
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Theoretical studies of energetics and binding isotope effects of binding a triazole-based inhibitor to HIV-1 reverse transcriptase†
A.
Krzemińska
a,
K. P.
Świderek
ab and
P.
Paneth
*a
aInstitute of Applied Radiation Chemistry, Lodz University of Technology, 90-924 Lodz, Poland. E-mail: paneth@p.lodz.pl
bDepartment de Quimica Fisica i Analitica, Universitat Jaume I, 12071 Castello, Spain
First published on 20th November 2015
Abstract
Understanding of protein-ligand interactions is crucial for rational drug design. Binding isotope effects, BIEs, can provide intimate details of specific interactions between individual atoms of an inhibitor and the binding pocket. We have applied multi-scale QM/MM simulations to evaluate binding energetics of a novel triazole-based non-nucleoside inhibitor of HIV-1 reverse transcriptase and to calculate associated BIEs. The binding sites can be distinguished based on the 18O-BIE.
Introduction
Human immunodeficiency virus (HIV-1) is a cytopathic human retrovirus that primarily infects CD+ helper T lymphocytes leading to the loss of their number and function. The progressive failure of the immune system associated with the loss of predominant T lymphocytes and other cells critical to this system, e.g. macrophages or dendritic cells, leads to acquired immune deficiency syndrome (AIDS). The progressive disappearance of immunity weakens the body, allows opportunistic infections to evolve, leads to cancers and ultimately to premature death.1 Nowadays, treatment of HIV-1 infected patients relies on cocktails of different drugs. Due to undesired side-effects, especially occurring during long-term treatments, there has been considerable interest in developing new inhibitors targeting three HIV-1 enzymes: integrase (IN), protease (PR) and reverse transcriptase (RT).2 Herein we focus on an inhibitor of the latter enzyme.HIV-1 RT is an asymmetric heterodimer, composed of p66 and p51 subunits, which catalyses the transcription of single-stranded viral RNA into viral DNA double helices.3 The enzyme contains one DNA polymerase active site and one ribonuclease H (RNase H) active site, both of which reside in the p66 subunit in spatially distinct regions while the smaller p51 chain is responsible for structural scaffolding. The hydrophilic polymerase active site is located between palm (light blue), finger (blue) and thumb (cyan) subdomains (see Fig. 1) and is made up of Asp110, Asp185 and Asp186 amino acids that can coordinate two Mg2+ or two Mn2+ cations.4 The allosteric cavity, composed of Lys103, Tyr181, Tyr188, Phe227, Trp229, Leu234 and Tyr318 residues5 (highlighted in Fig. 1), lies approximately 10 Å from the polymerase active site. The RNase H active site comprising Asp443, Asp498 and Asp549 amino acids and two bound Mg2+ cations6 is located 60 Å from the allosteric pocket. The two active sites and the allosteric cavity constitute a key target of antiviral research. Nucleoside reverse transcriptase inhibitors (NRTIs) inhibit the polymerase active site and need to be phosphorylated by cellular kinases to active triphosphate form, which competes with natural dNTPs and finally becomes incorporated into the growing primer. Nucleotide reverse transcriptase inhibitors (NtRTIs), in contrast to NRTIs, are structures already equipped with the phosphonate group and after the phosphorylation step are able to terminate DNA synthesis also by the competitive mechanism.7 Non-nucleoside RT inhibitors (NNRTIs), on the other hand, follow the non-competitive mechanism of inhibition. They are considered to be highly specific compounds that bind tightly in the allosteric pocket, acting on the enzyme subunits as a molecular wedge.8,9 Simultaneous inhibition by combining nucleoside and non-nucleoside inhibitors is also currently explored.10,11 To date eight NRTIs, two NtRTIs and five NNRTIs have been approved by FDA.12 In the present study we focus on a ligand that can bind either in the allosteric cavity or the RNase H active site of HIV-1 RT and compare it to ligands designed specifically for one of these sites (structural formulas of all ligands are presented in ESI,† Fig. S1).
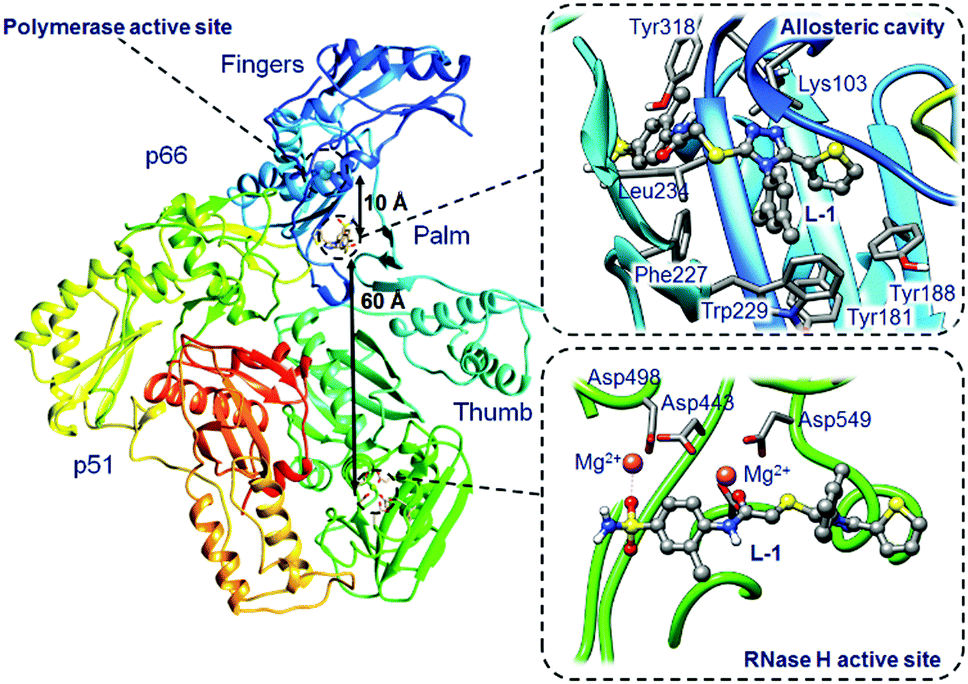 | ||
| Fig. 1 Representation of HIV-1 RT with two binding sites, allosteric cavity (upper right box) and RNase H active site (lower right box) with a bound L-1 inhibitor. | ||
An accurate description of binding affinity of an inhibitor to an enzyme is still a challenging task.13,14 Nevertheless, using Monte Carlo simulation, Jorgensen and co-workers estimated binding affinities of several NNRTIs.15–18 Binding energies of NNRTIs have also been studied by MM-PBSA19 and quantum calculations20 including the ONIOM method.21 Warshel and co-workers obtained the absolute binding free energies of NNRTIs by the PDLD/S-LRA/b method.22 Our own experience, first validated on the FDA approved drugs, indicates that binding free energies of HIV-1 RT complexes with NNRTIs and NRTIs can be efficiently obtained using a thermodynamic cycle from alchemical free energy perturbation (FEP) calculations.23,24
Recently, Schramm and co-workers showed that binding isotope effects, BIEs,25 exhibit different values upon binding an inhibitor, para-aminobenzoic acid, to an enzyme (dihydropteroate synthase) from different sources (Staphylococcus aureus and Plasmodium falciparum). In this communication we extend these findings to the identification of the binding site within a single enzyme using BIEs on the example of a novel potent HIV-1 RT triazole-based inhibitor.
