
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Is the boundary layer of an ionic liquid equally lubricating at higher temperature?†
Nicklas
Hjalmarsson
a,
Rob
Atkin
b and
Mark W.
Rutland
*ac
aSurface and Corrosion Science, KTH Royal Institute of Technology, SE-10044 Stockholm, Sweden. E-mail: mark@kth.se
bPriority Research Centre for Advanced Fluids and Interfaces, The University of Newcastle, Callaghan, NSW 2308, Australia
cChemistry, Materials and Surfaces, SP Technical Research Institute Sweden, SE-11428 Stockholm, Sweden
First published on 4th March 2016
Abstract
Atomic force microscopy has been used to study the effect of temperature on normal forces and friction for the room temperature ionic liquid (IL) ethylammonium nitrate (EAN), confined between mica and a silica colloid probe at 25 °C, 50 °C, and 80 °C. Force curves revealed a strong fluid dynamic influence at room temperature, which was greatly reduced at elevated temperatures due to the reduced liquid viscosity. A fluid dynamic analysis reveals that bulk viscosity is manifested at large separation but that EAN displays a nonzero slip, indicating a region of different viscosity near the surface. At high temperatures, the reduction in fluid dynamic force reveals step-like force curves, similar to those found at room temperature using much lower scan rates. The ionic liquid boundary layer remains adsorbed to the solid surface even at high temperature, which provides a mechanism for lubrication when fluid dynamic lubrication is strongly reduced. The friction data reveals a decrease in absolute friction force with increasing temperature, which is associated with increased thermal motion and reduced viscosity of the near surface layers but, consistent with the normal force data, boundary layer lubrication was unaffected. The implications for ILs as lubricants are discussed in terms of the behaviour of this well characterised system.
Introduction
Ionic liquids (ILs) are pure salts that are liquids at temperatures below 100 °C and have attracted significant scientific interest over the last two decades.1–3 This is because of their tunability to specific tasks and many attractive characteristics for various applications. These properties include but are not limited to: high thermal stability, low vapour pressure, high viscosity, ionic conductivity, controllable miscibility, low combustibility and a large electrochemical window. ILs are today being used, or considered for use, in a wide range of applications, such as CO2 capture,4 as electrolytes in batteries and solar cells,5 as lubricants,6 for replacement solvents in synthesis,7 and as solvents for polymers and surfactants.8,9 ILs are often classified into two groups; protic and aprotic.3,10 Protic ILs are synthesised by proton transfer from a Brønsted acid to a Brønsted base, hence creating proton donor or acceptor sites. These sites permit hydrogen bonding which influences the IL's properties.11–13 The protic IL used in this study (ethylammonium nitrate, EAN) has an extensive hydrogen bonding network14 and was first synthesised by Paul Walden in 1914.15ILs offer important advantages over conventional base-oil lubricants because of their ability to self-assemble (both in bulk and at interfaces)16–18 and their strong interactions with solid surfaces, meaning they resist “squeeze out”. This leads to improved, “intrinsic” boundary lubrication and has been demonstrated for several different surface combinations and ILs.19–23 Conversely, conventional lubricant oils depend on polar additives to adhere to surfaces and create a boundary layer.24 Also, ILs often have high thermal stabilities and low vapour pressures, two very important general properties when developing new lubricants due to the ever increasing pressure to perform at higher temperatures. The first investigation into boundary layer properties for ILs under confinement was performed using a surface forces apparatus (SFA).25 Oscillatory force profiles were collected and demonstrated strong interfacial ordering for EAN, which became weaker as the water content was increased. Two decades later, an atomic force microscope (AFM) with a sharp AFM tip was used to measure structural forces on mica, silica, and graphite. The surfaces were immersed in three different ILs: EAN, propylammonium nitrate (PAN) and 1-ethyl-3-methylimidazolium acetate.26 Structural forces were measured for all surface and liquid combinations but variations were noted, corresponding to the ion-pair size of the IL and surface type. Many articles have since been published on interfacial structuring of ILs at solid surfaces using different techniques, such as SFA,16,21,22,27–30 AFM,18,20,26,31–36 and X-ray reflectivity (XRR).37–41
