
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescent DNA probes at liquid/liquid interfaces studied by surface second harmonic generation†
Giuseppe
Licari
a,
Pierre-François
Brevet
b and
Eric
Vauthey
*a
aDepartment of Physical Chemistry, University of Geneva, 30 Quai Ernest-Ansermet, CH-1211 Geneva 4, Switzerland. E-mail: eric.vauthey@unige.ch
bInstitut Lumière Matière, UMR CNRS 5306, Université Claude Bernard, Lyon 1, Campus LyonTech – La Doua, 10 Rue Ada Byron, 69622 Villeurbanne cedex, France
First published on 22nd December 2015
Abstract
The properties of a series of oxazole yellow dyes, including the dicationic YOPRO-1 and its homodimeric parent YOYO-1 and two monocationic dyes (YOSAC-1 and YOSAC-3), have been investigated at the dodecane/water interface using stationary and time-resolved surface second harmonic generation (SSHG) combined with quantum chemical calculations. Whereas YOYO-1 exists predominantly as a H-dimer in aqueous solution, the stationary SSHG spectra reveal that such dimers are not formed at the interface. No significant H-aggregation was observed with YOPRO-1, neither in solution nor at the interface. In the case of the monocationic YOSAC dyes, a distinct SSHG band due to H-aggregates was measured at the interface, whereas only weak aggregation was found in solution. These distinct aggregation behaviors can be explained by the different orientations of the dyes at the interface, as revealed from the analysis of polarization-resolved experiments, the doubly-charged dyes lying more flat on the interface than the singly charged ones. Although YOYO-1 and YOPRO-1 do not form H-dimer/aggregates at the interface, time-resolved SSHG measurements point to the occurrence of intra- and intermolecular interactions, respectively, which inhibit the ultrafast non-radiative decay of the excited dyes via large amplitude motion, and lead to a nanosecond excited-state lifetime. The distinct behavior evidenced here for YOYO-1 and YOSAC dyes points to their potential use as fluorescent or SHG interfacial probes.
Introduction
The synthesis of oxazole yellow (YO) cyanine dyes and their homodimeric forms (YOYO) was a major breakthrough in molecular biology, because fluorescence detection could achieve, for the first time, sensitivity to DNA similar to that of radioactive probes.1,2 Spectroscopic investigations of these cyanine dyes in solution have evidenced two mechanisms responsible for their >100-fold increase of fluorescence intensity upon intercalation into ds-DNA.3–6 The first arises from the loss of mobility around the methine bridge upon binding to DNA, which inhibits the non-radiative deactivation of the excited state through large amplitude motion.3,4,7 The second mechanism mostly concerns the homodimeric dyes, which tend to self-aggregate into non-fluorescent H-dimers in aqueous solution.6–8 The excitonic interaction in the dimers results in delocalization of the electronic excitation and splitting of the energy levels into two excitonic states: a dark state at lower energy and a bright state at higher energy.9 As a consequence, H-dimers are characterized by an absorption band that is blue-shifted relatively to that of the monomer and by an absence of fluorescence, because photoexcitation is followed by an ultrafast internal conversion from the bright to dark excitonic state. Upon binding to DNA, the dimers are disrupted, both the excitonic interaction and the non-radiative decay via large amplitude motion are suppressed, and fluorescence is operative. The removal of the excitonic interaction upon bis-intercalation into DNA was suggested to be due to not only the larger distance between the two chromophoric units but also their quasi-orthogonal mutual orientation.8The aforesaid ability of cyanines to self-organize is invaluable for many applications and has been extensively studied in solution, where dimers and higher-order H- or J-type aggregates were observed.10–18 Moreover, the aggregation properties of several cyanine derivatives were also studied at liquid/water or air/water interfaces using surface second harmonic generation (SSHG).19–26 Steady-state, time-resolved (TR) SSHG as well as SHG microscopy were applied to obtain information about their adsorption, orientation and dynamic properties. These studies revealed that cyanines, like other surface-active organic molecules,27–32 form aggregates at the interface already at concentrations at which no aggregation takes place in the bulk phase.
To the best of our knowledge, YO and YOYO DNA probes have never been investigated so far at liquid/water interfaces. Additionally to exhibit fluorescence enhancement upon intercalation into DNA, a good fluorescent probe should be soluble in water and have a high affinity for the low-polarity environment created by the DNA base-pair stack. Therefore, fluorescence DNA intercalators can be expected to have a high affinity for liquid/water interfaces. To test this idea and evaluate whether such dyes could be good interfacial probes, we have carried out an investigation of four cyanine dyes belonging to the YO family (Chart 1) at the dodecane/water interface using both stationary and time-resolved SSHG measurements combined with quantum chemical calculations. These dyes differ in their electric charge, the number of chromophoric units and the hydrophobic substituents. YOPRO-1 bears two positive charges, whereas its homodimeric relative YOYO-1 has four positive charges. YOSAC-1 and YOSAC-3 are only singly charged and possess a hydrophobic chain that should confer them higher surface-activity. Here, we show that these dyes generate intense resonantly enhanced SSHG signal at the dodecane/water interface that could make them attractive two-photon probes for biological imaging in the near infrared region.33–35 Furthermore, their aggregation behaviour is strongly dye-dependent. With YOYO-1, the H-dimer form that predominates in aqueous solution is no longer present at the interfaces. On the other hand, the YOSAC dyes exhibit a much higher propensity to aggregate at the interface than in solution. These results will be rationalized in terms of different orientations of the dyes when adsorbed at the interface.
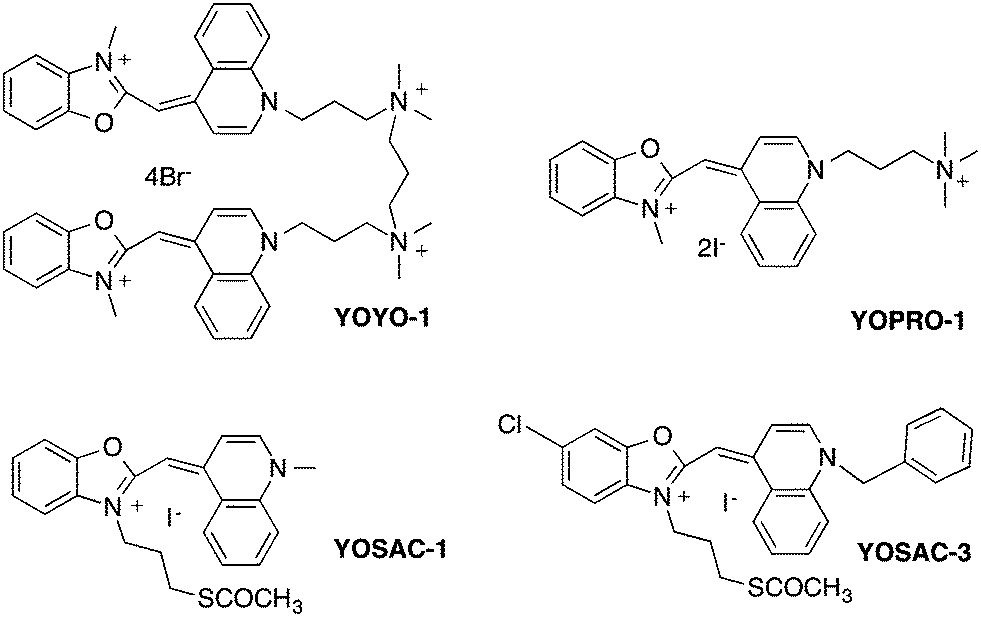 | ||
| Chart 1 Structure of the fluorescent DNA probes. | ||
Experimental
Samples
The dyes were obtained from Prof. T. Deligeorgiev (Faculty of Chemistry, University of Sofia) and were used as received. Their synthesis has been described in ref. 36 and 37. Stock solutions of the dyes were prepared in DMSO (Acros Organics, spectroscopic grade) at 2 mM concentration and the samples were freshly prepared from the stock solutions before each measurement. Dodecane (DOD, 99+%) was purchased from Alfa Aesar, phosphate-buffered saline (PBS, composition: 2.7 mM KCl, 1.5 mM KH2PO4, 137.9 mM NaCl, and 8.1 mM Na2HPO4·7H2O) from Invitrogen and ethylenediaminetetraacetic acid (EDTA) disodium salt dihydrate from AppliChem. The PBS solutions also contained 1 mM EDTA. Double-stranded salmon sperm DNA was purchased from Sigma.Absorption spectra were recorded on a Cary 50 spectrophotometer (Varian) using 1 cm quartz cells. The samples for SSHG measurements were prepared by pouring 10 mL of aqueous dye solution into a 4 × 4 × 4 cm2 quartz cell and then slowly adding 12 mL of dodecane. The experiments were performed at 20 ± 1 °C. For concentration-dependent SSHG measurements, the concentration was changed in situ by adding variable amounts of dye into the sample with a syringe. Such an addition leads to a rise of the interface plane by at most 60 μm. With the 500 mm lens used to focus the probe beam, the Rayleigh length is around 20 mm. Therefore, such a small change in the position of the interface has a negligible effect on the SSHG signal.
SSHG measurements
The SSHG setup has been already described in detail previously.38,39 Shortly, the near-infrared probe pulses (∼100 fs, ∼1 μJ at the interface) were generated using a collinear optical parametric amplifier (TOPAS-C, Light Conversion) and their polarization was controlled by a half-wave plate. They were focused onto the interface at an angle of incidence of about 70°. The pump pulses (∼80 fs, ∼1 μJ at the interface) used in TR-SSHG were produced by a non-collinear optical parametric amplifier (TOPAS White, Light Conversion) and were circularly polarized to avoid photoselection. Both TOPASs were fed by the output of a 1 kHz Ti:Sapphire amplified system (Solstice, Spectra-Physics) centred at 800 nm. The SSHG signal was focused onto the entrance slit of a Czerny–Turner spectrograph (Shamrock 163, Andor) and detected by a multipixel cooled CCD camera (Newton 920, Andor). The SSHG intensity was checked to scale quadratically with the probe intensity. The non-resonant signal coming from the bare interface was found to be negligibly small with respect to the resonant signal coming from the dye and was not subtracted. The raw TR-SSHG profiles were transformed into S(t) profiles by first taking the square root of the measured signal to obtain a quantity that scales linearly with the concentration of the probed molecule as well as with the modulus of the second-order nonlinear susceptibility, |χ(2)|. This procedure could be done here because the SSHG signal is purely resonant. The photoinduced change in the resulting intensity was then normalized to −1, since in the present measurements, photoexcitation always resulted in a decrease of the signal. The usual polarization nomenclature will be used: p for parallel polarization with respect to the plane of incidence and s for perpendicular polarization.The SSHG spectra were recorded by scanning the TOPAS-C with 2 or 2.5 nm steps. The SSGH spectra of the dyes were corrected for the wavelength dependence of the setup using as reference the SSHG signal measured with a silver mirror immersed in dodecane (Thorlabs, see reflectance in Fig. S11 in the ESI†).