Computational modelling and methods
System setup
Novel triazole derivative N-(2-chloro-4-sulphamoylphenyl)-2-((4-(2,4-dimethyl-phenyl)-5-(thiophen-2-yl)-4H-1,2,4-triazol-3yl)sulphanyl)-acetamide, L-1,26 which was found to be a potent NNRTI, has been docked to the allosteric cavity and the RNase H active site of HIV-1 RT based on the crystallographic structure from the Protein Data Bank with PDB ID 2RKI27 using Glide package as implemented in the Schrödinger program.28 Both complexes are depicted in Fig. 1. The 2RKI structure was crystalized with 4-benzyl-3-[(2-chlorobenzyl)sulfanyl]-5-thiophen-2-yl-4H-1,2,4-triazole mole-molecule bound in the allosteric cavity. The native ligand was removed from 2RKI and L-1 was docked to the allosteric cavity and to the active site using Glide XP.29 The pKa for titratable amino acids was calculated using PROPKA3.0.30–32 Assuming a physiological pH value, hydrogen atoms were added using the tLEAP module of AMBER program.33 Neutralization of the L-1-allosteric cavity and L-1-RNase H active site complexes was completed by adding 15 and 9 Cl− counterions, respectively. Finally, both complexes were placed in orthorhombic boxes of TIP3P34 water molecules of 144 × 160 × 144 Å3. Parameters for L-1 were obtained from GAFF35 as implemented in AMBER Tools. Afterwards, several optimization and dynamics simulations were performed using AMBER ff0336 implemented in NAMD37 program with a time step of 1 fs and periodic boundary conditions using the particle mesh Ewald method.38 The cut-off for nonbonding interactions was applied using a smooth switching function with a radius range from 14.5 to 16 Å. First energy optimizations were carried out by means of the conjugated gradient algorithm. The systems were heated by increasing temperature from 0 to 300 K with 0.001 K increment. The equilibration of the systems was achieved during 300 ps of Langevin–Verlet dynamics at 300 K. Then 2 ns of NVT MM MD were carried out. Subsequently, 200 ps for the L-1-cavity and 1 ns for L-1-RNase H complexes were simulated using QM/MM MD simulations at the AM139/AMBER:TIP3P theory level as implemented in fDynamo.40 All atoms beyond 20 Å from L-1 were kept frozen. In order to describe the binding energy of L-1 to HIV-1 RT, a reference system of L-1 in an aqueous solution of the equivalent size (144 × 160 × 144 Å3) was simulated during 200 ps of AM1/TIP3P dynamics.Alchemical free energy perturbation (FEP) method