Despite the fact that many lubrication applications require operation at high temperature, the effect of temperature on interfacial ordering on liquids has received little attention. This is related to experimental challenges associated with measuring structural forces at the nanoscale as temperature is increased. The first report of the effect of temperature variation on interfacial liquid structure was for octamethylcyclotetrasiloxane (OMCTS) confined between two mica surfaces between 22 and 40 °C,42 which showed that structural forces were insensitive to temperature over that range. Later, temperatures between 14 and 22 °C,43 which straddles the melting point of OMCTS (17.5 °C), were probed and it was found that structural forces were similarly unaffected. It was thus concluded ordering is not due to a surface induced local freezing. In the first AFM study on the temperature behaviour of solvation forces, two different surfaces, mica and graphite, were studied with n-alcohols (n = 8–12).44 The temperature was ramped from 25 to 60 °C and a strong effect was observed for mica but not for graphite. AFM imaging revealed that as the n-alcohols were heated above a critical temperature they changed conformation on the mica surface but not for graphite. The effect of temperature on solvation forces in OMCTS, n-hexadecane, and n-dodecanol on mica was also investigated by AFM in a more recent publication.45 Solvation layers were probed for all three liquids between 25 and 75 °C and there were significant decreases in the strength of the solvation forces and the number of layers as the temperature was increased. It was suggested that, while structural forces are only weakly temperature dependent, the squeeze out process on compression is thermally activated, which then accounted for the results. The discrepancy between these two papers for n-alcohols on graphite as a function of temperature is as yet unresolved, though as the authors in reference45 point out, the challenges of imaging adsorbed layers on surfaces18,35,36,46 are significant.
Only a few papers have previously investigated the temperature effects of IL ordering on a solid surface.34,39 Wakeham et al. used an AFM tip to study the interfacial forces as a function of temperature (14–30 °C) for five protic ILs, including EAN, on mica. For EAN, seven layers were observed at 14 °C and as the temperature was increased, the number of layers and the corresponding layer rupture force decreased. The authors attributed this to the increased thermal energy disrupting the solvophobic interactions between cation alkyl chains, as predicted in an early simulation paper.47 Mezger et al. studied three FAP-based ILs [FAP− being a tris(pentafluoroethyl)trifluorophosphate anion] between −15 and 110 °C using high-energy XRR at a charged sapphire surface and concluded that increased temperature only weakly decreased the interfacial structure, despite the measurements also straddling the melting point.39
To our knowledge, there are no previous studies of the effect of temperature on the nanotribology of ionic liquids. The only nanotribology study examining the effect of temperature was performed for dipalmitoylphosphatidylcholine bilayers.48 Friction was independent of temperature between 25 and 52 °C but the load-bearing capacity increased, which was attributed to a change of state from gel to disordered liquid. There are, however, a number of studies that have investigated the dependence of temperature on interfacial friction in the absence of lubricant.49–52 Schirmeisen et al.49 and Barel et al.51 have both investigated the effect of temperature on friction in ultrahigh vacuum and found a maximum in the friction coefficient at around 100 K which varied slightly with the material. At room temperature and above, the material dependence largely disappears and the results were explained in terms of the formation and rupture of atomic contacts. Zhao et al. studied friction over a much larger temperature range (140–750 K) on highly oriented pyrolytic graphite and found a thermally activated sliding mechanism that showed a large decrease in friction as temperature was increased from 140 to 400 K, followed by a much weaker dependence at higher temperatures.50
In this study, the nanotribological properties of EAN are investigated as a function of temperature using colloidal probe AFM.53 EAN serves as a useful model molecule for fundamental studies because more physical and structural data are available for EAN than any other IL,14,17,54–58 which facilitates data interpretation. Temperatures are examined between 25 °C and 80 °C, which is similar to the operating temperatures of car engines. The experiments reveal that while dynamic forces vary markedly with temperature, the boundary layers remain intact and thus the coefficient of friction remains essentially unchanged at high loads.