If not specified, the SSHG spectra were recorded at m/s polarization geometry, where m stands for mixed polarization (45° with respect to the plane of incidence) and designates the polarization of the probe field, whereas s is the measured polarization component of the SSHG signal. This geometry was chosen arbitrarily among the three most used polarization geometries (m/s, p/p and s/p). Furthermore, the raw SSHG spectra were transformed into S(λ) spectra by taking the square root of intensity and by subsequent normalization, so that S(λ) is proportional to |χ(2)|.
Quantum chemical calculations
All the calculations were done at the density functional theory (DFT) level using the Gaussian 09 (Rev. C) package.40 For geometry optimizations, the Becke's three-parameter exchange functional41 and the Lee–Yang–Parr (LYP) correlation functional in the form of the B3LYP functional,42 as well as tight convergence criteria were employed. For the prediction of electronic response properties, such as vertical excitations and hyperpolarizabilities, the Coulomb-attenuating hybrid exchange–correlation functional (CAM-B3LYP) with improved long-range properties was used with the following functional parameters: μ = 0.33, α = 0.15 and β = 0.37.43 This method has already been successfully used to compute the electronic (hyper)polarizability of organic compounds.44–47 Moreover, contrary to the B3LYP functional, it does not predict low-lying charge-transfer states for the molecules investigated here. As an extended basis set is required for computing hyperpolarizability,48–50 the Pople 6-311++G(d,p) basis set with diffuse and polarization functions was used throughout. Water solvation was treated implicitly through the SMD model proposed by Truhlar and coworkers based on the integral-equation-formalism polarized continuum model (IEF-PCM) implemented in Gaussian 09.51 The structure of the YO monomer was re-optimized at CAM-B3LYP/6-311++G(d,p) in order to obtain the ground-state transition dipole moment at the same level of theory as the hyperpolarizability.The electronic transition energies were obtained from time-dependent DFT (TD-DFT),52–54 including solvent effects with the PCM model. Frequency-dependent hyperpolarizabilities, β(−2ω;ω,ω), were computed in the EFISH orientation through the Coupled-Perturbed Hartree–Fock procedure.55–57 A comprehensive discussion on the second-order optical response can be found in ref. 50. For each hyperpolarizability tensor element, a total number of 80 frequencies were calculated in a range centred at the most intense vertical transition predicted by the TD-DFT calculations. The near-IR spectral ranges were adapted according to the system: 0.0520–0.0582 a.u. (1.41–1.58 eV) for YO, 0.0530–0.0595 a.u. (1.44–1.62 eV) for the H-dimer and 0.0516–0.0580 a.u. (1.40–1.58 eV) for the J-dimer. The real part of the hyperpolarizability obtained from the calculations was converted into the imaginary part using the Kramers–Kronig relationship. Only the electronic contribution to the first hyperpolarizability was considered and the pure vibrational and zero-point vibrational average contributions were neglected.
Results and discussion
SSHG spectra
The electronic absorption spectra of diluted aqueous solutions of YO cyanine dyes are characterized by a low-energy band arising from the S1 ← S0 transition with a maximum between 470 and 490 nm and with a shoulder at shorter wavelength due to a vibronic transition.3 Very similar spectra are observed with the homodimer YOYO-1 in organic solvents (Fig. 1 and 2).6–8 In aqueous solution, however, the absorption band culminates at 458 nm with a secondary maximum at 485 nm. The 458 nm band has been shown to originate from intramolecular YOYO-1 H-dimers absorbing coincidentally at a wavelength similar to that of the vibronic transition of the non-aggregated form.6–8 Addition of ds-DNA results in the suppression of the H-dimer band and in a spectrum close to that measured in organic solvents. This effect arises from the bis-intercalation of YOYO-1 into DNA and the ensuing elimination of the excitonic interaction between the two chromophoric units.8 Aggregation can also be observed, although to a smaller extent, with the YOSAC dyes as a weak growth of the relative absorbance on the blue side of the absorption band upon increasing concentration (Fig. S4, ESI†).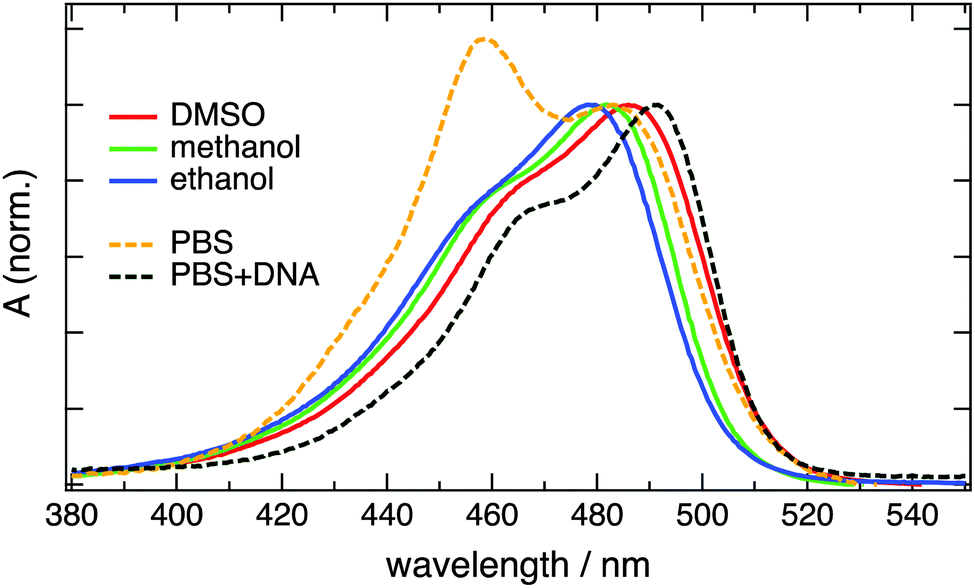 | ||
| Fig. 1 Electronic absorption spectra measured with 2 μM YOYO-1 in various solvents. The concentration of DNA corresponds to a base-pair/dye ratio >5. | ||
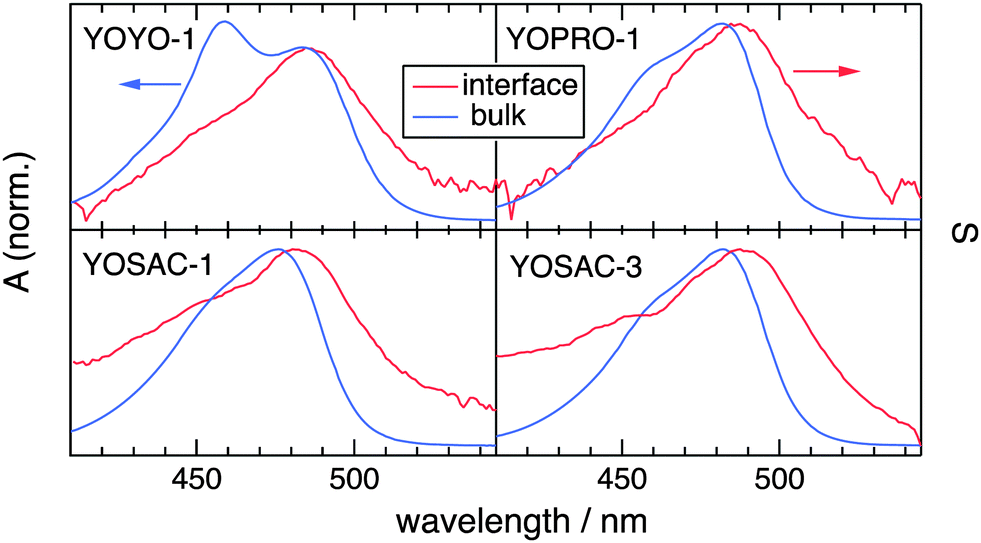 | ||
| Fig. 2 Comparison of the electronic absorption and SSHG spectra, S(λ), of the YO dyes in aqueous solution and at the dodecane/water interface, respectively. The aqueous phase is water for all compounds except for YOPRO-1 for which PBS was used. The concentration was 10 μM for YOPRO-1 and 5 μM for the other dyes. | ||
Fig. 2 shows a comparison of the electronic absorption and SSHG spectra, S(λ), of the dyes in aqueous solution and at the dodecane/water interface, respectively. The measurements with YOPRO-1 were performed in PBS to obtain a higher SSHG signal (see below). In principle, the SSHG spectrum should be compared with the product of the one- and two-photon absorption spectra.58 However, one- and two-photon absorption spectra are very similar for molecules far from being centrosymmetric,59–61 which are thus polar and characterized by a significant hyperpolarizability, β. Since the cyanines studied here are polar and have a substantial β (see below), the SSHG spectra can be safely compared with the linear absorption spectra.62
Fig. 2 reveals that the SSHG spectrum of YOYO-1 lacks the 458 nm band of the H-dimer and resembles the electronic absorption spectrum measured in organic solvents or in the presence of DNA, where YOYO-1 is in the non-aggregated form. This indicates that YOYO-1 does not form H-dimers when adsorbed at the interface. An alternate explanation, which can be dismissed on the basis of quantum-chemical calculations described below, would be that the interfacial H-dimers make a negligibly small contribution to the SSHG signal. The SSHG spectrum recorded with YOPRO-1 is very similar to that of YOYO-1. The small bathochromic shift of the band maximum relative to the absorption band could be due to a solvatochromic effect, in agreement with the solvent dependence of the absorption spectrum shown in Fig. 1. This indicates that, like in bulk aqueous solution, YOPRO-1 does not form H-aggregates at the interface.
A different behaviour is observed with the YOSAC dyes (Fig. 2): in addition to a red-shifted maximum and a broadening on the long-wavelength side that is larger than that observed for the two other cyanines, the SSHG band is markedly more intense at wavelengths corresponding to the absorption of the H-dimer, pointing to the formation of H aggregates at the interface.
To support the assignment of this spectral broadening to aggregation, the SSHG spectra of all four dyes were recorded at different bulk concentrations and are depicted in Fig. 3, and Fig. S3 and S4 (ESI†). The shape of the SSHG spectra recorded with YOYO-1 is independent of concentration (Fig. 3A), indicating that this dye forms neither intramolecular H-dimers nor intermolecular H-aggregates at the interface, at least at those concentrations. In the case of YOPRO-1, the relative intensity of the blue side of the band does not depend on concentration either, confirming the absence of H-aggregation at the interface. A small increase of the relative intensity on the red side of the spectrum appears (>500 nm) when going from 10 to 15 μM YOPRO-1. This difference is however small and not far from the noise level of the signal and vanishes if the aqueous solution is stirred during measurements. Such stirring decreases the signal to noise ratio and was thus not systematically performed as it requires longer acquisition time.