The enzyme–ligand binding affinity is a main goal of drug design. Among the different methods developed for this purpose, the alchemical free energy perturbation (FEP) combined with QM/MM molecular dynamics, while not providing detailed information about the full binding process, delivers reliable values of binding free energy, ΔΔGbind, at a reasonable computational cost. The FEP method by means of the thermodynamic cycle was first introduced by Warshel and co-workers.41 Herein, a series of QM/MM MD have been carried out at 300 K in the NVT ensemble with two parameters λ and γ, which correspond to electrostatic and van der Waals interactions, respectively: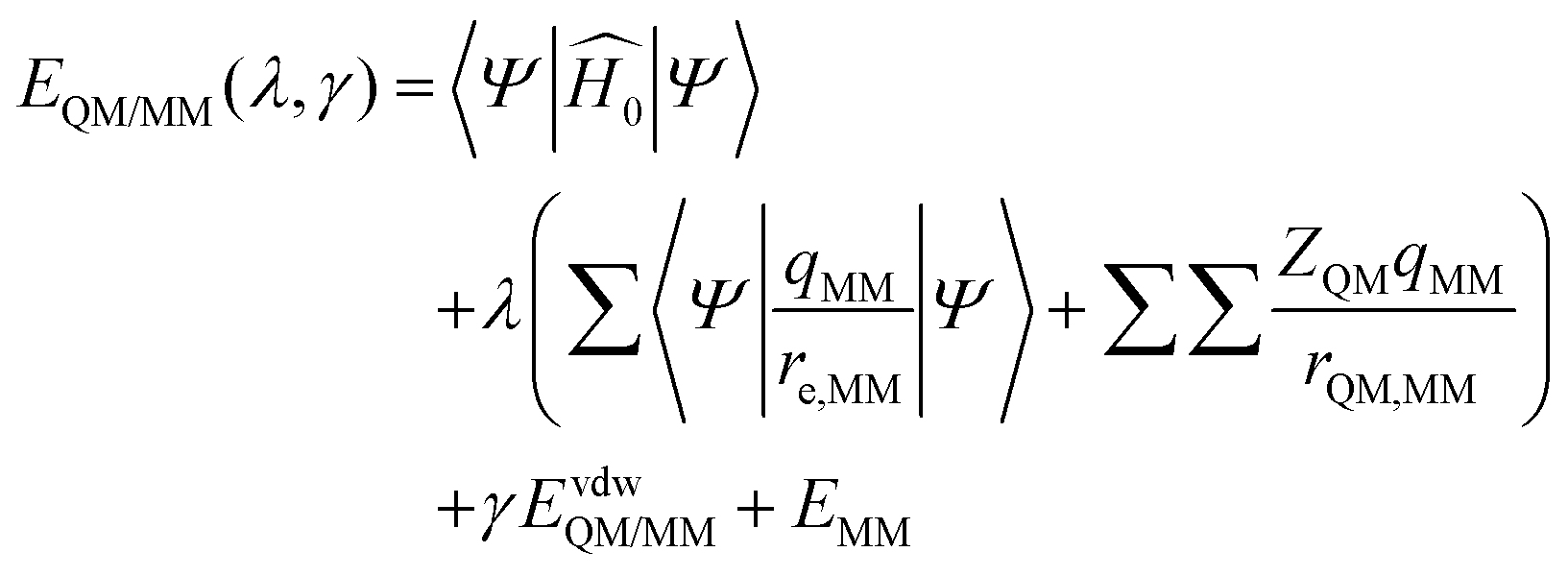 | (1) |
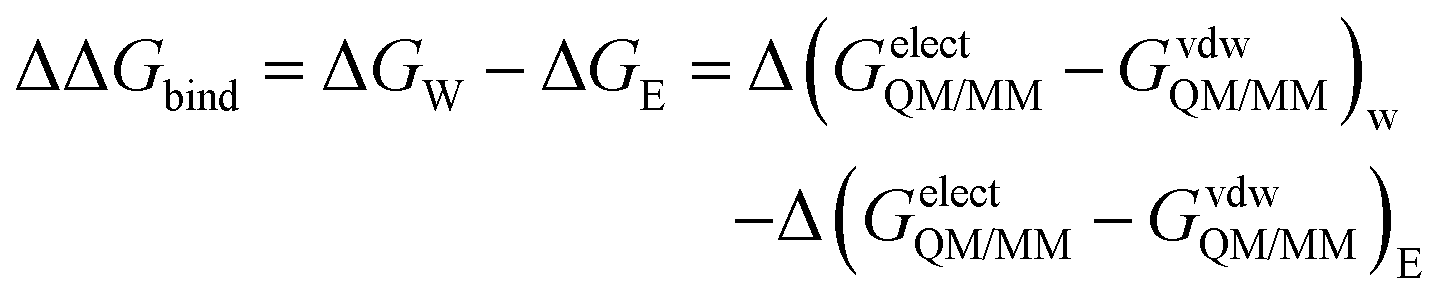 | (2) |
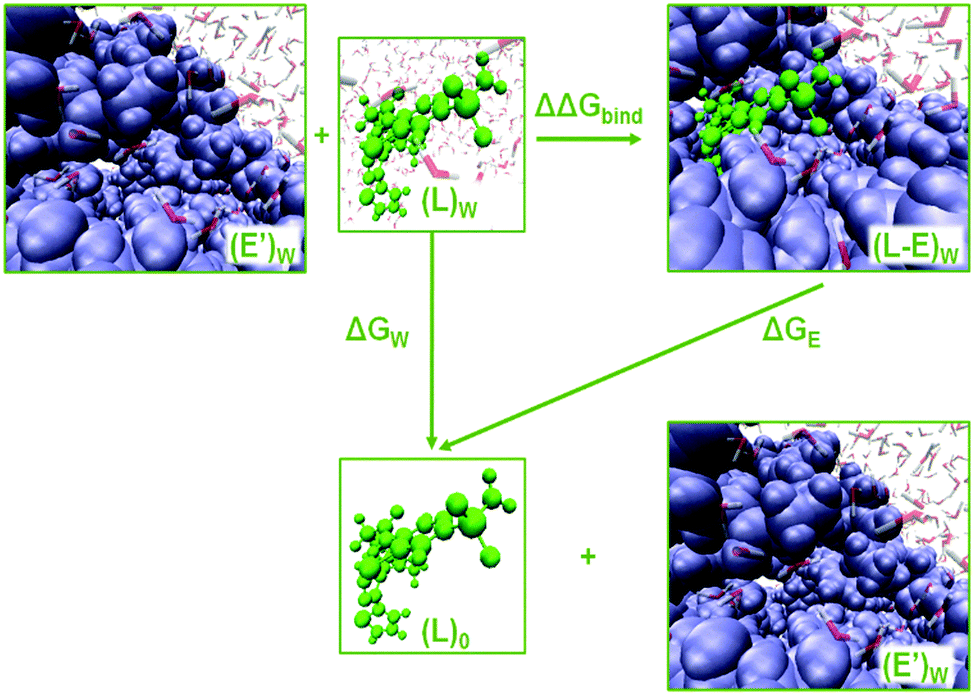 | ||
| Fig. 2 Thermodynamic cycle to compute enzyme–ligand binding free energies from the FEP method, where (E) is the enzyme with ligand (L) in its binding site, E′ is the apo form of enzyme, and (L)0 is the ligand in the gas phase. | ||
The potential energy
In order to describe individual interactions between allosteric cavity amino acids and the triazole molecule, the analysis of averaged potential energy has been performed through QM/MM MD based on eqn (3)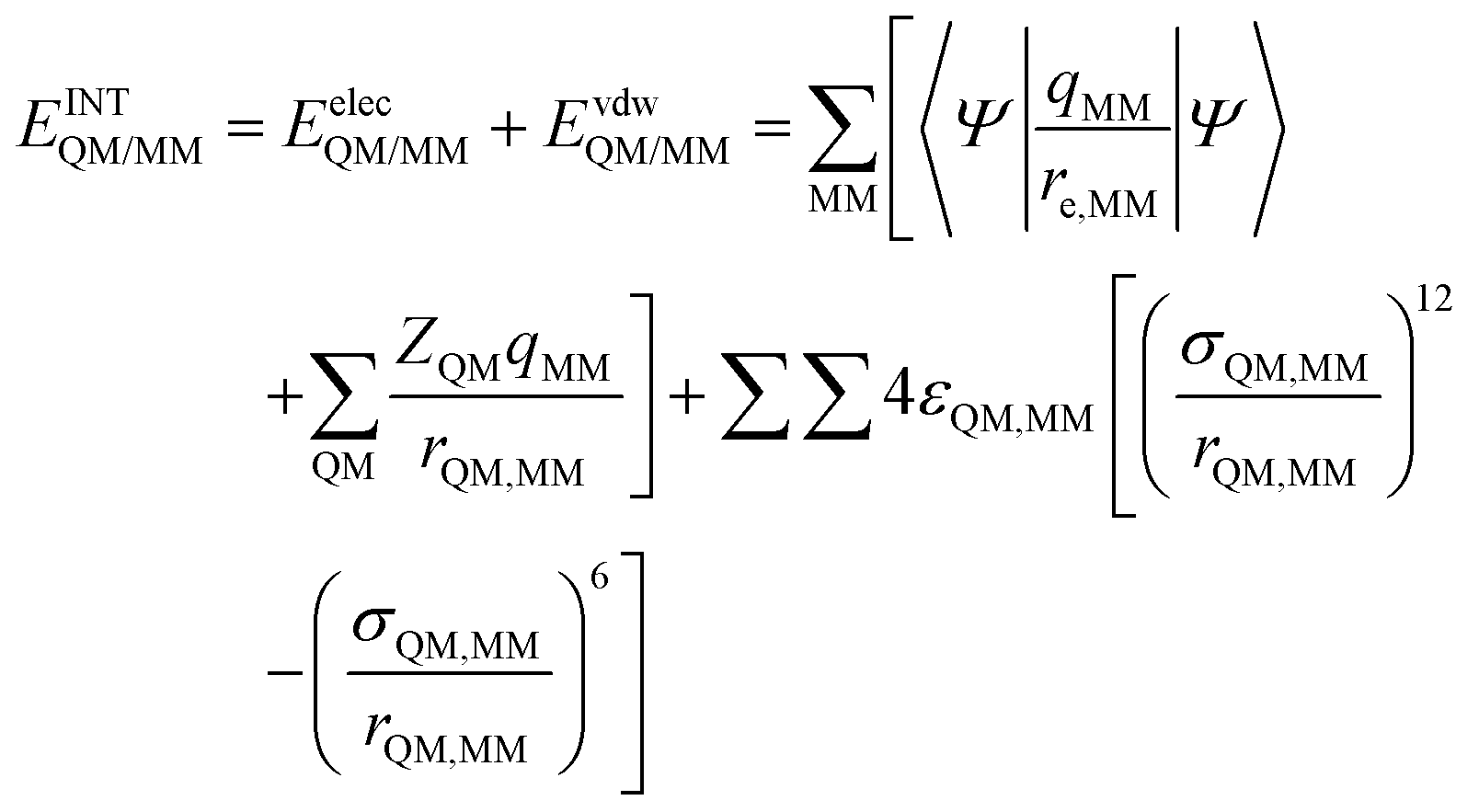 | (3) |
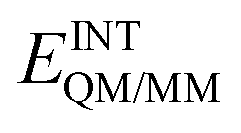 is composed of electrostatic,
is composed of electrostatic, 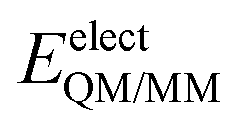 , and van der Waals,
, and van der Waals, 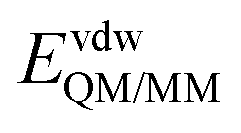 , interaction terms as a function of interaction energy between QM and MM parts. The polarized QM subsystem includes the coulombic interaction of the QM nuclei (ZQM) and the electrostatic interaction of the polarized electronic wave-function (Ψ) with enzyme amino acid charges (qMM), while the MM part brings non-polarizable potential contribution.