Materials and methods
Ethylamine (66 wt%) and concentrated nitric acid (70 wt%) were purchased from Sigma Aldrich (Munich, Germany) and the water used was purified to a resistivity of 18.2 MΩ cm by employing a Milli-Q Purification System (Millipore, Malsheim, France). EAN was synthesized by drop-wise addition of concentrated nitric acid to a solution of ethylamine until equimolar amounts were mixed with constant agitation. This was done with excess water and the system was cooled to a temperature below 10 °C with ice during the reaction stage to prevent oxide formation. Rotary evaporation was used for 2 hours at 45–50 °C after the reaction was completed to remove remaining water. However, this treatment did not completely remove all water from the solution and it was therefore purged with nitrogen and heated to 110 °C overnight. Karl-Fischer titration showed small concentrations of water (<100 ppm) at the start of every experiment and a maximum of 1000 ppm measured after several hours. While ammonium nitrate is a well known explosive, ethylammonium nitrate is thermally stable, and does not demonstrate similar behaviour, despite the fact that other nitrate based ammonium salts may potentially do so.59–61 Thus caution should be employed in the choice of ILs for high temperature applications. All glassware and tubing used was thoroughly cleaned prior to experiments. The AFM liquid cell and O-ring were rinsed with copious amounts of water, followed by high purity ethanol (99.9%, Kemetyl, Haninge Sweden), and then dried with nitrogen. Tubing, connectors, tweezers and other accessories were stored in a glass beaker containing 70% ethanol and ultrasonicated for at least 30 minutes and thereafter treated the same way as the liquid cell and the O-ring prior to use. The cantilever used for carrying out the experiment was cleaned in the same way as the liquid cell and the O-ring, but upon completion of that cleaning stage it was also subjected to plasma treatment (Plasma Cleaner/Sterilizer PDC-32G, Harrick Plasma, Ithaca, NY, USA) for 15 seconds.A silica particle (Bangs Laboratories, Fishers, IN, USA) was attached to a tipless cantilever (CSC12, Mikromasch, Tallinn, Estonia) with AralditeTM epoxy glue (Hunstman LLC, Duxford, United Kingdom). An optical microscope connected to a micromanipulator was used to put the colloidal particle in place. The particle radius, the length and width of the cantilever were obtained using ImageJ software (NIST, Gaithersburg, MD, USA). These values were then used to calculate the normal spring constant, following Sader’s method of thermal vibration.62 The torsional spring constant was determined using an approach recently devised in our group,63 together with the acquired cantilever dimensions:
 | (1) |
All normal force and friction measurements were performed using a Nanoscope IIIa Multimode AFM (Bruker, Santa Barbara, CA, USA) equipped with a PicoForce scanner in contact mode and a PicoForce controller. The normal force curves were obtained using a ramp size of 500 nm at six different scan rates (0.1, 0.4, 0.6, 1.1, 2.2, and 4.4 Hz) with a maximum deflection of 2 V and a 5 s delay before ramping another force curve. The data recorded by the AFM are deflections of the cantilever against the piezo movement which are then converted to forces as a function of apparent separation.64 A total of 25 curves were obtained and then averaged to create an averaged force curve for each case with calculated standard deviations. For friction, the slow scan axis was disabled and a scan size of 5 μm (512 points per line, 32 lines) with five different scan rates (0.5, 1, 2, 3, and 4 Hz) was used to study friction as a function of velocity as well as friction as a function of load. The applied load was increased from 0 to 2 V, and then unloaded to −0.2 V, with a constant increment of 0.2 V. The friction coefficient was determined by the slope when plotting the friction force against the applied load, provided the relationship is linear.65 The experiments were conducted at three different temperatures, room temperature (25 °C), 50 °C, and 80 °C. The temperature changes were achieved using a Nanoscope High Temperature Heater Controller (Bruker, Santa Barbara, CA, USA).
Rheological measurements were performed on a Malvern Kinexus Rheometer (Worcestershire, UK) using a cup and a concentric cylinder configuration, with a distance of 5.12 mm separating them. The shear rate was swept from 1 to 4000 s−1 and an average of the measured viscosity was obtained in the mid-range of the Newtonian response. Rheological data was measured at six temperatures between 25 and 80 °C and the system was allowed a 5 min equilibrium period each time it had reached the set temperature.