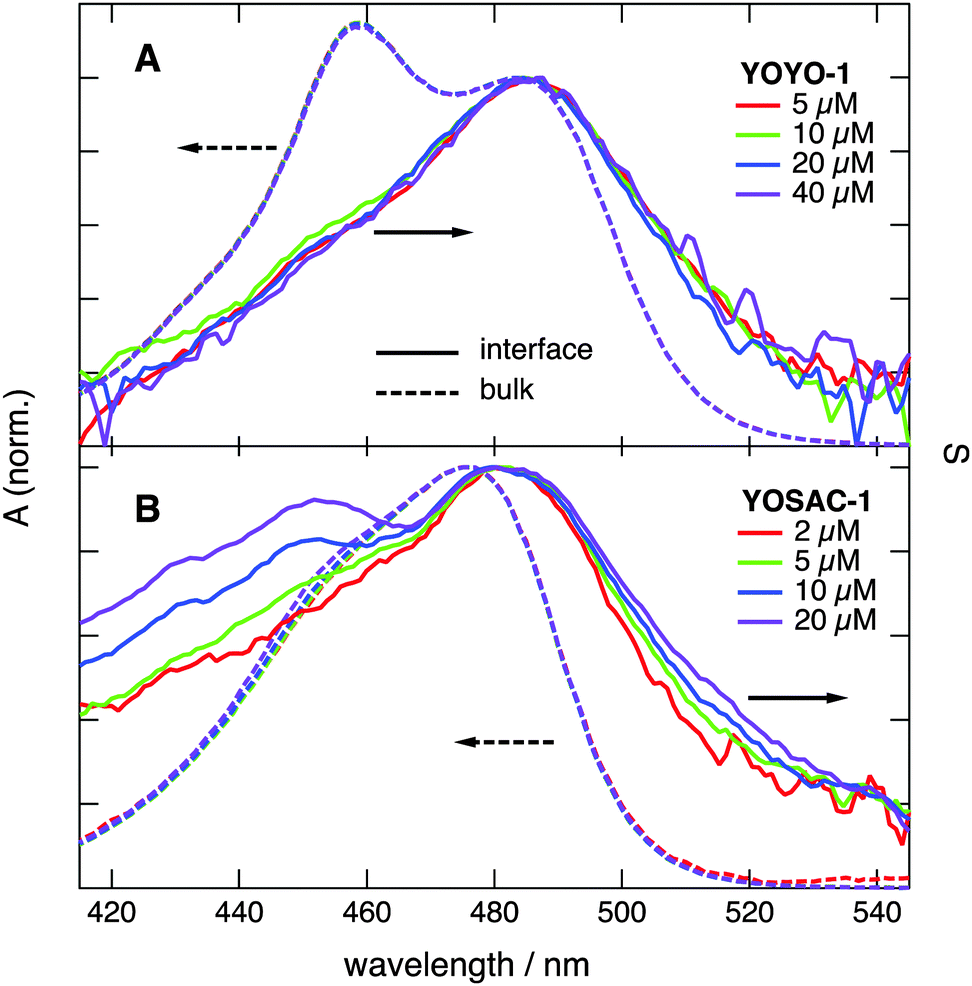 | ||
| Fig. 3 Concentration dependence of the SSHG spectra, S(λ), measured with (A) YOYO-1 and (B) YOSAC-1 at the dodecane/water interface. The electronic absorption spectra in water at the same concentrations are shown for comparison. | ||
Fig. 3B reveals that the SSHG spectrum recorded with YOSAC-1 changes dramatically with increasing concentration and, at 20 μM, exhibits a distinct secondary maximum at around 453 nm that coincides with the H-dimer absorption band measured with YOYO-1 in aqueous solution. Additionally, the relative SSHG intensity on the red side of the band exhibits a weaker but distinct growth with increasing concentration. As this increase seems to follow the same concentration dependence on both sides of the band, it should be associated with a single type of aggregate rather than to the coexistence of both H- and J-type aggregates at the interface. H- and J-aggregates are two extreme cases where the arrangement of the chromophores is such that it leads to either cancellation or addition of the dipolar strength, i.e. to totally dark and bright excitonic states.9 The weaker increase of the red side of the band relative to the blue side could be due to aggregates with a mutual orientation of the chromophores departing from that in an ideal H-aggregate, and which, therefore, does not lead to totally dark lower excitonic states.
Increasing YOSAC-3 concentration affects only the blue-side of the SSHG band (Fig. S4A, ESI†), but to a much smaller extent compared to YOSAC-1. Indeed, whereas a clear secondary maximum is observed with 20 μM YOSAC-1, only a shoulder can be seen with the same concentration of YOSAC-3.
The higher propensity of the YOSAC dyes compared to YOYO-1 and YOPRO-1 to form H-aggregates at the interface can be explained by their lower electric charge, which first decreases their hydrophilic character, favouring their adsorption at the interface, and second, decreases Coulombic repulsion, abetting aggregation. The higher relative contribution of the H-aggregate to the SSHG signal observed with YOSAC-1 relative to YOSAC-3 could reflect a higher propensity of the former to aggregate. This could arise from the bulky phenyl-methylene substituent on YOSAC-3, which albeit it enhances the hydrophobic character of this dye and most likely its affinity for the interface, most probably disfavours aggregation through steric hindrance. However a large hyperpolarizability of the YOSAC-1 aggregate or a different orientation at the interface cannot be excluded.
The SSHG measurements with YOPRO-1 were performed in PBS, because the signal in pure water was too weak to record reliable spectra, in agreement with the above discussion about the influence of electric charge on the interfacial activity. The SSHG intensity was found to substantially increase for all four dyes when going from pure water to PBS. This effect has already been reported,30,38,63 and is due to the higher affinity of some ions, like Cl−, for the interface and thus the Coulombic attraction of the cationic cyanine dyes toward the interfacial region.
Using the buffer instead of pure water has no significant effect on the electronic absorption and SSHG spectra of YOYO-1 and YOSAC-1 (Fig. S1 and S2, ESI†). On the other hand, the electronic absorption spectrum of YOSAC-3 in PBS exhibits an additional band at around 535 nm whose relative intensity increases with dye concentration, and which can be attributed to a J-aggregate (Fig. S4B, ESI†). This effect, not observed with other dyes, confirms the more hydrophobic nature of YOSAC-3. However, this red-shifted band does not appear in the SSHG spectrum at the dodecane/PBS interface (Fig. S4B, ESI†). The shoulder at around 450 nm, due to the H-aggregates, can still be observed but is weaker and increases less with concentration than at the dodecane/water interface. These results indicate that the buffer does not affect very significantly the aggregation behaviour of the YO dyes at the interface.
Whereas the enhanced aggregation of the YOSAC dyes at the interface can be explained in terms of higher interfacial population due to their superior amphiphilic character, the absence of YOYO-1 H-dimers cannot be accounted for by concentration effects, because this H-dimer is intramolecular and its formation does not depend on concentration. Therefore, the lack of a H-dimer band should be rather due to the mutual orientation of the two YO units when adsorbed at the interface that does not lead to significant excitonic coupling.
Polarization-resolved SSHG and orientation at the interface
To obtain a deeper insight into the interfacial orientation of the dyes, polarization-resolved SSHG measurements were carried out with YOYO-1 and YOSAC-1. In a first step, SSHG spectra were recorded at different polarizations to evidence possible differences between the nonlinear susceptibility tensors associated with the aggregated and non-aggregated forms of the dye,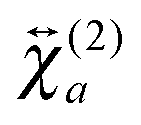 and
and 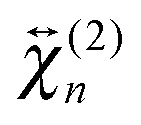 , respectively.
, respectively.
The shape of the SSHG spectrum measured with YOYO-1 does not exhibit a significant dependence on the polarization of the probe beam (s, p, or m) and the measured polarization component of the signal (s or p) (Fig. S5, ESI†). This indicates that, unless the relative magnitudes of the tensor elements of 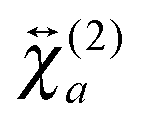 and
and 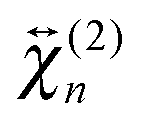 are identical, which is improbable as shown below, only one species, namely the non-aggregated form, contributes to the SSHG signal. Much stronger polarization dependence, which increases substantially with concentration, is observed with YOSAC-1 as shown in Fig. 4. At 2 μM, the H-aggregate band is absent and the shape of the SSHG spectrum is the same for all three polarization geometries, indicating that the signal originates from one species only. At higher concentrations, a strong polarization dependence of the spectral shape can be observed. These changes are the largest below 480 nm, but can also be detected on the long-wavelength side of the band. As already suggested above, the SSHG intensity on the red side of the band could be due to a resonance with the transition to the lowest excitonic state of the aggregate, which is not totally dark due to small departure from the ideal H-aggregate geometry. In the p/p geometry and above 10 μM, the H-aggregate band at around 450 nm is substantially more intense than the band due to the non-aggregated dyes.
are identical, which is improbable as shown below, only one species, namely the non-aggregated form, contributes to the SSHG signal. Much stronger polarization dependence, which increases substantially with concentration, is observed with YOSAC-1 as shown in Fig. 4. At 2 μM, the H-aggregate band is absent and the shape of the SSHG spectrum is the same for all three polarization geometries, indicating that the signal originates from one species only. At higher concentrations, a strong polarization dependence of the spectral shape can be observed. These changes are the largest below 480 nm, but can also be detected on the long-wavelength side of the band. As already suggested above, the SSHG intensity on the red side of the band could be due to a resonance with the transition to the lowest excitonic state of the aggregate, which is not totally dark due to small departure from the ideal H-aggregate geometry. In the p/p geometry and above 10 μM, the H-aggregate band at around 450 nm is substantially more intense than the band due to the non-aggregated dyes.