, interaction terms as a function of interaction energy between QM and MM parts. The polarized QM subsystem includes the coulombic interaction of the QM nuclei (ZQM) and the electrostatic interaction of the polarized electronic wave-function (Ψ) with enzyme amino acid charges (qMM), while the MM part brings non-polarizable potential contribution.
Molecular electrostatic potential (MEP)
The molecular electrostatic potential, MEP, describes the interaction energy between the charge distribution of a molecule and a unit positive charge. This method allows predicting nuclear and electronic charge distribution of a given molecule. Despite the fact that polarization is not included, charge distribution remains unperturbed by the external test charge, and the MEP is still effective for the interpretation and prediction of chemical reactivity.42 Three-dimensional MEP surfaces for L-1 extracted from the allosteric cavity and the RNase H active site were obtained at the B3LYP/6-31+G(d,p) theory level using Gaussian09.43 Negative electronic potential corresponds to the most nucleophilic regions (red), while positive electronic potential indicates the most electrophilic regions (blue).Binding isotope effects (BIEs)
Binding isotope effects can provide information on conformational changes and ligand–enzyme interactions.44 In combination with the experimental values they can also be used to determine the actual binding site. We have already successfully used BIEs to analyse interactions between ligands and HIV-1 RT.24 BIEs can be expressed by the binding free energy of the light, L, and heavy, H, isotopologs considering the relationship between the binding free energy and the equilibrium constant: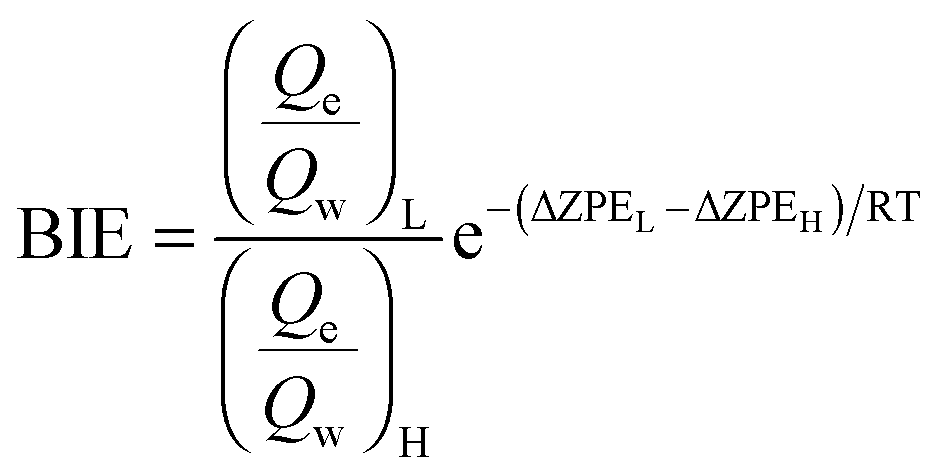 | (4) |
Results and discussion
The obtained total free energies of binding inhibitors to HIV-1 RT binding sites are collected in Table 1. A more extensive comparison is presented in Fig. 3. There are some difficulties in the validation of ΔΔGbind using experimental data. Our experience shows that IC50 measurements are not suitable for this purpose23,24 in contrast to the reciprocal of the dissociation constant (KI) of the protein–ligand complex.47 Unfortunately, no systematic study of KI values of different NNRTIs measured under the same experimental conditions can be found in the literature. The evaluation of our FEP calculations was made by using NRTI23 and NNRTI24 drugs approved by FDA, where structures of NRTIs correspond to 2,7-dihydroxy-4-1(methylethyl)-2,4,6-cycloheptatrien-1-one (LNA-1), 3-cyclopentyl-1,4-dihydroxy-1,8-naphthyridin-2(1H)-one (LNA-2), ethyl 1,4-di-hydroxy-2-oxo-1,2-dihydro-1,8-naphthyridine-3-carboxylate (LNA-3), and 3-[4-(diethylamino)phenoxy]-6-(ethoxycarbonyl)-5,8-dihydroxy-7-oxo-7,8-dihydro-1,8-naphthyridin-1-ium (LNA-4), while structures of NNRTIs correspond to nevirapine (LNN-1), efavirenz (LNN-2), rilpivirine (LNN-3), etravirine (LNN-4), and delavirdine (LNN-5), for more details see Fig. S1 in the ESI.† In a hydrophobic pocket van der Waals interactions are the most favourable, as observed for NNRTIs. van der Waals interactions are responsible for the strong (−29.2 kcal mol−1) binding of L-1 in the allosteric cavity. It is worth noticing that electrostatic interactions between L-1 and both water molecules and the allosteric site have the same ΔGelect value which indicates that the electrostatic term does not influence protein–ligand interactions. In this case the magnitude of the total energy of binding of L-1 to the allosteric cavity is thus solely due to the van der Waals term (see the green bars in Fig. 3), which is preferable in the hydrophobic pocket. Usually, the van der Waals term is much smaller than the electrostatic term, hence the relative ΔΔGvdW value obtained for L-1 is the highest among NNRTIs. This makes L-1 one of the most potent NNRTIs. Binding of L-1 to the RNase H active site is due to preferable electrostatic forces with ΔΔGbind equal to −36.5 kcal mol−1.| Inhibitor | Cavity | ΔGelect-aq | ΔGelect-HIV | ΔΔGelect | ΔGvdW-aq | ΔGvdW-HIV | ΔΔGvdW | ΔΔGbind |