Results and discussion
Measured viscosity is shown as a function of temperature for EAN in Fig. 1. The data indicates that viscosity decreases with increasing temperature, in broad agreement with previously published viscosity data for EAN over the range of coincidence up to 40 °C (green triangles)56 and 50 °C (red squares),54 respectively. The small discrepancies are likely due to variable water content in EAN, which was recently shown to reduce the viscosity.66 This suggests that the EAN used in this work is drier than that in the earlier work. The natural logarithm of the viscosity is plotted against the inverse temperature in the inset of Fig. 1 and is linear (R2 > 0.995), which reveals Arrhenius-type behaviour (a single activation energy).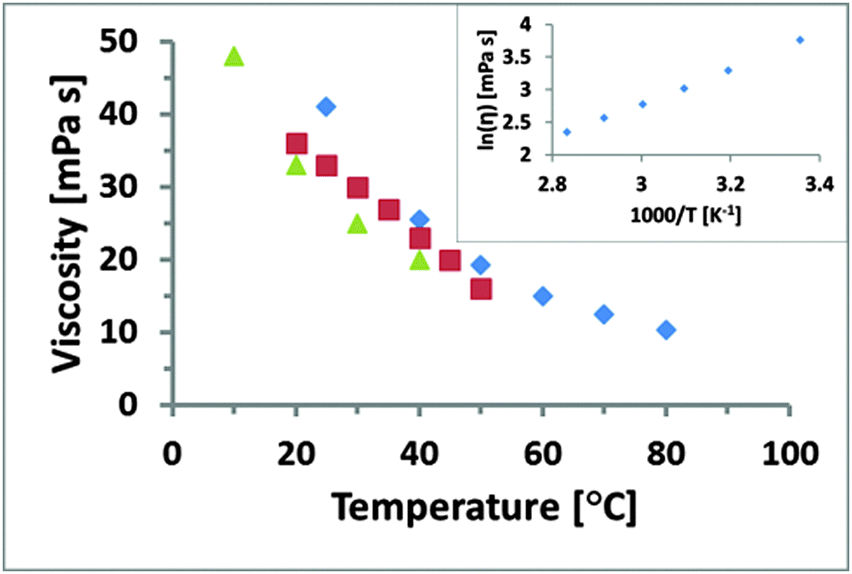 | ||
| Fig. 1 Rheological data for EAN obtained in different studies. The data for blue diamonds was collected in this study in a temperature range from 25 to 80 °C, red squares was acquired from 20 to 50 °C by Smith et al.54 and green triangles from 10 to 40 °C by Capelo et al.56 The natural logarithm of the viscosity is plotted against the inverse absolute temperature in the inset and shows a linear Arrhenius behaviour. | ||
Normal forces – room temperature
Fig. 2 shows the averaged normalised force measured as a function of apparent separation as the mica surface approaches the silica probe in EAN. The force data reveal an increasing repulsive force as a function of increased rate, demonstrating the influence of fluid dynamics on the system. The shear viscosity of EAN at room temperature is 40 mPa s, which qualifies it as a liquid of “high viscosity” (Fig. 1 and ref. 54 and 56), and masks the structural forces at these approach rates. Previous papers have demonstrated that much slower approach rates/smaller geometries are required to unveil structural forces in high viscosity liquids.19–22,26 For reasons of clarity, the three approach rates shown in Fig. 2 are systematically used throughout the paper. The experiments were, however, performed using six different approach rates within this range and Fig. S1 in the ESI† shows the force profiles for all rates at room temperature. The distance axis in the force profiles has been chosen to emphasise the interaction forces at small separations but the measurements were performed over much greater distances (500 nm). Error bars (shown in Fig. S1, ESI†) calculated for both the “sensed” force and the apparent separation demonstrate hardly any variation between the 25 individual force profiles, which contribute to each of the averaged force curves. | ||
| Fig. 2 Averaged AFM approach force profiles for three different approach rates in a mica–EAN–silica system at room temperature. The experiment was performed using a tipless cantilever with a normal spring constant of 0.92 N m−1 and a silica colloidal probe attached (radius 3.4 μm). There is a rate dependence due to fluid dynamics. Each curve is the average of 25 force curves. | ||
The effect of viscosity is enhanced in this study because a colloidal probe is used rather than a sharp AFM tip (used in most prior studies of EAN), the radius of which is several orders of magnitude smaller. Eqn (2) illustrates the relationship between probe radius, R, and fluid dynamic force, F, if the viscosity, η, relative velocity, ν, and separation, h, are known:67
 | (2) |
 | (3) |
 | (4) |
In Fig. 2, the different rates lead to different absolute forces as probe and surface approach each other, which is consistent with a “squeeze force” acting on the system. The slowest scan rate shown in Fig. 2 has a repulsive force with a detectable force range of approximately 5 nm and as the velocity increases, the range increases. Eqn (2) was used to fit the force profiles in Fig. 2 with and without a correction for slip. The best fits employed a non-zero slip using the viscosity measured in bulk (b = 18–20 nm, μb = 40 mPa s, see Fig. S2 (ESI†) for an example fit). The use of a non-zero slip to fit the data is consistent with recent measurements in an analogous ionic liquid where the slip length was of comparable magnitude.69Eqn (4) shows that, if b > 0 and the bulk viscosity is constant, there are two possible physical origins for this observation. (1) The quotient between bulk and interfacial viscosity is small and the boundary layer is thick or (2) the quotient is large and the boundary layer is thin. There is currently no means to distinguish between these two extremes, though perhaps in the future it may be feasible to perform neutron reflectance experiments under confinement. Finally, it is also implicit in the approach that there is a single layer with a single viscosity, which for a multi-layered system might be an oversimplification.
