), beyond 1.5 nm and 2.7 nm were filtered out. Moreover, steric clashes between helices II and III were prevented. The plugin, “Show bumps”, was implemented in PyMOL to calculate the van-der Waals repulsion between the helices (http://www.pymolwiki.org/index.php/Show_bumps). Again all structural models with steric conflicts caused by the repulsion between the two helices were discarded. Using these three filter threshold conditions reduces the number of possible structures.
Conclusions
In summary, we studied the conformational changes of two doubly Ç-spin-labeled cocaine aptamer constructs upon cocaine binding by PELDOR spectroscopy. A lack of orientation dependence in the PELDOR experiment of aptamer 1 in the absence of cocaine in combination with a strong damping of the oscillation showed that the aptamer is rather flexible. However, the flexibility was reduced upon addition of cocaine, indicating a change in the global conformation. Quantitative simulations of the 2D PELDOR data as a function of the offset frequency, together with a priori knowledge of the secondary structure of the aptamer, allowed determination of the conformational flexibility of the aptamer in its cocaine-bound state.This study is the first example of applying PELDOR with rigid spin labels to study the changes in the structure and conformational flexibility of a nucleic acid junction upon ligand binding. The results clearly show the advantage of using rigid spin labels to investigate such systems, since the flexibility of the spin label itself does not need to be taken into account, allowing more accurate distance determination and unravelling additional information about the relative orientation of the two spin labels with respect to each other. This yielded conclusive information on the structure and flexibility of an aptamer from a single pair of spin label positions. In spite of no available atomic resolution structure of the investigated cocaine aptamer, a molecular model describing the global folding and flexibility of the cocaine aptamer using the helices as rigid bodies was obtained. This work lays the foundation to develop an efficient EPR structure calculation program for large nucleic acids or nucleic acid–protein complexes.
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft DFG (SFB 902 Molecular Principles of RNA-based Regulation, Cluster of Excellence Frankfurt Macromolecular Complexes) and the Icelandic Research Fund [120001022]. T. F. Prisner and C. M. Grytz gratefully acknowledge support by the Fond der Chemischen Industrie. We thank Dnyaneshwar B. Gophane for help in sample preparation.References
- D. H. J. Bunka and P. G. Stockley, Nat. Rev. Microbiol., 2006, 4, 588–596 CrossRef CAS PubMed.
- G. Mayer, Angew. Chem., Int. Ed., 2009, 48, 2672–2689 CrossRef CAS PubMed.
- S. P. Song, L. H. Wang, J. Li, J. L. Zhao and C. H. Fan, TrAC, Trends Anal. Chem., 2008, 27, 108–117 CrossRef CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed.
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CAS.
- A. D. Ellington and J. W. Szostak, Nature, 1992, 355, 850–852 CrossRef CAS PubMed.
- A. Serganov and E. Nudler, Cell, 2013, 152, 17–24 CrossRef CAS PubMed.
- D. M. J. Lilley, Curr. Opin. Struct. Biol., 2005, 15, 313–323 CrossRef CAS PubMed.
- D. J. Patel, Curr. Opin. Chem. Biol., 1997, 1, 32–46 CrossRef CAS PubMed.
- M. N. Stojanovic, P. de Prada and D. W. Landry, J. Am. Chem. Soc., 2000, 122, 11547–11548 CrossRef CAS.
- M. N. Stojanovic, P. de Prada and D. W. Landry, J. Am. Chem. Soc., 2001, 123, 4928–4931 CrossRef CAS PubMed.
- P. Cekan, E. O. Jonsson and S. Th. Sigurdsson, Nucleic Acids Res., 2009, 37, 3990–3995 CrossRef CAS PubMed.
- M. A. D. Neves, O. Reinstein and P. E. Johnson, Biochemistry, 2010, 49, 8478–8487 CrossRef CAS PubMed.
- N. Barhate, P. Cekan, A. P. Massey and S. Th. Sigurdsson, Angew. Chem., Int. Ed., 2007, 46, 2655–2658 CrossRef CAS PubMed.
- P. Cekan and S. Th. Sigurdsson, Chem. Commun., 2008, 3393–3395 RSC.
- P. Cekan, A. L. Smith, N. Barhate, B. H. Robinson and S. Th. Sigurdsson, Nucleic Acids Res., 2008, 36, 5946–5954 CrossRef CAS PubMed.
- A. D. Milov, K. M. Salikohov and M. D. Shirov, Fiz. Tverd. Tela, 1981, 23, 975–982 CAS.
- A. D. Milov, A. B. Ponomarev and Y. D. Tsvetkov, Chem. Phys. Lett., 1984, 110, 67–72 CrossRef CAS.
- R. G. Larsen and D. J. Singel, J. Chem. Phys., 1993, 98, 5134–5146 CrossRef CAS.
- O. Schiemann and T. F. Prisner, Q. Rev. Biophys., 2007, 40, 1–53 CrossRef CAS PubMed.
- G. Jeschke, Annu. Rev. Phys. Chem., 2012, 63, 419–446 CrossRef CAS PubMed.
- O. Schiemann, N. Piton, Y. G. Mu, G. Stock, J. W. Engels and T. F. Prisner, J. Am. Chem. Soc., 2004, 126, 5722–5729 CrossRef CAS PubMed.