|---|---|---|---|---|---|---|---|---|
| L-1 | RNase H | −33.6 | −56.2 | −22.5 | −37.2 | −51.2 | −14.0 | −36.5 |
| L-1 | Allosteric | −33.6 | −33.6 | 0.0 | −37.2 | −66.4 | −29.2 | −29.2 |
| LNA-3 | RNase H | −8.2 | −37.7 | −29.5 | −23.1 | −23.8 | −0.7 | −30.2 |
| LNN-3 | Allosteric | −7.1 | −14.9 | −7.8 | −29.5 | −50.9 | −21.4 | −29.2 |
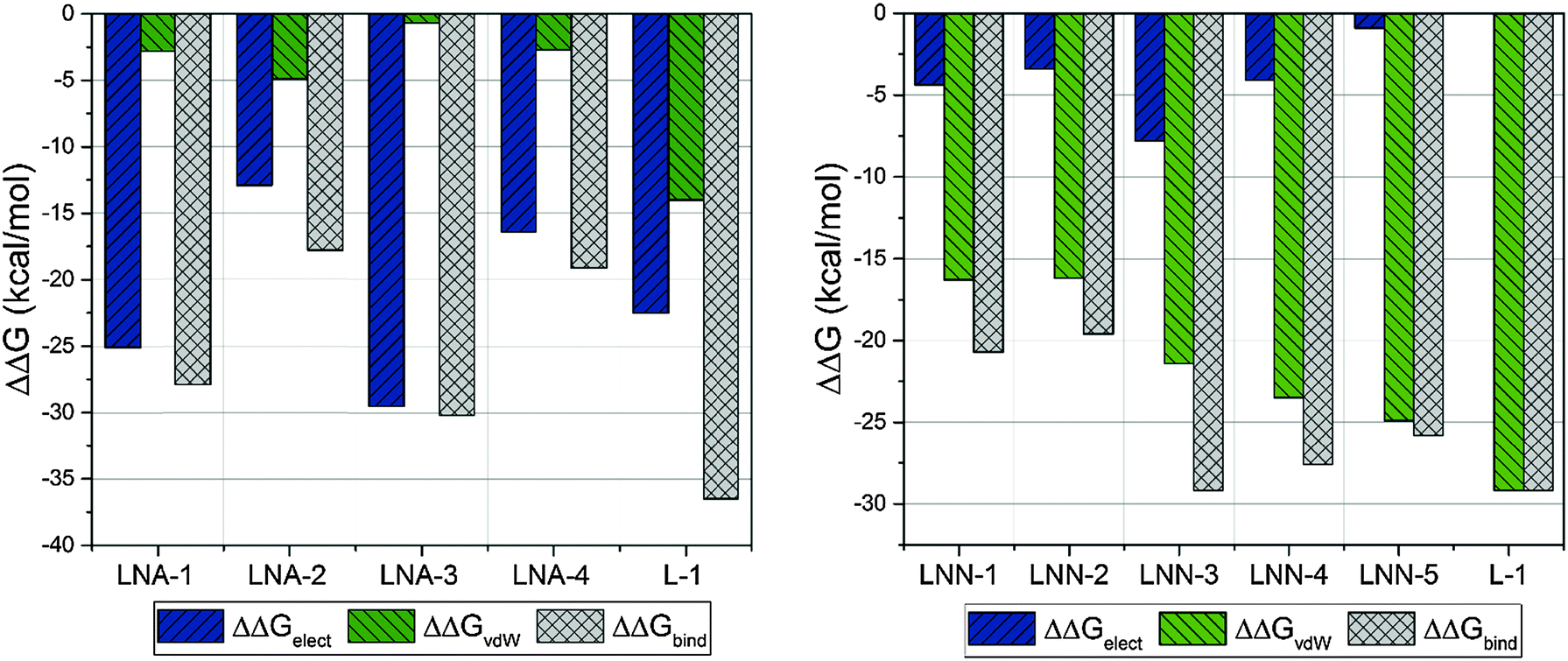 | ||
| Fig. 3 Contributions of electrostatic and van der Waals terms to total binding free energy obtained from FEP calculations. | ||
There is a meaningful difference between L-1 interactions in aqueous solution and the enzyme environment in both electrostatic and van der Waals terms. NRTIs show the same behaviour in the case of electrostatic interactions, but opposite in van der Waals interactions. Consequently, binding L-1 to the RNase H site is accompanied by both strong electrostatic and van der Waals terms, and indicates that L-1 can successfully bind into the RNase H active site. The potential energy of electrostatic and van der Waals interactions between L-1 and allosteric cavity amino acids has been obtained along the QM/MM dynamics; the averaged results are displayed in Fig. 4. Negative values correspond to favourable interactions and stabilize the enzyme–ligand complex, while positive values represent unfavourable interactions. The strength of interactions depends on the nature of the ligand (size, stereochemistry, number and type of functional groups) and differs among NNRTIs. The crystallographic structures5,48 indicate that Leu100, Lys101, Lys103, Val106, Thr107, Val108, Val179, Tyr181, Tyr188, Val189, Gly190, Phe227, Trp229, Leu234, and Tyr318 all from chain A and Glu138 from chain B are usually responsible for inhibitor–enzyme interactions. The L-1-allosteric cavity complex has meaningful interactions with Leu100, Lys103, Lys104, Val106 and Pro236 as shown in Fig. 4. L-1 interacts also with Phe227, His235 and Tyr318. Residues Lys101, Thr107, Val108, Val189, Gly190, Trp229, Leu234, and Tyr318 are too far, while Val179, Tyr181, Tyr188, and Glu138 from chain B exercise negligible bending affinity toward L-1. The main contribution of favourable electrostatic interactions, shown in Fig. 4 and 5, is observed between 1,2-triazole nitrogen atoms and Lys103. However, these interactions do not enhance binding affinities, because interactions of L-1 with hydrogen atoms of solvent molecules fully compensate them. Significantly weaker are favourable electrostatic interactions with Lys104 and Tyr318, wherein carbonyl oxygen from Lys104 interaction with the hydrogen atom from the aromatic ring is further stabilized by the π-stacking with Tyr318. These positive electrostatic interactions are compensated by the electrostatic repulsion of Asp192. The determinant factor of L-1-RT attraction comes from the van der Waals interactions, not present in water, especially with Val106, Leu100, Pro236, and His235. Residues Tyr181 and Tyr188 are stabilizing one aromatic ring of L-1 with two methyl groups and Phe227 interacts by π-stacking with another aromatic ring neighbouring the sulphate group. The electrostatic positive and negative terms are equal, therefore the binding process is a result of van der Waals interactions between L-1 and the allosteric cavity; the potential energy of van der Waals interactions is sufficient for incorporating and maintaining L-1 in this hydrophobic pocket.