Normal forces – 50 and 80 °C
The force curves change dramatically upon increasing the temperature to 50 °C and 80 °C (Fig. 3). At all approach rates, the fluid dynamic force is either small or non-existent. Eqn (2) indicates that this is consistent with much lower viscosity compared to room temperature. This correlates well with the measured bulk viscosity data in Fig. 1 where the shear viscosity of EAN at 50 and 80 °C decreases to 19 and 10 mPa s, respectively. Fitting of the obtained force profiles in Fig. 3, using the same method as above (see Fig. S2, ESI†) indicates slip length values in the same range as for room temperature (typically 20 to 25 nm). Once again the fits return viscosity values at large separation agreeing well with the rheology data in Fig. 1 (within 2 mPa s). The same limitation on the interpretation of the slip length applies, i.e. it is not clear whether the viscosity changes gradually over a larger layer or if there is a sudden drop in viscosity for a thin boundary film. The fact that the slip lengths are the same and the “bulk” viscosity is appropriate irrespective of temperature suggests that there is no transition occurring with temperature. | ||
| Fig. 3 Force curves are displayed for a mica–EAN–silica system at 50 (a, c and e) and 80 °C (b, d and f), employing three different approach rates: 0.4 Hz (a and b), 1.1 Hz (c and d), and 4.4 Hz (e and f). A colloidal probe cantilever (spring constant 0.92 N m−1 and radius 3.4 μm) was used to sample 25 force curves per dataset and all force profiles were averaged with corresponding error bars. The insets of the force curves show clear structural forces at surface separations of 0.25–0.3 nm and 0.75 nm, respectively. | ||
In Fig. 3, distinct force barriers are observed at two different separations. These barriers, often referred to as “steps”, generally reflect the size of ion pairs as they are excluded from the contact.21,25,26,28 The step size of the barrier at larger separation (5 Å) is consistent with this idea. The step at shortest separation is of considerably smaller size (2.5–3 Å) and is less than the ion-pair size of EAN, which has previously been determined to be 5 Å.26 However, both the mica surface and the silica colloidal probe have a localised negative surface charge (the level of dissociation is unknown) and it is therefore logical that, for electrostatic reasons, a layer of predominantly cations is surface bound to both the surfaces. Previous studies, employing contact mode and amplitude modulated AFM,18,26,34,36 have shown that the cation of primary ammonium ILs (including EAN) strongly binds to the mica lattice. Therefore, it is concluded that the cation binds more strongly to the mica surface than the silica. The “half-step” at shortest separation is thus the layer of surface bound cations at the silica probe being removed from contact, which is consistent with previous AFM studies.18,19,26,34,36
The similarities of the force profiles in Fig. 3 prompts the question of how temperature affects the interfacial structuring of EAN. In a recent study, the bulk nanostructure of EAN was investigated between 25 and 60 °C and no changes were detected in this range.57 Another study reported that the density of EAN decreased with 2% between 10 and 40 °C, which suggests that there may be small changes in the average separation of the ion pairs.56 As previously discussed, solvation forces at interfaces for conventional molecular liquids appear to be insensitive to temperature42–45 whereas there is limited dependence for ILs.34,39 Irrespective of whether the steps in the force profiles in Fig. 3 are regarded as solvation forces (ion-pairs), or as a near-surface structure, which was recently suggested,36 or whether these two concepts can even be distinguished under confinement, there is clearly no dramatic effect of temperature on the interfacial structuring of EAN. Nonetheless, small differences can be observed between the two temperatures. At 50 °C the steps are defined by an almost vertical barrier, which gives way at a well-defined value of both force and distance and is followed by a “jump” to the next force wall. The larger point spacing in the “jump” indicates a higher speed. At 80 °C the force barriers have a lower slope, indicating a more compressible layer. The absolute forces at which the barriers collapse are also systematically slightly lower than those at 50 °C. Both of these observations are consistent with a loss of order caused by the higher thermal energy – analogous to a broadening of a pair distribution function with increasing temperature (which was also observed using high energy X-ray reflectivity in a different IL system39).