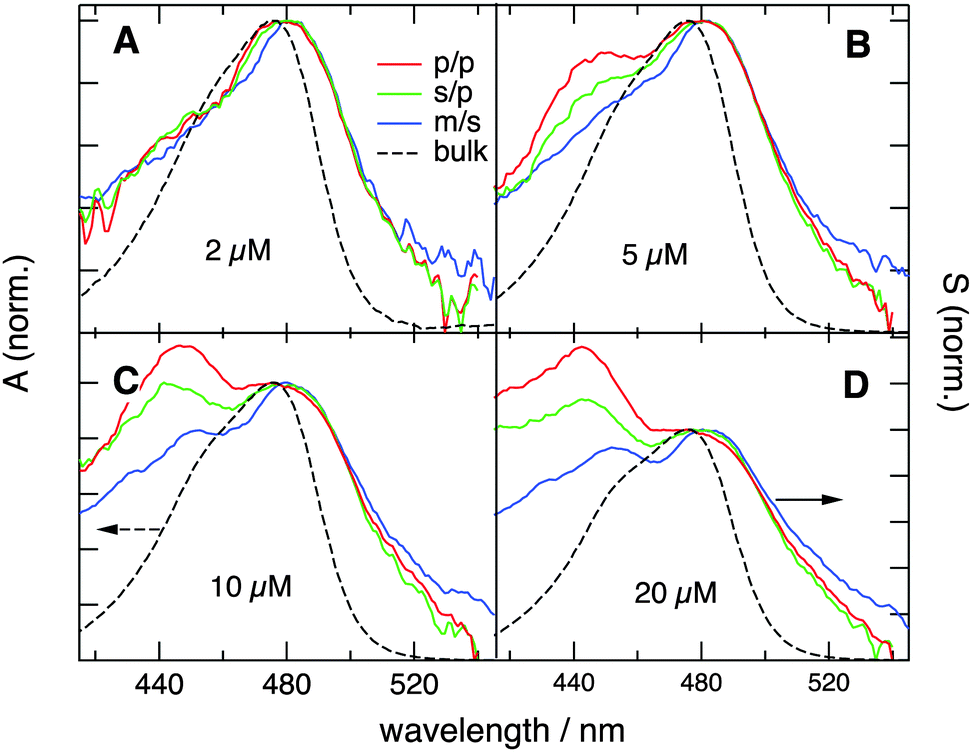 | ||
| Fig. 4 SSHG spectra measured with YOSAC-1 at different concentrations and different polarization geometries. The electronic absorption spectra are shown for comparison. | ||
The three polarization geometries probe different elements of the susceptibility tensor. In the case of an isotropic interface with C∞v symmetry, the nonlinear susceptibility tensor contains only three independent sets of nonzero elements: χ(2)ZZZ, χ(2)ZXX = χ(2)ZYY and χ(2)XXZ = χ(2)XZX = χ(2)YZY = χ(2)YYZ where the subscripts are the Cartesian coordinates in the laboratory frame, X and Y being in the interfacial plane and Y and Z in the plane of incidence of the probe beam. With the geometry used here, the dependence of the SSHG intensity on the polarization angle of the probe beam, γ (γ = 0°, 45° and 90° for p, m and s polarization), and of the signal component, Γ (Γ = 0 and 90° for p and s components) is given by:64,65
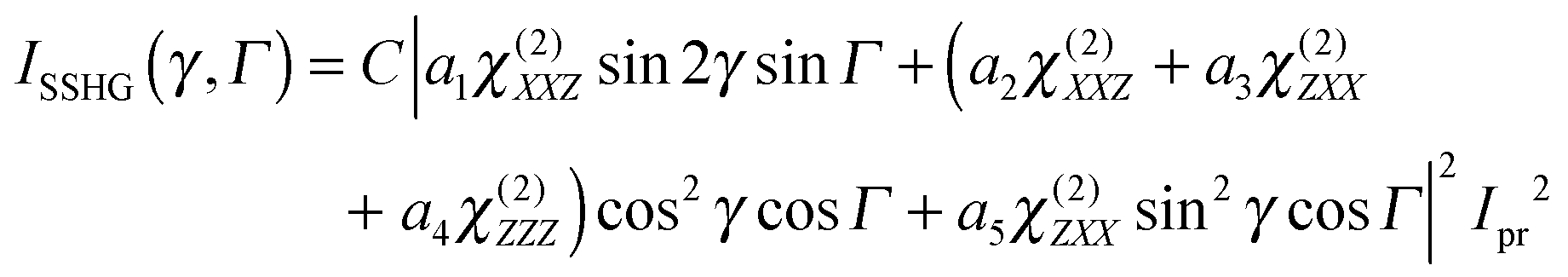 | (1) |
| Im/sSSHG = ISSHG (45°, 90°) ∝ |a1χ(2)XXZ|2 | (2a) |
| Ip/pSSHG = ISSHG (0°, 0°) ∝ |a4χ(2)ZZZ|2 | (2b) |
| Is/pSSHG = ISSHG (90°, 0°) ∝ |a5χ(2)ZXX|2 | (2c) |
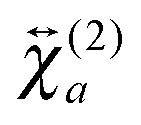 and
and 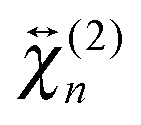 , are not identical and differ by the relative size of their elements, χ(2)ZZZ being comparatively larger for the aggregate. The nonlinear susceptibility is a macroscopic quantity that depends on the population, Ni, and hyperpolarizability, βi, of different species i adsorbed at the interface and contributing to the signal:64
, are not identical and differ by the relative size of their elements, χ(2)ZZZ being comparatively larger for the aggregate. The nonlinear susceptibility is a macroscopic quantity that depends on the population, Ni, and hyperpolarizability, βi, of different species i adsorbed at the interface and contributing to the signal:64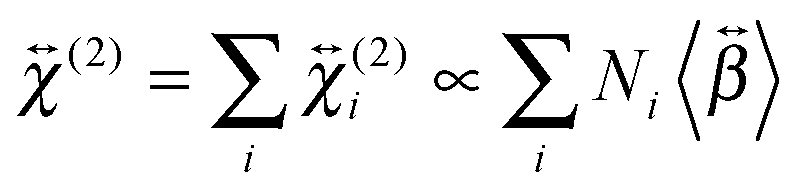 | (3) |
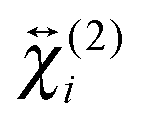 and
and 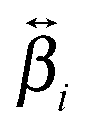 . The
. The 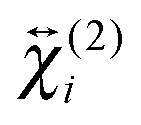 elements can be determined from polarization-resolved SSHG measurements.28,64–69 In a standard experiment, one records the intensity of several polarization components of the SSHG signal, e.g. Γ = 0° (p) and 90° (s), as a function of the polarization of the probe beam γ, (Fig. 5), and analyses the resulting intensity profiles using eqn (1). Analysis of an additional polarization profile measured at e.g. Γ = 45° gives access to the relative sign of the susceptibility tensor elements.
elements can be determined from polarization-resolved SSHG measurements.28,64–69 In a standard experiment, one records the intensity of several polarization components of the SSHG signal, e.g. Γ = 0° (p) and 90° (s), as a function of the polarization of the probe beam γ, (Fig. 5), and analyses the resulting intensity profiles using eqn (1). Analysis of an additional polarization profile measured at e.g. Γ = 45° gives access to the relative sign of the susceptibility tensor elements.
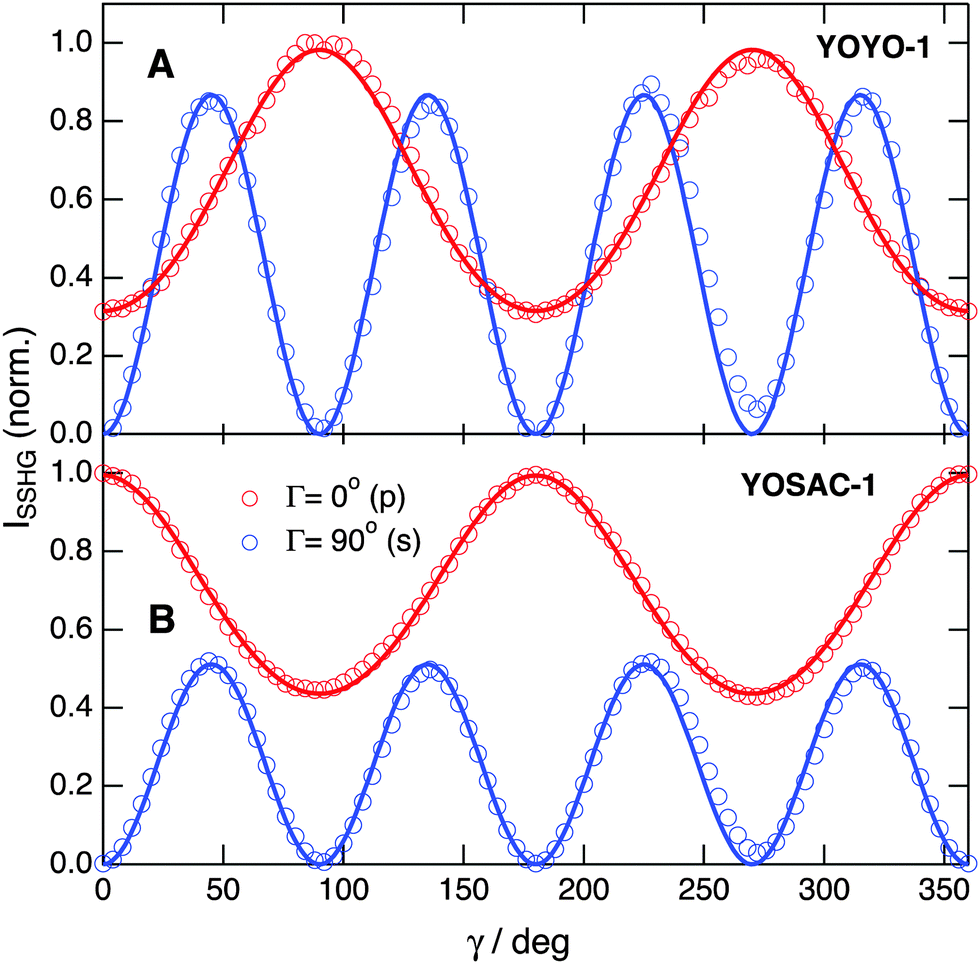 | ||
| Fig. 5 Polarization profiles measured with 10 μM YOYO-1 (A) and YOSAC-1 (B) and recorded at Γ = 0° (p, red) and Γ = 90° (s, blue) upon probing at 1000 nm. | ||
The relative magnitude of the 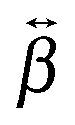 elements can be determined using quantum-chemical calculations or can be estimated on the basis of the structure and symmetry of the molecule. Here, we assume that
elements can be determined using quantum-chemical calculations or can be estimated on the basis of the structure and symmetry of the molecule. Here, we assume that 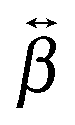 is dominated by a single element, namely βzzz, where the subscript corresponds to the Cartesian coordinates in the molecular frame, and where the z axis coincides with the S1 ← S0 transition dipole moment, which itself is aligned along the long molecular axis (see Fig. 7). The validity of this assumption is supported by the quantum-chemical calculations described in the next section and by the ratio of the hyperpolarizability tensor elements extracted from the polarization profiles (Table S2, ESI†). Polarization profiles similar to those shown in Fig. 5 have been recorded with YOYO-1 and YOSAC-1 at four different concentrations and at 880 and 1000 nm, where the aggregated and non-aggregated forms of the dyes should contribute most to the signal, respectively, (Fig. S6 and S7, ESI†).
is dominated by a single element, namely βzzz, where the subscript corresponds to the Cartesian coordinates in the molecular frame, and where the z axis coincides with the S1 ← S0 transition dipole moment, which itself is aligned along the long molecular axis (see Fig. 7). The validity of this assumption is supported by the quantum-chemical calculations described in the next section and by the ratio of the hyperpolarizability tensor elements extracted from the polarization profiles (Table S2, ESI†). Polarization profiles similar to those shown in Fig. 5 have been recorded with YOYO-1 and YOSAC-1 at four different concentrations and at 880 and 1000 nm, where the aggregated and non-aggregated forms of the dyes should contribute most to the signal, respectively, (Fig. S6 and S7, ESI†).