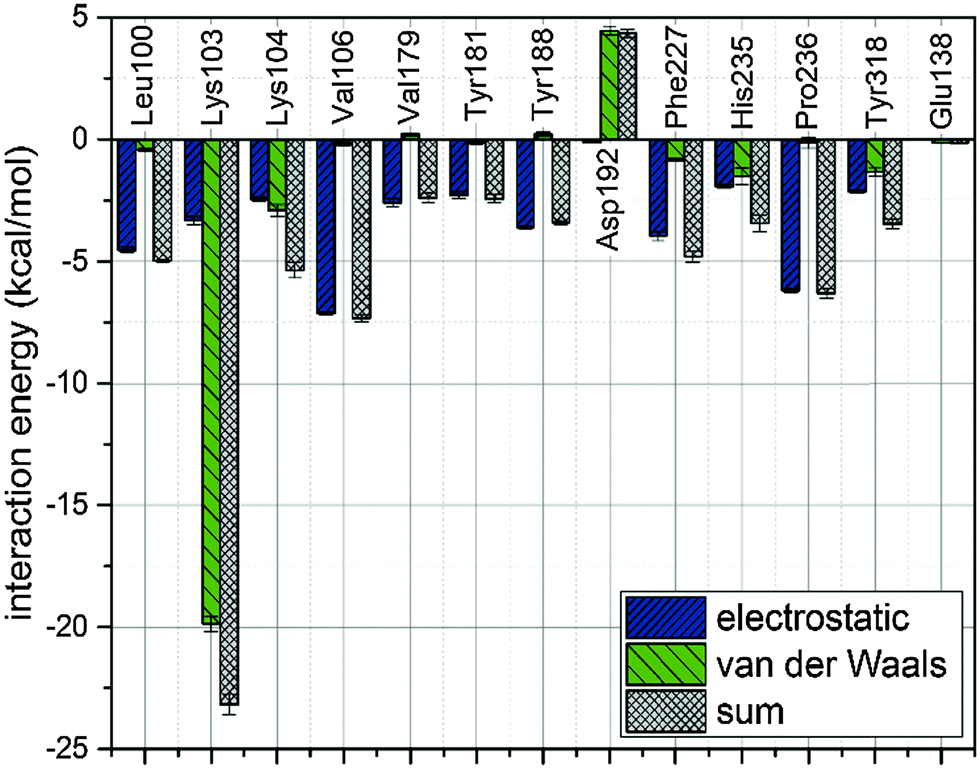 | ||
| Fig. 4 Analysis of potential energy of interactions between L-1 and allosteric cavity amino acids. | ||
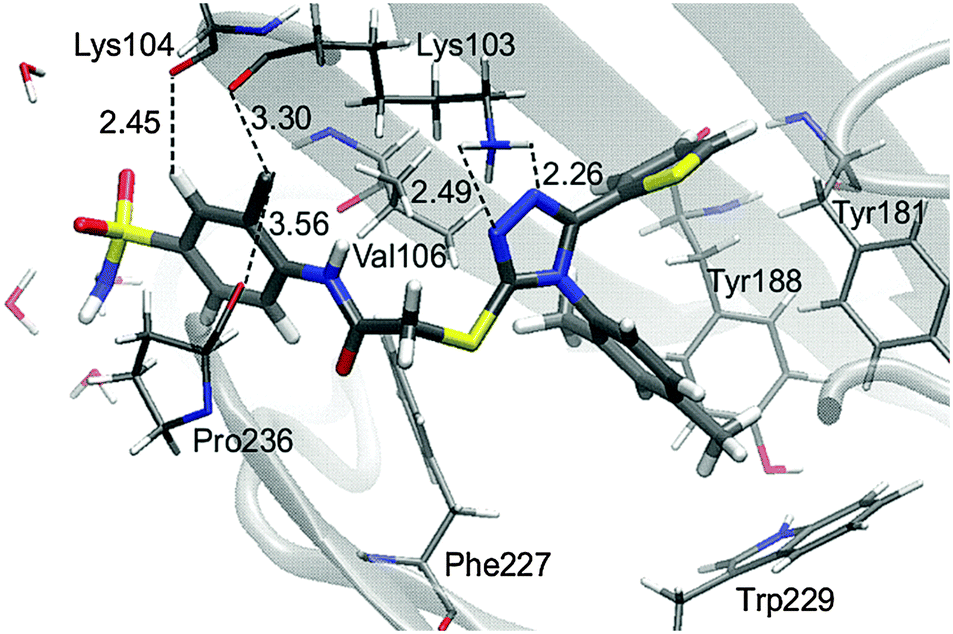 | ||
| Fig. 5 Arrangement of L-1 at the allosteric cavity with the average of key distances in Å. | ||
In contrast to the L-1-allosteric cavity complex, the electrostatic interactions between L-1 and RNase H active sites are crucial to hold the ligand in the active site centre. The RNase H active site is made up of Asp443, Glu478, Asp498 and Asp549 amino acids, which coordinate two Mg2+. The magnesium cation tends to create six coordination bonds but in the active site usually only five are occupied. Hence, two cations can interact with ligands by two relatively strong coordination bonds (Fig. 6).
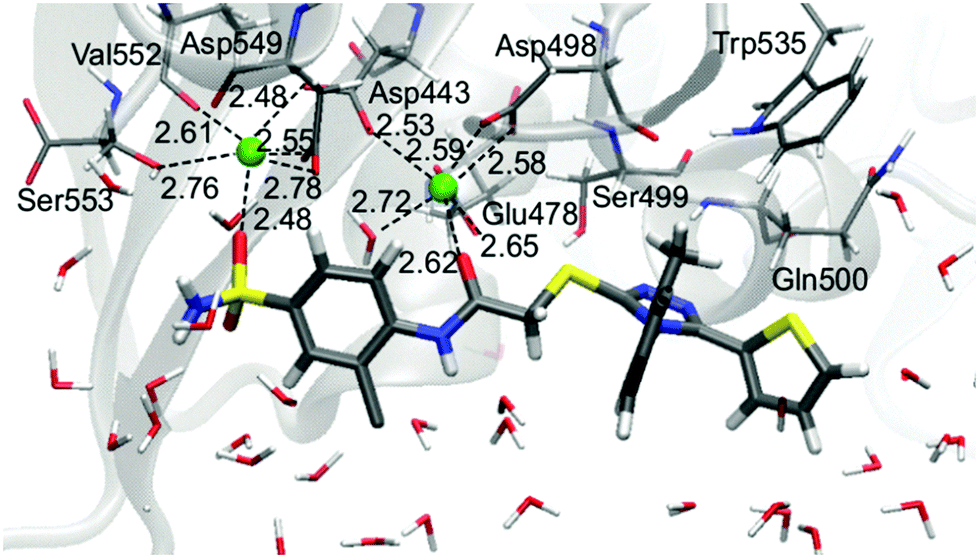 | ||
| Fig. 6 Arrangement of L-1 at the RNase H active site with average key distances in Å. Magnesium cations are highlighted as green balls. | ||
Small ligands, like LNA-1,23 interact with both Mg2+ without reorganizing active sites. An opposite situation is observed for bigger molecules such as LNA-4 and L-1. Both alter the RNase H active site in the same way, causing an increase of the distance between magnesium cations and forcing coordination of Val552 and Ser553 by one Mg2+. The open and flexible character of the RNase H region supports the reorganization of the active site. This dynamic effect, observed along MM MD, supports binding of a bigger ligand such as L-1.
Recently, effective medical treatment of HIV infection has been focused on the composite approach combining different targets (enzymes) of the virus as well as different sites within a single enzyme. It is assumed that inhibitors of different types bind to different sites of the enzyme. However, recent reports on finding several alternative binding sites within HIV-1 RT shed new light on the actual binding sites of particular inhibitors.49 In order to analyse the specificity of the triazole-based inhibitors we have performed calculations of the binding energetics of L-1 in two binding spots: the allosteric centre and the RNase H active site. After QM/MM MD simulations we have extracted the last structure of L-1 from the L-1-cavity and L-1-RNaseH complexes, and calculated three-dimensional MEP surfaces. The obtained MEP surfaces are shown in Fig. 7, where the most nucleophilic regions are in red (−0.1 a.u.) and the most electrophilic regions are in blue (+0.1 a.u.). Both structures have similar distribution of nuclear and electronic charges. As expected, the triazole ring and the sulphate group exhibit negative electronic potential. The sulphate group is dangling almost outside of the hydrophobic pocket, thus it can interact with water molecules. In the L-1-RNase H complex, the active site is open to the aqueous environment and also in this case the sulphate group can interact with solvent molecules. The L-1 nitrogen atoms are deep inside the allosteric cavity and in the presence of Lys103 they act as nucleophiles, while in the RNase H active site they interact electrostatically with Asp498, Gln500 and Tyr501. Consequently, there is no big difference between the charge distribution of L-1 extracted from an allosteric cavity and from the RNase H active site. These results indicate that L-1 can bind to both binding sites.