Friction forces
The lateral friction force for EAN was systematically measured for load/unload cycles at different temperatures and scan rates (Fig. 4). The friction loops at room temperature have the same features as in previous publications, with two distinct regimes.19,20 Low loads (<15 nN) correspond to the probe shearing against the more loosely ordered near surface structure of the liquid, which is removed from the contact at higher loads and manifested as “steps” in the force curves; this has earlier been shown to be rate dependent.19 At higher loads (>20 nN) the probe penetrates the near surface layer and shears against the highly ordered IL on the surface. In this high load region, the absolute friction force is considerably larger than in the low load region. Only a single transition is seen (as opposed to two steps in the force curves at least at high T). The resolution of the friction data is insufficient to determine whether there is another regime at lower loads corresponding to the step at around 0.7 nm in the force curve. The force barrier of that step corresponds to a load of around 2 nN, at which the friction forces are almost immeasurably small. The transition between these regions is associated with the removal of the near surface layer from the contact, with solvophobic interactions with the cation rich boundary layer responsible for the sharpness of this transition.70 In this case, a contributing effect to the friction will be the work required to strip the solvophobically bound layer from the surface during sliding, which affects the magnitude, but not the gradient in the higher load regime. It should also be remembered that, unlike in the surface force apparatus, where the glue that holds the surfaces in place deforms to form a parallel plate configuration of essentially constant thickness, the surface deformation is much smaller for colloidal probe experiments. Thus the friction measured at higher forces consists of contributions both from the flattened boundary layer contact (typically a few μm2) and also from an annulus around the contact where the separation is consistent with that of the lower load regime. | ||
| Fig. 4 AFM friction loops were collected with the same system (mica–EAN–silica) using the same colloidal probe cantilever (spring constant 0.92 N m−1 and radius 3.4 μm). In this graph, three different temperatures (blue diamonds – room temperature, red squares – 50 °C, green triangles – 80 °C) are shown for three scan rates: 1 (a), 2 (b), and 3 Hz (c). | ||
The rate dependent frictional forces in the lower load regime are expected to have some correlation with the liquid viscosity, since the dissipation mechanisms are essentially analogous. The normal forces in Fig. 3 confirm that the viscosity is dramatically reduced at elevated temperatures, as expected from bulk measurements (Fig. 1). This is also reflected in the magnitude of the friction forces at low loads – at room temperature the friction is higher. The transition to the higher load regime appears to be roughly independent of both speed and temperature, which is entirely consistent with the observation that the height of the force barriers seen in Fig. 3 are also rate and temperature independent.
The friction coefficients in the higher load regime, obtained from the slope of the linear region, are approximately temperature independent, consistent with the fact that in this region the load dependence is of a Tomlinson-like contact on the boundary layer and lubrication is thus maintained. The density and conformation of this boundary film is determined by the charge interaction with the surface and solvophobic interactions within the layer, and are thus not expected to change significantly over this temperature interval. The magnitude of the forces is much lower at higher temperature in Fig. 4, reflecting that the dissipation forces in the annulus around contact are much lower, due to reduced viscosity and increased thermal motion which will facilitate the removal of the near surface layer and the surface bound cation layer at the silica probe from the contact, as described earlier for the normal forces at 50 and 80 °C. The magnitude of the forces are also rate dependent in this region, the viscous resistance in the annulus region is of course also rate dependent.
Conclusion
Both the friction and the normal forces measured between solids in this ionic liquid are strongly temperature dependent. As expected, the fluid dynamic interactions become much less significant at elevated temperatures due to the strong dependence of the viscosity on temperature. The viscosities extracted from force data, and corresponding to larger separations, are entirely consistent with the bulk rheology measurements. The analysis, nonetheless, reveals a nonzero slip length, indicating either a larger region of somewhat reduced viscosity associated with either confinement or with the interface, or a narrow boundary layer of very different viscosity. The oscillatory force profile associated with the interfacial ordering of ionic liquids is maintained even at elevated temperatures and does not reflect the dramatic changes that are seen in the viscosity. Both the physical location and the height of the force barriers associated with the different layers are maintained, and the effect of temperature is rather to broaden the barrier, or make it less rigid, reflecting increased thermal motion.The friction forces essentially confirm the robustness of the layering towards a temperature increase, since the characteristic two regime friction behaviour is maintained. However, the friction forces at higher temperatures are much reduced, emphasising the fact that these forces, measured with a colloid probe, reflect both the contact friction and the pre-contact friction, which is strongly correlated with the viscosity. As such, the colloid probe provides a more realistic model of an asperity contact than does the surface force apparatus, which in turn is to be preferred for parallel plate studies of the relationship between separation and friction.