These profiles were analysed as described in detail in the ESI† to determine the orientation parameter, D = 〈cos3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ〉/〈cosθ〉, where θ is the tilt angle, i.e. the angle between the z molecular axis and the normal of the interface. The tilt angle θ was then calculated assuming a Dirac delta-function distribution of orientations at the interface. Fig. 6 shows the dependence of θ on the concentration of YOYO-1 and YOSAC-1 determined from the SSHG measurements at 880 and 1000 nm. The absolute values of θ should not be taken too literally and should be considered as higher-limit values as they were determined assuming a Dirac delta-function distribution of orientations. The same orientation parameter D can be obtained assuming a Gaussian distribution of the tilt angle centred around a smaller θ. Despite this, the following information can be extracted from this figure:
θ〉/〈cosθ〉, where θ is the tilt angle, i.e. the angle between the z molecular axis and the normal of the interface. The tilt angle θ was then calculated assuming a Dirac delta-function distribution of orientations at the interface. Fig. 6 shows the dependence of θ on the concentration of YOYO-1 and YOSAC-1 determined from the SSHG measurements at 880 and 1000 nm. The absolute values of θ should not be taken too literally and should be considered as higher-limit values as they were determined assuming a Dirac delta-function distribution of orientations. The same orientation parameter D can be obtained assuming a Gaussian distribution of the tilt angle centred around a smaller θ. Despite this, the following information can be extracted from this figure:
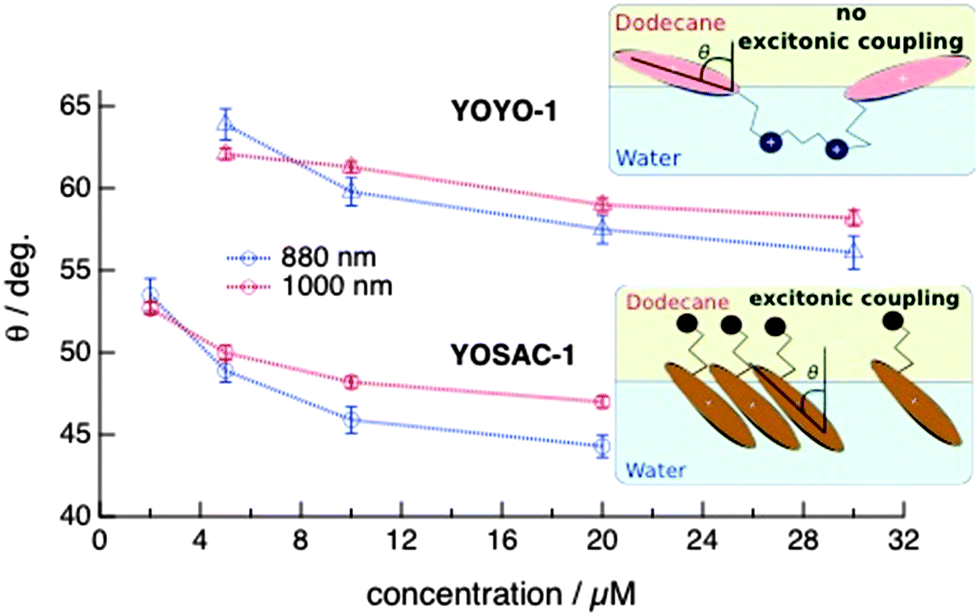 | ||
| Fig. 6 Tilt angle, θ, determined from polarization-resolved SSHG measurements at 880 and 1000 nm as a function of the concentration of YOYO-1 and YOSAC-1 in the aqueous phase. The dotted lines are guide for the eye. The insets show schematic representations of the interfacial orientation. | ||
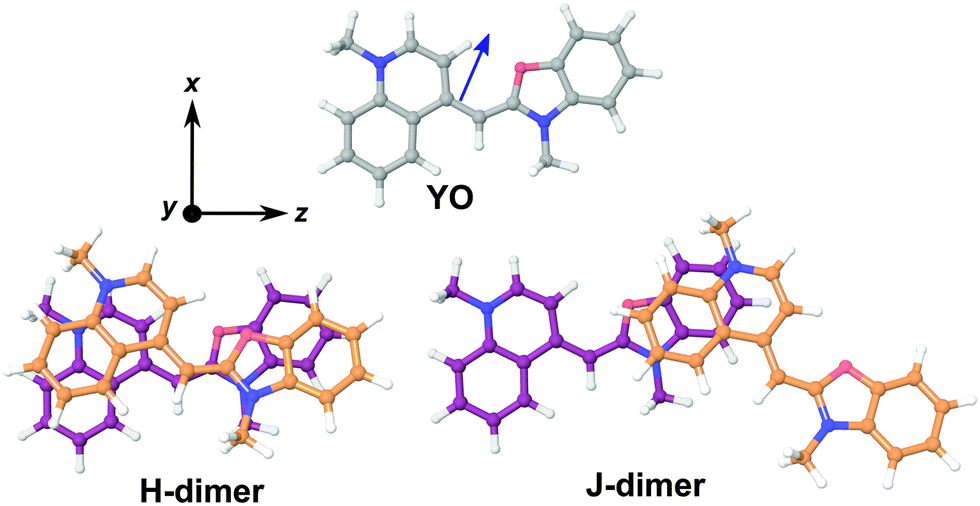 | ||
| Fig. 7 Optimized structures of YO and the H- and J-dimers. The z-axis coincides with the S1 ← S0 transition dipole moment of the YO monomer. For the dimers, the same orientation has been used for the YO unit in purple. The ground-state dipole moment (2.85 D) of YO is shown by the blue arrow. | ||
(1) At all concentrations and probe wavelengths, the tilt angle of YOYO-1 is substantially larger than that of YOSAC-1, indicating that the two chromophoric units of YOYO-1 lie flatter on the dodecane/water interface than YOSAC-1. This can be understood by considering the distribution of the electric charges on YOPRO-1, which corresponds to one half of YOYO-1, with one positive charge located on the ammonium group and the other delocalized over the whole aromatic system. Consequently, the ammonium end should be attracted toward the aqueous phase, whereas the aromatic system with its delocalized charge is neither highly hydrophilic nor lipophilic. A flat orientation at the interface of the chromophoric unit can accommodate this charge distribution.
By contrast, YOSAC-1 has a larger amphiphilic character as it contains a lipophilic side-chain that should be attracted toward the apolar phase. The presence of this chain most probably favours a more perpendicular orientation with respect to the interface.
(2) For both dyes, increasing concentration leads to a decrease of θ, i.e. to a more perpendicular orientation. This could be explained by considering that a smaller tilt angle allows a denser packing of adsorbates at the interface. This effect is less pronounced with YOYO-1, most probably because of the charge distribution discussed above that prevents the perpendicular orientation. The latter would require a hydrophilic fragment of the molecule to reside in the apolar phase.
(3) In the case of YOYO-1, the wavelength dependence of θ and of its variation with concentration are small and close to the limit of error. It could arise from the neglect of dispersion in the calculation of the optical coefficients ai (see ESI†). In any case, this confirms that the SSHG signal is mainly due to a single form of the molecule and that H-dimers do not play a significant role at the interface. The more distinct wavelength dependence that can be observed with YOSAC-1 above ∼5 μM is in full agreement with the SSHG spectra, which exhibits the H-aggregate band at around 445 nm at 5 μM and higher concentrations. The tilt angle obtained from the 880 nm data is significantly smaller than that deduced from the polarization profiles at 1000 nm, pointing to a more perpendicular orientation of the aggregates relatively to the monomeric YOSAC-1 molecules. This is in excellent agreement with H-aggregation, which requires a face-to-face arrangement of the dyes that cannot be achieved with a large tilt angle, i.e. with the molecules lying on the interface.
Calculations of the frequency-dependent hyperpolarizability
In order to have a better insight into the SSHG response of the aggregates, quantum-chemical calculations of the hyperpolarizability tensor,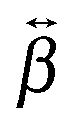 , of the monomeric and dimeric forms of the YO dyes were performed at the DFT level, using a YO analogue with methyl substituents on the two nitrogen atoms (Fig. 7). A solvation model for water was included in the calculation to better describe the experimental conditions (see Experimental section). The molecular coordinates were fixed in all calculations with the z-axis aligned along the S1 ← S0 transition dipole moment of YO. Geometries of two dimers among the optimized ones were identified as H- and J-dimers (Fig. 7). The rotational offset (i.e. the angle between the long axes of the two YO units) amounts to 20° and 39° for the H- and J-dimers, whereas the slip angle (i.e. the angle between the stacking axis and the long molecular axis) is equal to 77° and 38°, respectively. These mutual orientations found for both dimers result in substantial excitonic coupling, as shown by the TD-DFT calculations described below. Moreover, both dimers are far from centro-symmetric and thus their SSHG spectrum should be close to their linear absorption spectrum.
, of the monomeric and dimeric forms of the YO dyes were performed at the DFT level, using a YO analogue with methyl substituents on the two nitrogen atoms (Fig. 7). A solvation model for water was included in the calculation to better describe the experimental conditions (see Experimental section). The molecular coordinates were fixed in all calculations with the z-axis aligned along the S1 ← S0 transition dipole moment of YO. Geometries of two dimers among the optimized ones were identified as H- and J-dimers (Fig. 7). The rotational offset (i.e. the angle between the long axes of the two YO units) amounts to 20° and 39° for the H- and J-dimers, whereas the slip angle (i.e. the angle between the stacking axis and the long molecular axis) is equal to 77° and 38°, respectively. These mutual orientations found for both dimers result in substantial excitonic coupling, as shown by the TD-DFT calculations described below. Moreover, both dimers are far from centro-symmetric and thus their SSHG spectrum should be close to their linear absorption spectrum.
The assignment of these complexes to H- and J-dimers is based, besides geometrical considerations, on the results of TD-DFT calculations, which predict blue- and red-shifted electronic transitions relative to that of the monomer, respectively, (Fig. 8). Whereas the first vertical electronic transition of YO is calculated at 3.00 eV with an oscillator strength of f = 1.06, an intense absorption with f = 1.86 is predicted at 3.08 eV for the H-dimer. On the other hand, the transition to the lowest excitonic state of the H-dimer is calculated at 2.90 eV with negligible intensity (f = 0.05). In the case of the J-dimer, an intense transition to the lower excitonic state with f = 1.84 is predicted at 2.97 eV, whereas the transition to the upper excitonic state is at 3.02 eV and is much weaker (f = 0.27). The non-vanishing oscillator strength of the transition to the upper excitonic state is due to an imperfect alignment of the two dyes in the J-dimer. The nature and structure of aggregates have been shown to critically depend on factors such as electrostatic interactions, size and geometry.70 For example, highly polar merocyanine dyes form π-stacks with a 3.5 Å distance between the aromatic planes, a slip angle close to 60° and an antiparallel arrangement, as revealed by crystallographic and 2D NMR measurements.70 In our optimized H-dimer model, the distance between the aromatic planes is 4.9 Å, larger than in the merocyanine aggregate, most probably due to the electrostatic repulsions between the two positively charged YO units. The larger predicted slip angle of 77°, i.e. close to the value of 90° for a perfect face-to-face geometry, can be explained by the less sterically hindered YO model. The departure from 90° minimizes the electrostatic repulsion associated with the permanent electric dipole of YO.