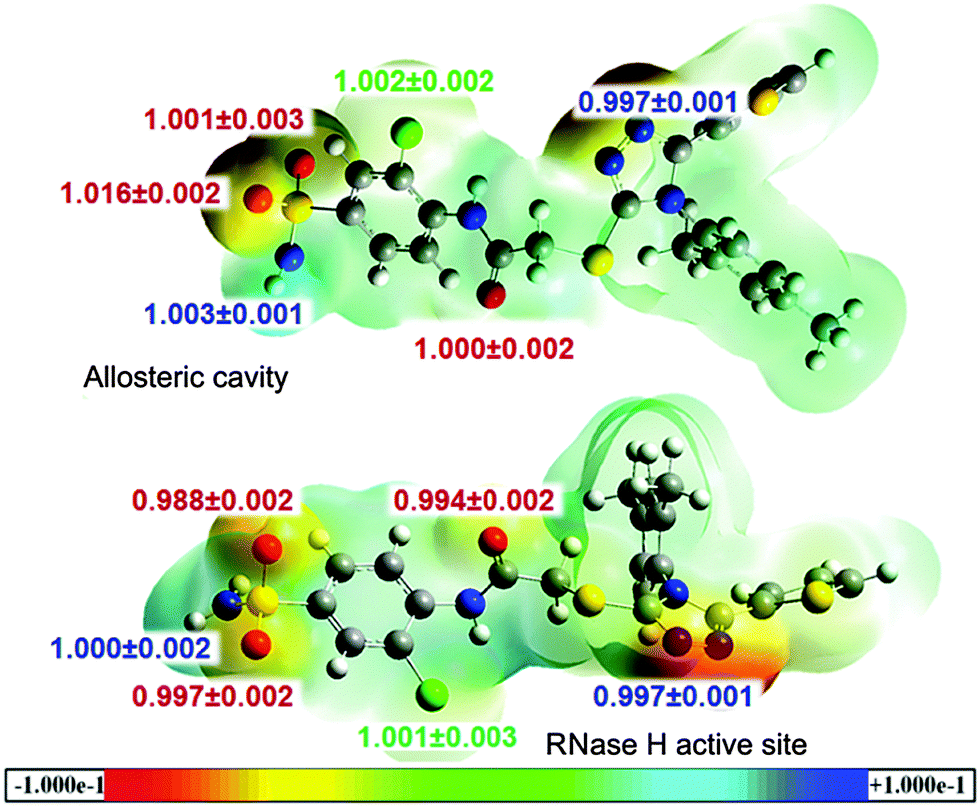 | ||
| Fig. 7 Three-dimensional MEP surfaces for the L-1 extracted from the allosteric cavity and the RNase H active site with heavy atoms BIEs. Negative electronic potential corresponds to the most nucleophilic regions (red), while positive electronic potential indicates the most electrophilic regions (blue). | ||
Another analysis of interactions of L-1 in complexes with the allosteric cavity and the RNase H active site has been based on heavy atom (13C, 15N, 18O, 37Cl and 34S) BIEs. The calculated values are collected in Fig. 7. BIEs can provide information on differences in interactions between L-1 dissolved in water and bound in RT binding sites. Despite the dissimilarity in the molecular character of the hydrophobic cavity and the hydrophilic RNase H active site, in both RT binding sites lack of BIEs for most of the atoms has been observed. The largest isotope effects were obtained for 18O of the sulphate group and apparently this isotope effect can be experimentally used to distinguish the place in which L-1 is bound; a normal 18O-BIE (1.016 ± 0.002) is expected when L-1 binds in the allosteric cavity, where oxygen atom interacts (see Fig. 4) with the hydrogen atom from Val106 (the distance is 4.23 Å) and is also surrounded by three water molecules. On the other hand an inverse BIE (0.988 ± 0.002) for sulphate group 18O atoms is expected for the complex of L-1 with RNase H, originating from strong interactions with one of the active site Mg2+ cations (the distance is 2.41 Å) and additionally from stabilization by the hydrogen atom (the distance is also 2.41 Å) from the methyl group of Ala445 and one water molecule. Isotope effect differentiation of the other oxygen of the sulphonyl group is not possible as it is only slightly inverse (0.997 ± 0.001) when L-1 is bound in the RNase H active site in contrast to the lack of the isotope effect of this oxygen atom in the allosteric cavity. This slightly inverse value of BIE originates from the interaction with the second Mg2+ cation (the distance is 2.56 Å). Although electrostatic interaction between the nitrogen atom of the triazole ring and the hydrogen atom from Lys103 (the distance is 2.52 Å) is unfavourable in the allosteric cavity, while in the RNase H active site electrostatic interactions of this atom with Gln500 and Tyr501 hydrogen atoms tend to stabilize the complex (the distances are 2.55 Å and 2.10 Å, respectively), there is no reflection of these differences in the isotopic binding patterns with both 15N-BIEs being slightly inverse (0.997 ± 0.001) and equal. Finally, a negligible normal BIE (1.002 ± 0.002) was calculated for the 37Cl-BIE when L-1 binds in the RNase H active site, and a similar value was obtained when L-1 is complexed in the allosteric cavity. Although it should be kept in mind that equilibrium isotope effects, such as BIEs, are generally small and hard to measure experimentally, the above analysis illustrates that it is possible to distinguish binding sites based on their values in favourable cases.
Conclusions
The novel inhibitor of HIV-1 RT, L-1, has been proposed and has been examined based on theoretical simulations. The L-1 molecule was docked to both RT binding sites: allosteric cavity and RNase H active site. Final complexes of the L-1-binding site have been obtained after long QM/MM dynamics. Once the stability of both systems was confirmed, the interactions between L-1 and RT binding sites have been quantified by free energy of binding computed using the FEP method. The analysis of this affinity has been also enriched by potential energy calculations, three-dimensional MEP isosurfaces, structural analysis and heavy atom (13C, 15N, 18O, 37Cl and 34S) BIEs. The obtained results confirmed the differences between the allosteric cavity and the RNase H binding sites, as well as dissimilarity in the ligand binding pathways. In the allosteric cavity the inhibitor binds by van der Waals affinities, which are preferable in this hydrophobic pocket, while the electrostatic interactions are unfavourable. The opposite situation was observed for binding the ligand to the RNase H active site, where binding occurs by electrostatic forces.Inhibition is accompanied by the release of standard free energy, ΔΔGbind. The values of ΔΔGbind were obtained by the alchemical FEP method. Comparison of different NNRTIs, and NRTIs, L-1 bound in the allosteric cavity and the RNase H active site shows that L-1 has the largest negative standard free energy of binding, indicating that L-1 is able to successfully inhibit both the allosteric cavity and RNase H sites. Also the three-dimensional MEP isosurface display shows that there is no large difference between L-1 charge distribution in the allosteric cavity and the RNase H active site.
BIE values are almost identical for both L-1-enzyme complexes, with one exception. In contrast to the L-1-allosteric cavity complex, where a normal (larger than unity) 18O-BIE is expected, in the L-1-RNase H active site an inverse BIE for two oxygens from the sulphate group is predicted. This occurs due to the strong interactions with two Mg2+ originally belonging to the RNase H active site. Thus BIEs allow us to distinguish to which site L-1 binds. Experimental determination of this 18O-BIE is being currently carried out in our laboratory.
Acknowledgements
This work has been supported by the grants 2011/02/A ST4/00246 (Maestro) from the Polish National Research Center (NCN) and 0478/IP3/2015/73 (Iuventus Plus) from the Polish Ministry of Science and Higher Education. The authors also acknowledge the Servei d'Informatica, Universitat Jaume I for generous allotment of the computer time.References
- R. A. Weiss, Science, 1993, 260, 1273–1279 CAS.
- J. A. Moss, Radiol. Technol., 2013, 84, 247–267 Search PubMed.
- J. M. Coffin, S. H. Hughes and H. E. Varmus, Retroviruses, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 1997 Search PubMed.