- Q. Cai, A. K. Kusnetzow, W. L. Hubbell, I. S. Haworth, G. P. C. Gacho, N. Van Eps, K. Hideg, E. J. Chambers and P. Z. Qin, Nucleic Acids Res., 2006, 34, 4722–4730 CrossRef CAS PubMed.
- G. Sicoli, G. Mathis, S. Aci-Seche, C. Saint-Pierre, Y. Boulard, D. Gasparutto and S. Gambarelli, Nucleic Acids Res., 2009, 37, 3165–3176 CrossRef CAS PubMed.
- N. A. Kuznetsov, A. D. Milov, N. P. Isaev, Y. N. Vorobjev, V. V. Koval, S. A. Dzuba, O. S. Fedorova and Y. D. Tsvetkov, Mol. BioSyst., 2011, 7, 2670–2680 RSC.
- O. Schiemann, P. Cekan, D. Margraf, T. F. Prisner and S. Th. Sigurdsson, Angew. Chem., Int. Ed., 2009, 48, 3292–3295 CrossRef CAS PubMed.
- T. F. Prisner, A. Marko and S. Th. Sigurdsson, J. Magn. Reson., 2015, 252, 187–198 CrossRef CAS PubMed.
- A. Godt, M. Schulte, H. Zimmermann and G. Jeschke, Angew. Chem., Int. Ed., 2006, 45, 7560–7564 CrossRef CAS PubMed.
- A. Marko, V. Denysenkov, D. Margraf, P. Cekan, O. Schiemann, S. Th. Sigurdsson and T. F. Prisner, J. Am. Chem. Soc., 2011, 133, 13375–13379 CrossRef CAS PubMed.
- G. Jeschke and Y. Polyhach, Phys. Chem. Chem. Phys., 2007, 9, 1895–1910 RSC.
- K. M. Salikhov and I. T. Khairuzhdinov, Appl. Magn. Reson., 2015, 46, 67–83 CrossRef CAS.
- B. E. Bode, D. Margraf, J. Plackmeyer, G. Durner, T. F. Prisner and O. Schiemann, J. Am. Chem. Soc., 2007, 129, 6736–6745 CrossRef CAS PubMed.
- A. Marko and T. F. Prisner, Phys. Chem. Chem. Phys., 2013, 15, 619–627 RSC.
- D. M. J. Lilley, Q. Rev. Biophys., 2000, 33, 109–159 CrossRef CAS PubMed.
- D. R. Duckett and D. M. J. Lilley, EMBO J., 1990, 9, 1659–1664 CAS.
- F. Stuhmeier, J. B. Welch, A. I. H. Murchie, D. M. J. Lilley and R. M. Clegg, Biochemistry, 1997, 36, 13530–13538 CrossRef CAS PubMed.
- L. S. Shlyakhtenko, V. N. Potaman, R. R. Sinden, A. A. Gall and Y. L. Lyubchenko, Nucleic Acids Res., 2000, 28, 3472–3477 CrossRef CAS PubMed.
- V. Thiviyanathan, B. A. Luxon, N. B. Leontis, N. Illangasekare, D. G. Donne and D. G. Gorenstein, J. Biomol. NMR, 1999, 14, 209–221 CrossRef CAS PubMed.
- B. Wu, F. Girard, B. van Buuren, J. Schleucher, M. Tessari and S. Wijmenga, Nucleic Acids Res., 2004, 32, 3228–3239 CrossRef CAS PubMed.
- X. Zhang, A. C. Dantas Machado, Y. Ding, Y. Chen, Y. Lu, Y. Duan, K. W. Tham, L. Chen, R. Rohs and P. Z. Qin, Nucleic Acids Res., 2014, 42, 2789–2797 CrossRef CAS PubMed.
- C. Abe, D. Klose, F. Dietrich, W. H. Ziegler, Y. Polyhach, G. Jeschke and H. J. Steinhoff, J. Magn. Reson., 2012, 216, 53–61 CrossRef CAS PubMed.
- R. E. Martin, M. Pannier, F. Diederich, V. Gramlich, M. Hubrich and H. W. Spiess, Angew. Chem., Int. Ed., 1998, 37, 2834–2837 CAS.
- G. Jeschke, V. Chechik, P. Ionita, A. Godt, H. Zimmermann, J. Banham, C. R. Timmel, D. Hilger and H. Jung, Appl. Magn. Reson., 2006, 30, 473–498 CrossRef CAS.
- B. Endeward, A. Marko, V. P. Denysenkov, S. Th. Sigurdsson and T. F. Prisner, Methods Enzymol., 2015, 564, 403–425 CAS.
- A. Marko, D. Margraf, H. Yu, Y. Mu, G. Stock and T. Prisner, J. Chem. Phys., 2009, 130, 064102 CrossRef CAS PubMed.
- G. H. Zheng, X. J. Lu and W. K. Olson, Nucleic Acids Res., 2009, 37, W240–W246 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-and Q-band data for aptamer 1 in deuterated solvent, details about the analysis, structural models, and twist and bend angle calculations. See DOI: 10.1039/c5cp06158j |
| This journal is © the Owner Societies 2016 |