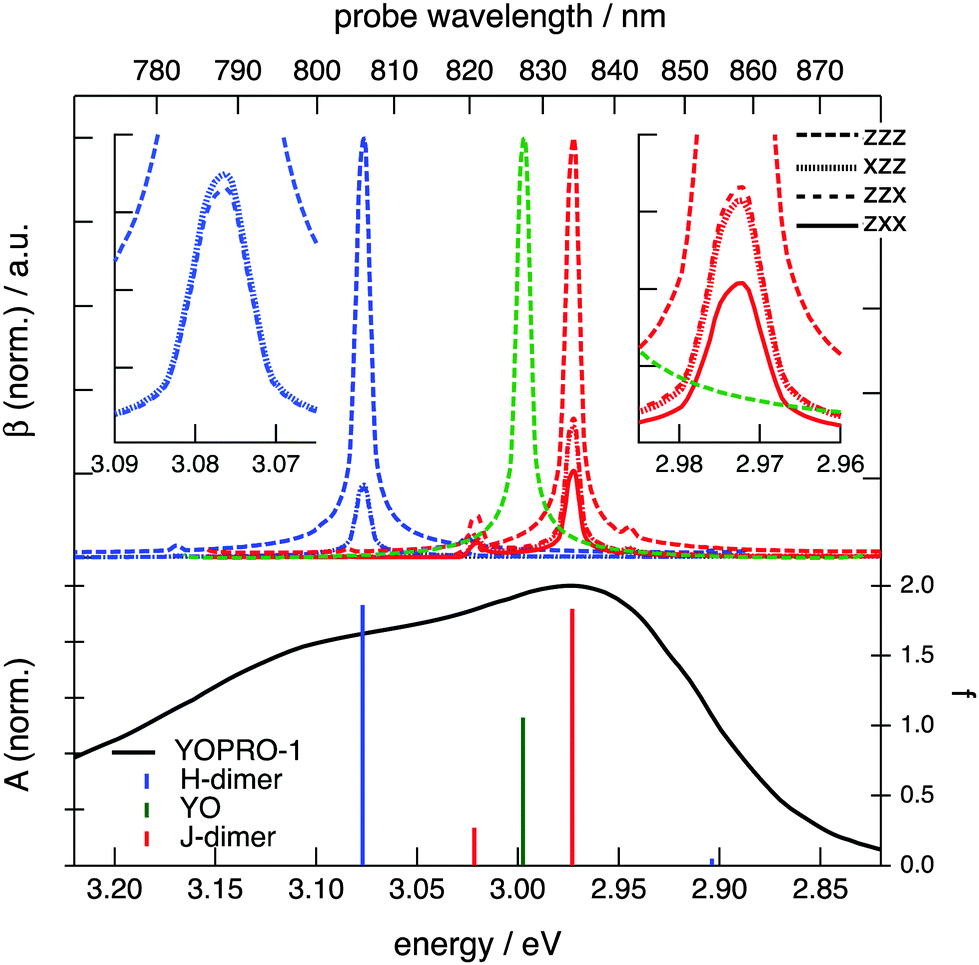 | ||
| Fig. 8 (bottom) Calculated electronic transitions of YO, H- and J-dimers in water. The solid line is the absorption spectrum of a 20 μM solution of YOPRO-1 in water shifted to higher energy by 0.40 eV. (top) Spectral dependence of the main elements of the hyperpolarizability tensors of the monomer and dimers. | ||
The  tensor elements of YO and the H- and J-dimers were calculated using fixed coordinates as presented in Fig. 7. Fig. 8 shows the spectral dependence of the modulus of all major non-zero elements, |βijk|, normalized to the maximum of the most intense element of each tensor:
tensor elements of YO and the H- and J-dimers were calculated using fixed coordinates as presented in Fig. 7. Fig. 8 shows the spectral dependence of the modulus of all major non-zero elements, |βijk|, normalized to the maximum of the most intense element of each tensor:
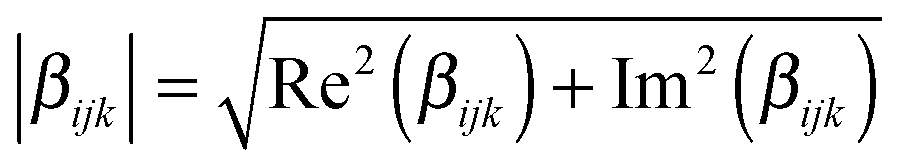 | (4) |
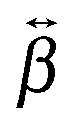 tensor of YO is dominated by a single element, βzzz, confirming the assumption made above in the analysis of the polarization-resolved experiments. This element is aligned along the S1 ← S0 transition dipole moment and exhibits an intense two-photon resonance with this transition. The βzzz element is also the major one for the dimers, although other elements, namely βxzz and βzzx, have a non-negligible magnitude. In this case as well, these tensor elements show a two-photon resonance with the transition to the higher and lower excitonic states of the H- and J-dimers, respectively.
tensor of YO is dominated by a single element, βzzz, confirming the assumption made above in the analysis of the polarization-resolved experiments. This element is aligned along the S1 ← S0 transition dipole moment and exhibits an intense two-photon resonance with this transition. The βzzz element is also the major one for the dimers, although other elements, namely βxzz and βzzx, have a non-negligible magnitude. In this case as well, these tensor elements show a two-photon resonance with the transition to the higher and lower excitonic states of the H- and J-dimers, respectively.
The ratio between the total intrinsic hyperpolarizability of the dimers and the monomer, βtot(dim)/βtot(mon) has been estimated using βtot defined as:50
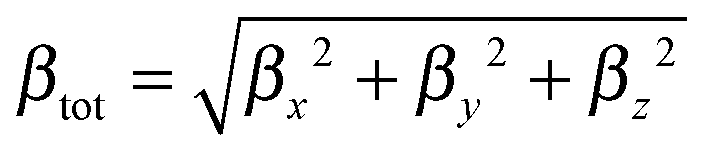 | (4a) |
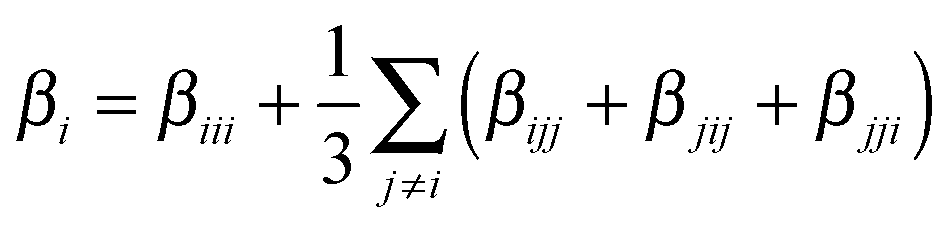 | (4b) |
These calculations confirm that both H- and J-type YO dimers have a second-order optical response, which is comparable in magnitude with that of a single YO molecule. Therefore, the SSHG band observed at around 450 nm with the YOSAC dyes can unambiguously be ascribed to a H-dimer or a higher H-aggregate.
The absence of a H-dimer band in the SSHG spectrum of YOYO-1 cannot be ascribed to a vanishingly small nonlinear response, as this would require a mutual orientation of the chromophoric units resulting in a H-dimer with a centre of inversion. Such geometry cannot be realized with YOYO-1 because of the constraints imposed by the linker. Therefore, the lack of aggregate SSHG band indicates that YOYO-1 does not exist in the H-dimer form at the interface. This is due to the distribution of electric charges in the molecule that favors a rather flat orientation of the chromophore on the interface and thus prevents the formation of H-dimers. The latter would require a smaller tilt angle, similar to that found for YOSAC-1.