- B. A. Larder, D. J. Purifoy, K. L. Powell and G. Darby, Nature, 1987, 327, 716–717 CrossRef CAS PubMed.
- J. Ding, K. Das, Y. Hsiou, S. G. Sarafianos, A. D. Jr Clark, A. Jacobo-Molina, C. Tantillo, S. H. Hughes and E. Arnold, J. Mol. Biol., 1998, 284, 1095–1111 CrossRef CAS PubMed.
- E. Rosta, M. Nowotny, W. Yang and G. Hummer, J. Am. Chem. Soc., 2011, 23, 8934–8941 CrossRef PubMed.
- Y. Mehellou and E. D. Clercq, J. Med. Chem., 2010, 2, 521–538 CrossRef PubMed.
- F. Esposito, A. Corona and E. Tramontano, Mol. Biol. Int., 2012, ID 586401, 23 Search PubMed.
- S. G. Sarafianos, B. Marchand, K. Das, D. M. Himmel, M. A. Parniak, S. H. Hughes and E. Arnold, J. Mol. Biol., 2009, 385, 693–713 CrossRef CAS PubMed.
- K. Das, S. E. Martinez, J. D. Bauman and E. Arnold, Nat. Struct. Mol. Biol., 2012, 19, 253–259 CAS.
- R. Meleddu, S. Distinto, A. Corona, G. Bianco, V. Cannas, F. Esposito, A. Artese, S. Alcaro, P. Matyus, D. Bogdan, F. Cottiglia, E. Tramontano and E. Maccioni, Eur. J. Med. Chem., 2015, 93, 452–460 CrossRef CAS PubMed.
- http://www.fda.gov .
- A. R. Leach, B. K. Shoichet and C. E. Peishoff, J. Med. Chem., 2006, 49, 5851–5855 CrossRef CAS PubMed.
- C. J. Woods, M. Malaisree, S. Hannongbua and A. J. Mulholland, J. Chem. Phys., 2011, 134, 054114 CrossRef PubMed.
- R. H. Jr. Smith, W. L. Jorgensen, J. Tirado-Rives, M. L. Lamb, P. A. Janssen, C. J. Michejda and M. B. Kroeger Smith, J. Med. Chem., 1998, 41, 5272–5286 CrossRef PubMed.
- R. C. Rizzo, J. Tirado-Rives and W. L. Jorgensen, J. Med. Chem., 2001, 44, 145–154 CrossRef CAS PubMed.
- W. L. Jorgensen, M. Bollini, V. V. Thakur, R. A. Domaoal, K. A. Spasov and K. A. Anderson, J. Am. Chem. Soc., 2011, 133, 15686–15696 CrossRef CAS PubMed.
- D. J. Cole, J. Tirado-Rives and W. L. Jorgensen, Biochim. Biophys. Acta, 2015, 1850, 966–971 CrossRef CAS PubMed.
- J. Wang, P. Morin, W. Wang and P. A. Kollman, J. Am. Chem. Soc., 2001, 123, 5221–5230 CrossRef CAS PubMed.
- X. He, Y. Mei, Y. Xiang, D. W. Zhang and J. Z. H. Zhang, Proteins: Struct., Funct., Bioinf., 2005, 61, 423–432 CrossRef CAS PubMed.
- S. Saen-oon, M. Kuno and S. Hannongbua, Proteins: Struct., Funct., Bioinf., 2005, 61, 859–869 CrossRef CAS PubMed.
- N. Singh and A. Warshel, Proteins: Struct., Funct., Bioinf., 2010, 78, 1705–1723 CAS.
- K. Świderek, A. Martí and V. Moliner, Phys. Chem. Chem. Phys., 2012, 14, 12614–12624 RSC.
- A. Krzemińska, P. Paneth, V. Moliner and K. Świderek, J. Phys. Chem. B, 2015, 119, 917–927 CrossRef PubMed.
- C. F. Stratton, H. A. Namanja-Magliano, S. A. Cameron and V. L. Schramm, ACS Chem. Biol. DOI:10.1021/acschembio.5b00490.
- T. Fraczek, A. Paneth, R. Kaminski, A. Krakowiak and P. Paneth, J. Enzyme Inhib. Med. Chem., 2015 DOI:10.3109/14756366.2015.1039531 [Epub ahead of print].
- T. A. Kirschberg, M. Balakrishnan, W. Huang, R. Hluhanich, N. Kutty, A. C. Liclican, D. J. McColl, N. H. Squires and E. B. Lansdon, Bioorg. Med. Chem. Lett., 2008, 18, 1131–1134 CrossRef CAS PubMed.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196 CrossRef CAS PubMed.
- T. Fraczek, A. Siwek and P. Paneth, J. Chem. Inf. Model., 2013, 53, 3326–3342 CrossRef CAS PubMed.
- H. Li, A. D. Robertson and J. H. Jensen, Proteins: Struct., Funct., Bioinf., 2005, 61, 704–721 CrossRef CAS PubMed.
- D. C. Bas, D. M. Rogers and J. H. Jensen, Proteins: Struct., Funct., Bioinf., 2008, 73, 765–783 CrossRef CAS PubMed.
- M. H. M. Olsson, C. R. Sndergard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Goetz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- Y. Duan, C. Wu, S. Chowdhury, M. C. Lee, G. Xiong, W. Zhang, R. Yang, P. Cieplak, R. Luo, T. Lee, J. Caldwell, J. Wang and P. Kollman, J. Comput. Chem., 2003, 24, 1999–2012 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902–3909 CrossRef CAS.
- M. J. Field, M. Albe, C. Bret, F. Proust-De Martin and A. Thomas, J. Comput. Chem., 2000, 21, 1088–1100 CrossRef CAS.
- A. Warshel, F. Sussman and G. King, Biochemistry, 1986, 25, 8368–8372 CrossRef CAS PubMed.
- C. K. Bagdassarian, V. L. Schramm and S. D. Schwartz, J. Am. Chem. Soc., 1996, 118, 8825–8836 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- K. Świderek and P. Paneth, Chem. Rev., 2013, 113, 7851–7879 CrossRef PubMed.
- G. D. Ruggiero, S. J. Guy, S. Martí, V. Moliner and I. H. Williams, J. Phys. Org. Chem., 2004, 17, 592–601 CrossRef CAS.
- J. J. Ruiz-Pernía and I. H. Williams, Chem. – Eur. J., 2012, 18, 9405–9414 CrossRef PubMed.
- Y. Cheng and W. H. Prusoff, Biochem. Pharmacol., 1973, 22, 3099–3108 CrossRef CAS PubMed.
- L. A. Kohlstaedt, J. Wang, J. M. Friedman, P. A. Rice and T. A. Steitz, Science, 1992, 256, 1783–1790 CAS.
- J. D. Bauman, D. Patel, C. Dharia, M. W. Fromer, S. Ahmed, Y. Frenkel, R. S. K. Vijayan, J. T. Eck, W. C. Ho, K. Das, A. J. Shatkin and E. Arnold, J. Med. Chem., 2013, 56, 2738–2746 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Structural formulas for the NRTIs and NNRTIs, computed values of BIEs, the average of key distances for L-1 bound in the allosteric cavity and in the RNase H active site, and evolution of key distances during dynamic simulation for L-1 bound in the allosteric cavity and in the RNase H active site. See DOI: 10.1039/c5cp06050h |
| This journal is © the Owner Societies 2016 |