Time-resolved SSHG
Further insight into the properties of the YO dyes adsorbed at the interfaces was obtained from TR-SSHG measurements. Fig. 9A shows the time-profiles of the SSHG signal intensity, S(t), recorded at 485 nm upon 500 nm excitation using s/p and m/s polarizations. As the SSHG signal at this wavelength is due to a two-photon resonance with the S1 ← S0 transition, the decrease of the SSHG intensity upon photoexcitation reflects the depletion of the ground-state population. The recovery of the signal can be due to two processes: (i) the decay of the excited-state population and (ii) the out-of-plane motion that suppresses the orientational anisotropy due to the excitation with circularly polarized light. Such out-of-plane motion has been shown to take place on hundreds of picoseconds timescale.71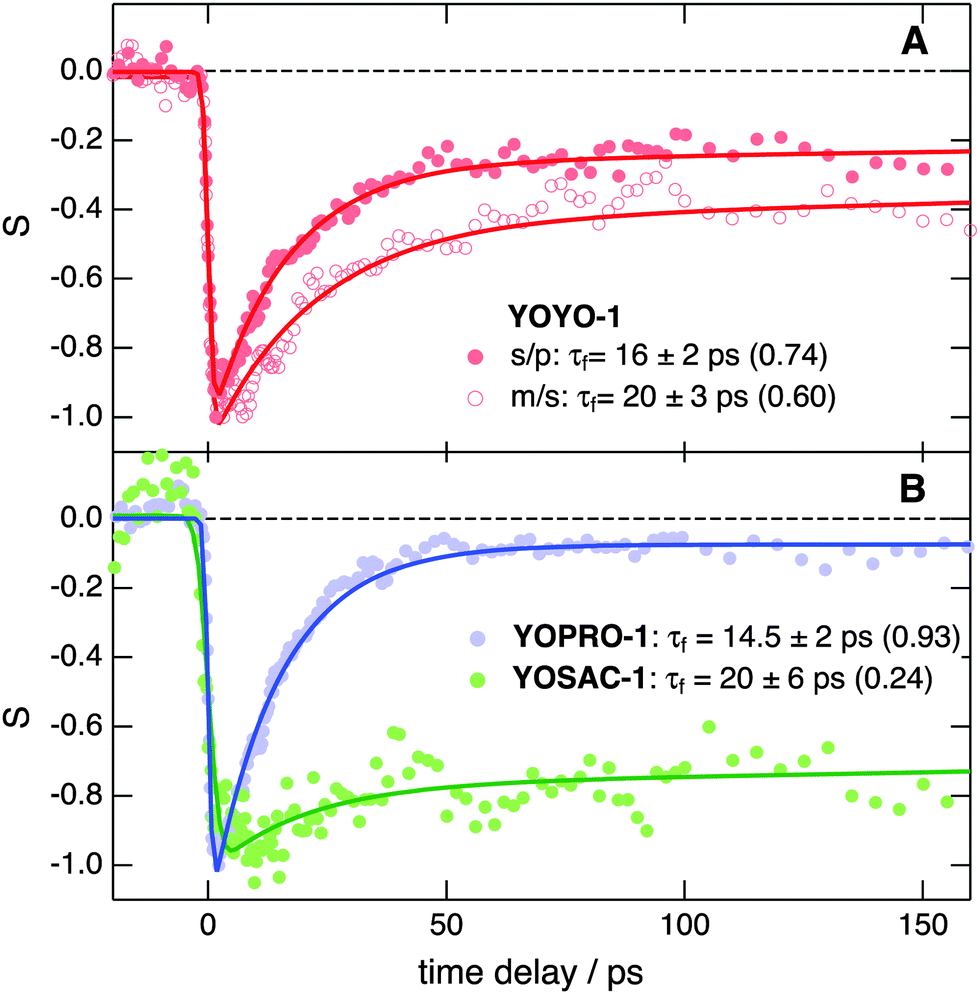 | ||
| Fig. 9 (A) TR-SSHG profiles, S(t), measured with (A) YOYO-1 (10 μM) at 485 nm at the dodecane/water interface using two polarization geometries and (B) a comparison of the TR-SSHG profiles measured at 485 nm using the s/p polarization with YOSAC-1 (10 μM) at the dodecane/water interface and with YOPRO-1 (10 μM) at the dodecane/PBS interface. The pump pulses were at 500 nm. The solid lines are the best bi-exponential fits (τf is the time constant of the fast component and its relative amplitude is given in parentheses; the time constant of the slow component is >1 ns). | ||
In the present case, the signal recovery occurs on different timescales and can be reproduced with the sum of two exponential functions (solid lines), with time constants of ∼15–20 ps and >1 ns. Although these time constants are similar for both polarization geometries, the relative amplitudes differ, with that associated with the fast component decreasing from 0.74 to 0.60 when going from s/p to m/s geometry. According to eqn (2), the SSHG intensity at those polarizations should reflect χ(2)ZXX and χ(2)XXZ, respectively. As the quantum chemical calculations evidenced that the hyperpolarizability tensor of YO is dominated by a single element, both χ(2)ZXX and χ(2)XXZ should be identical, even in the presence of orientational anisotropy,71 and given by:68
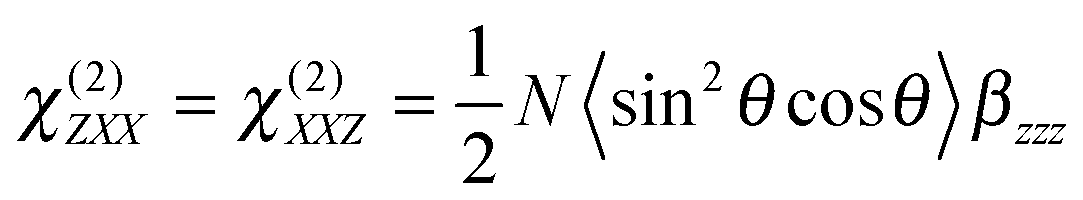 | (5) |
 tensor that differs from that with non-interacting units. However, excitonic coupling should not be very significant, as testified by the negligible polarization dependence of the SSHG spectrum of YOYO-1 (Fig. S5, ESI†) and by the TR-SSHG profile measured at 460 nm, which is similar to that at 485 nm but with a slightly larger amplitude of the fast component, i.e. 0.78 vs. 0.74 (Fig. S10, ESI†). For example, the YOYO-1 structure with close to perpendicular mutual orientation of the chromophores, as that observed when the dye is intercalated into DNA, would result in a small excitonic coupling. The
tensor that differs from that with non-interacting units. However, excitonic coupling should not be very significant, as testified by the negligible polarization dependence of the SSHG spectrum of YOYO-1 (Fig. S5, ESI†) and by the TR-SSHG profile measured at 460 nm, which is similar to that at 485 nm but with a slightly larger amplitude of the fast component, i.e. 0.78 vs. 0.74 (Fig. S10, ESI†). For example, the YOYO-1 structure with close to perpendicular mutual orientation of the chromophores, as that observed when the dye is intercalated into DNA, would result in a small excitonic coupling. The  tensor associated with this structure should differ from that of the monomer but the resonances should be essentially the same.
tensor associated with this structure should differ from that of the monomer but the resonances should be essentially the same.
The fast recovery component is ascribed to the YOYO-1 population with non-interacting units, whereas the slow component is attributed to the X form. This assignment is based on previous investigations of YOYO-1 in aqueous solutions that reported excited-state lifetimes of 4 ps and 1.1 ns for the non-aggregated and H-dimer forms, respectively.8 The longer excited-state lifetime of the dimer originates from the hindrance that arises upon π stacking of the large amplitude motion responsible for the non-radiative decay. A similar explanation can be proposed here to account for the slow recovery dynamics of the X form.
The 4–5 times larger time constant found here for the excited-state decay of the non-interacting form is probably due to the intrinsic properties of the interface. As observed with many other flexible chromophores,72 the excited-state dynamics of YOYO-1 and other YO derivatives in solution is dominated by a non-radiative decay process involving intramolecular modes with large amplitude motion. The time constant associated with this process, τnr, varies with the viscosity of the environment, η, as τnr ∝ ηα, where α depends on the nature of the intramolecular modes involved in the non-radiative decay, and ranges usually between ∼0.3 and 1.73 According to the data reported for YOPRO-1 and YOSAC-1 in four different solvents,7α can be estimated to be around 1. Therefore, the friction experienced by YOYO-1 at the dodecane/water interface can be estimated to be around 4–5 times as large as in bulk water. This observation agrees well with the lifetime increase reported for malachite green when going from bulk water (τ = 0.6 ps) to the dodecane/water interface (τ = 2.5 ps).74 Considering that dodecane is more viscous than water by a factor of 1.5 only, the larger excited-state lifetimes at the interface cannot be accounted by the protrusion of the adsorbed dyes in the apolar phase,26 and should reflect an intrinsic property of the interface. Higher interfacial friction has also been observed at the air/water interface,63,75,76 and has been ascribed to the stiffer nature of interfacial water, owing to a stronger hydrogen-bond network.
Fig. 9B reveals that the TR-SSHG profile at 485 nm measured with YOPRO-1 at the dodecane/PBS interface is almost entirely dominated by the fast component (relative amplitude of 0.93) with a time constant of 14.5 ± 2 ps, similar to that associated with the fast recovery component of YOYO-1. Upon doubling the bulk concentration of YOPRO-1 from 10 to 20 μM, the amplitude of this slow component increases to 0.1, but the short time constant remains essentially unchanged (Fig. S11, ESI†). At 475 nm, the slow component is no longer present and the time profile recorded at the same concentration can be reproduced with a single exponential function with a 15.5 ± 2 ps time constant (Fig. S12, ESI†). These results are in good agreement with the stationary SSHG measurements showing that YOPRO-1 does not form H-aggregates at the interface. However, the presence of a slow recovery component whose amplitude increases with bulk concentration points to the formation of another type of aggregate having properties similar to those found for the X form of YOYO-1.
Finally, Fig. 9B indicates that, contrary to YOYO-1 and YOPRO-1, the ground-state recovery dynamics of YOSAC-1 is dominated by the slow component, whose relative amplitude amounts to 0.76. This result is in accordance with the SSHG spectrum (Fig. 4C) that points to a large population of H-aggregates at the interface.
In summary, these TR-SSHG measurements confirm the high propensity of YOSAC-1 to form H-aggregates at the interface. They also reveal that, although YOYO-1 and YOPRO-1 do not exist as a H-dimer/aggregate at the interface, they exist in two different forms: a predominant one where the two chromophores are far apart and do not interact and another where the mutual orientation of the chromophoric units is such that it minimizes the excitonic interaction and inhibits the non-radiative decay of the excited state by large-amplitude motion.
Conclusions
We have presented here the first SSHG study, to the best of our knowledge, of the well-known YO fluorescent DNA probes at liquid/liquid interfaces. As anticipated from their remarkable performance as DNA intercalators, these dyes, especially the singly charged ones, exhibit a high affinity for the dodecane/water interface, and generate a large SSHG signal in the 440–500 nm region owing to a two-photon electronic resonance. The ensemble of experimental data reveals an interfacial behavior that depends strongly on the structure of the dye, especially its electric charge. The only homodimeric dye investigated here, YOYO-1, exhibits a behavior upon adsorption that is similar to that observed in bulk aqueous solutions upon addition of DNA. Indeed, the YOYO-1 H-dimer, which is predominant in water, is disrupted upon adsorption at the interface as well as upon intercalation into DNA. Because of the absence of a strong lipophilic group, the YO units adopt a rather flat orientation at the interface that does not favor π-stacking and thus inhibits the formation of H-dimers. However, some interaction between the YO units that hinders large-amplitude motion around the monomethine bond and hence lengthens the excited-state lifetime of the dye, can still take place at the interface. As a consequence, these results predict an increase of the fluorescence quantum yield of YOYO-1 upon adsorption at the interface. This aspect, which still needs to be verified, would confer YOYO-1 interesting properties as a fluorescent interfacial probe.On the other hand, the interfacial behavior of the monomeric dyes, i.e. YOPRO-1, YOSAC-1 and YOSAC-3, does not differ much from that observed in aqueous solution. In both cases, YOPRO-1 exhibits little tendency for H-aggregation, which can be explained by its doubly charged nature that favors neither adsorption nor aggregation. Some intermolecular interactions, which seem to be of a similar nature than the intramolecular interactions measured with YOYO-1, are operative with this dye at the interface. Being singly charged, the YOSAC dyes exhibit a higher affinity for the interface, where they adopt a more perpendicular orientation than the doubly charged dyes. Both this orientation and the smaller charge promote π stacking and the formation of H-aggregates. As the latter are not emissive, the YOSAC dyes should not be selected as fluorescent interfacial probes. However, given their large SSHG signal and high interfacial activity, they could be used as probes in SHG microscopy.
Very preliminary SSHG measurements in the presence of DNA in the aqueous phase indicate that these YO dyes can also be used to study the interfacial properties of nucleic acids. This approach might prove to be useful for investigating biomolecules in heterogeneous environments.
Acknowledgements
The authors wish to thank Prof. T. Deligeorgiev (Faculty of Chemistry, University of Sofia) for supplying the fluorescent dyes. This work was supported by the Swiss National Science Foundation through project no. 200020-147098 and the University of Geneva.References
- A. N. Glazer and H. S. Rye, Nature, 1992, 359, 859–861 CrossRef CAS PubMed.
- H. S. Rye, S. Yue, M. A. Quesada, R. P. Haugland, R. A. Mathies and A. N. Glazer, Methods Enzymol., 1993, 217, 414–431 CAS.
- C. Carlsson, A. Larsson, M. Jonsson, B. Albinsson and B. Norden, J. Phys. Chem., 1994, 98, 10313–10321 CrossRef CAS.
- T. L. Netzel, K. Nafisi, M. Zhao, J. R. Lenhard and I. Johnson, J. Phys. Chem., 1995, 99, 17936–17947 CrossRef CAS.
- L. Mikelsons, C. Carra, M. Shaw, C. Schweitzer and J. C. Scaiano, Photochem. Photobiol. Sci., 2005, 4, 798–802 CAS.
- A. Fürstenberg, T. G. Deligeorgiev, N. I. Gadjev, A. A. Vasilev and E. Vauthey, Chem. – Eur. J., 2007, 13, 8600–8609 CrossRef PubMed.
- A. Fürstenberg and E. Vauthey, J. Phys. Chem. B, 2007, 111, 12610–12620 CrossRef PubMed.
- A. Fürstenberg, M. D. Julliard, T. G. Deligeorgiev, N. I. Gadjev, A. A. Vassilev and E. Vauthey, J. Am. Chem. Soc., 2006, 128, 7661–7669 CrossRef PubMed.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
- V. Sundstroem and T. Gillbro, J. Chem. Phys., 1985, 83, 2733–2743 CrossRef CAS.
- I. Renge and U. P. Wild, J. Phys. Chem. A, 1997, 101, 7977–7988 CrossRef CAS.
- R. F. Khairutdinov and N. Serpone, J. Phys. Chem. B, 1997, 101, 2602–2610 CrossRef CAS.
- J. L. Seifert, R. E. Connor, S. A. Kushon, M. Wang and B. A. Armitage, J. Am. Chem. Soc., 1999, 121, 2987–2995 CrossRef CAS.
- M. Wang, G. L. Silva and B. A. Armitage, J. Am. Chem. Soc., 2000, 122, 9977–9986 CrossRef CAS.
- T. D. Slavnova, A. K. Chibisov and H. Gorner, J. Phys. Chem. A, 2005, 109, 4758–4765 CrossRef CAS PubMed.
- S. Gadde, E. K. Batchelor, J. P. Weiss, Y. Ling and A. E. Kaifer, J. Am. Chem. Soc., 2008, 130, 17114–17119 CrossRef CAS PubMed.
- S. M. Mooi, S. N. Keller and B. Heyne, Langmuir, 2014, 30, 9654–9662 CrossRef CAS PubMed.
- N. A. Toropov, P. S. Parfenov and T. A. Vartanyan, J. Phys. Chem. C, 2014, 118, 18010–18014 CAS.
- J. S. Schildkraut, T. L. Penner, C. S. Willand and A. Ulman, Opt. Lett., 1988, 13, 134–136 CrossRef CAS PubMed.
- N. Kato, K. Saito and Y. Uesu, Thin Solid Films, 1999, 338, 5–8 CrossRef CAS.
- Y. Niidome, H. Ayukawa and S. Yamada, J. Photochem. Photobiol., A, 2000, 132, 75–80 CrossRef CAS.
- G. Janssens, F. Touhari, J. W. Gerritsen, H. van Kempen, P. Callant, G. Deroover and D. Vandenbroucke, Chem. Phys. Lett., 2001, 344, 1–6 CrossRef CAS.
- R. Adachi, T. Inoue and T. Ogawa, Anal. Sci., 2001, 17, 449–451 CrossRef CAS PubMed.
- A. Yamaguchi, M. Nakano, K. Nochi, T. Yamashita, K. Morita and N. Teramae, Anal. Bioanal. Chem., 2006, 386, 627–632 CrossRef CAS PubMed.
- G. Martin-Gassin, E. Benichou, G. Bachelier, I. Russier-Antoine, C. Jonin and P. F. Brevet, J. Phys. Chem. C, 2008, 112, 12958 CAS.
- G. Martin-Gassin, D. Villamaina and E. Vauthey, J. Am. Chem. Soc., 2011, 133, 2358–2361 CrossRef CAS PubMed.
- D. A. Steinhurst, A. P. Baronavski and J. C. Owrutsky, J. Phys. Chem. B, 2002, 106, 3160–3165 CrossRef CAS.
- H. Nagatani, A. Piron, P.-F. Brevet, D. J. Fermin and H. H. Girault, Langmuir, 2002, 18, 6647–6652 CrossRef CAS.
- K. Fujiwara, S. Wada, H. Monjushiro and H. Watarai, Langmuir, 2006, 22, 2482–2486 CrossRef CAS PubMed.
- M. Fedoseeva, P. Fita, A. Punzi and E. Vauthey, J. Phys. Chem. C, 2010, 114, 13774–13781 CAS.
- M. Fedoseeva, R. Letrun and E. Vauthey, J. Phys. Chem. B, 2014, 118, 5184–5193 CrossRef CAS PubMed.
- S. Yamaguchi and T. Tahara, J. Chem. Phys., 2006, 125, 194711 CrossRef PubMed.
- J. Massin, A. Charaf-Eddin, F. Appaix, Y. Bretonnière, D. Jacquemin, B. van der Sanden, C. Monnereau and C. Andraud, Chem. Sci., 2013, 4, 2833 RSC.
- D. P. Mahoney, E. A. Owens, C. Fan, J. C. Hsiang, M. M. Henary and R. M. Dickson, J. Phys. Chem. B, 2015, 119, 4637–4643 CrossRef CAS PubMed.
- H. Chen, W. Lin, H. Cui and W. Jiang, Chem. – Eur. J., 2015, 21, 733–745 CrossRef CAS PubMed.
- T. Deligeorgiev, N. Gadjev, A. Vasilev, K. Drexhage and S. Yarmoluk, Dyes Pigm., 2006, 70, 185–191 CrossRef CAS.
- N. I. Gadjev, T. G. Deligeorgiev, I. Timcheva and V. Maximova, Dyes Pigm., 2003, 57, 161–164 CrossRef CAS.
- M. Fedoseeva, P. Fita and E. Vauthey, Langmuir, 2013, 29, 14865–14872 CrossRef CAS PubMed.
- S. Richert, S. Mosquera Vazquez, M. Grzybowski, D. T. Gryko, A. Kyrychenko and E. Vauthey, J. Phys. Chem. B, 2014, 118, 9952–9963 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (revision C1), Wallingford, CT, 2010 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- D. Jacquemin, E. A. Perpete, G. Scalmani, M. J. Frisch, R. Kobayashi and C. Adamo, J. Chem. Phys., 2007, 126, 144105 CrossRef PubMed.
- P. A. Limacher, K. V. Mikkelsen and H. P. Luthi, J. Chem. Phys., 2009, 130, 194114 CrossRef PubMed.
- M. Medved’, Š. Budzák and T. Pluta, Chem. Phys. Lett., 2011, 515, 78–84 CrossRef.
- A. Alparone, Adv. Phys. Chem., 2013, 2013, 1–8 CrossRef.
- G. Maroulis and A. J. Thakkar, J. Chem. Phys., 1989, 90, 366 CrossRef CAS.
- E. F. Archibong and A. J. Thakkar, Chem. Phys. Lett., 1993, 201, 485–492 CrossRef CAS.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195–242 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, J. Chem. Phys., 2006, 124, 94107 CrossRef PubMed.
- H. Sekino and R. J. Bartlett, J. Chem. Phys., 1986, 85, 976–989 CrossRef CAS.
- J. E. Rice and N. C. Handy, J. Chem. Phys., 1991, 94, 4959–4971 CrossRef CAS.
- J. E. Rice and N. C. Handy, Int. J. Quantum Chem., 1992, 43, 91–118 CrossRef CAS.
- S. Yamaguchi and T. Tahara, J. Phys. Chem. B, 2004, 108, 19079–19082 CrossRef CAS.
- N. S. Makarov, M. Drobizhev and A. Rebane, Opt. Express, 2008, 16, 4029–4047 CrossRef CAS PubMed.
- C. Xu and W. W. Webb, J. Opt. Soc. Am. B, 1996, 13, 481–491 CrossRef CAS.
- L. Goodman and R. P. Rava, Acc. Chem. Res., 1984, 17, 250–257 CrossRef CAS.
- I. Benjamin, Chem. Rev., 2006, 106, 1212–1233 CrossRef CAS PubMed.
- A. Punzi, G. Martin-Gassin, J. Grilj and E. Vauthey, J. Phys. Chem. C, 2009, 113, 11822–11829 CAS.
- K. B. Eisenthal, Chem. Rev., 1996, 96, 1343–1360 CrossRef CAS PubMed.
- A. A. Tamburello-Luca, P. Hebert, P. F. Brevet and H. H. Girault, J. Chem. Soc., Faraday Trans., 1996, 92, 3079–3085 RSC.
- G. J. Simpson, S. G. Westerbuhr and K. L. Rowlen, Anal. Chem., 2000, 72, 887–898 CrossRef CAS PubMed.
- B. Doughty, Y. Rao, S. W. Kazer, S. J. Kwok, N. J. Turro and K. B. Eisenthal, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5756–5758 CrossRef CAS PubMed.
- P.-F. Brevet, Surface Second Harmonic Generation, Presses polytechniques et universitaires romandes, Lausanne, 1997 Search PubMed.
- J. Rinuy, A. Piron, P. F. Brevet, M. Blanchard-Desce and H. H. Girault, Chem. – Eur. J., 2000, 6, 3434–3441 CrossRef CAS.
- Z. Chen, A. Lohr, C. R. Saha-Moller and F. Wurthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- D. Zimdars, J. I. Dadap, K. B. Eisenthal and T. F. Heinz, J. Phys. Chem. B, 1999, 103, 3425–3433 CrossRef CAS.
- A. Rosspeintner, B. Lang and E. Vauthey, Annu. Rev. Phys. Chem., 2013, 64, 247–271 CrossRef CAS PubMed.
- S. P. Velsko and G. R. Fleming, Chem. Phys., 1982, 65, 59–70 CrossRef CAS.
- P. Fita, A. Punzi and E. Vauthey, J. Phys. Chem. C, 2009, 113, 20705–20712 CAS.
- X. Shi, E. Borguet, A. N. Tarnovsky and K. B. Eisenthal, Chem. Phys., 1996, 205, 167–178 CrossRef CAS.
- P. Sen, S. Yamaguchi and T. Tahara, Faraday Discuss., 2010, 145, 411–428 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional SSHG spectra, polarization-resolved SSHG data, analysis of the polarization profiles, determination of the interfacial orientation, and quantum-chemical calculations. See DOI: 10.1039/c5cp06151b |
| This journal is © the Owner Societies 2016 |