While this ionic liquid is not a candidate lubricant for commercialisation, its simplicity of preparation, and the extensive body of knowledge around its physical chemical characteristics, renders it a useful model. In this work, understanding of the relationship between ordering, viscosity, friction and temperature relies on these detailed studies. The reduction in viscosity at higher temperature will obviously favour lubrication in the full film lubrication regime, but may well delay its onset. On the other hand the force barrier associated with the interfacial ordering appears to be largely unaffected by temperature, thus maintaining the boundary lubrication properties, and in fact lowering the absolute value of the friction. (The interfacial layering of ILs appears to be a general phenomenon independent of IL category and so it would seem reasonable to assume that the observations made here should be rather general. Nevertheless the IL employed here is protic and is thus by no means representative of all ILs, and for example lacks pi bonding possibilities. It is not absolutely certain that the findings are necessarily quantitatively applicable to all IL systems.) It should be stressed that no conventional liquid, such as a base oil, is capable of displaying this behaviour, without specific additives. The intrinsic van der Waals adhesion and the reduction in viscosity lead to increased friction in such liquids. No attempt has been made to study wear, and indeed the substrates used are not conventionally relevant in lubrication, but the physicochemical conclusions suggest a cautious optimism towards the pursuit of ionic liquid based lubricants.
Acknowledgements
The Swedish Research Council (VR 621-2011-4361) and the Knut and Alice Wallenberg Foundation (KAW 2012.0078) are gratefully acknowledged for their financial support. R. A. thanks the Australian Research Council Future Fellowship (FT120100313) and Discovery Project (DP120102708). MR further acknowledges the Nanofriction COST Action MP1303.References
- R. D. Rogers, Nature, 2007, 447, 917–918 CrossRef CAS PubMed.
- R. D. Rogers and K. R. Seddon, Science, 2003, 302, 792–793 CrossRef PubMed.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS PubMed.
- A. Maiti, ChemSusChem, 2009, 2, 628–631 CrossRef CAS PubMed.
- P. Wang, S. M. Zakeeruddin, J. E. Moser and M. Gratzel, J. Phys. Chem. B, 2003, 107, 13280–13285 CrossRef CAS.
- B. Bhushan, Philos. Trans. R. Soc., A, 2008, 366, 1499–1537 CrossRef CAS PubMed.
- R. A. Sheldon, Green Chem., 2005, 7, 267–278 RSC.
- R. Atkin, L. M. De Fina, U. Kiederling and G. G. Warr, J. Phys. Chem. B, 2009, 113, 12201–12213 CrossRef CAS PubMed.
- O. Werzer, G. G. Warr and R. Atkin, J. Phys. Chem. B, 2011, 115, 648–652 CrossRef CAS PubMed.
- T. Welton, Chem. Rev., 1999, 99, 2071–2084 CrossRef CAS PubMed.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2008, 108, 206–237 CrossRef CAS PubMed.
- J.-P. Belieres and C. A. Angell, J. Phys. Chem. B, 2007, 111, 4926–4937 CrossRef CAS PubMed.
- M. Yoshizawa, W. Xu and C. A. Angell, J. Am. Chem. Soc., 2003, 125, 15411–15419 CrossRef CAS PubMed.
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Angew. Chem., Int. Ed., 2013, 52, 4623–4627 CrossRef CAS PubMed.
- P. Walden, Bull. Acad. Imp. Sci. St. – Petersbourg, 1914, 8, 405–422 Search PubMed.
- S. Perkin, L. Crowhurst, H. Niedermeyer, T. Welton, A. M. Smith and N. N. Gosvami, Chem. Commun., 2011, 47, 6572–6574 RSC.
- R. Atkin and G. G. Warr, J. Phys. Chem. B, 2008, 112, 4164–4166 CrossRef CAS PubMed.
- J. J. Segura, A. Elbourne, E. J. Wanless, G. G. Warr, K. Voitchovsky and R. Atkin, Phys. Chem. Chem. Phys., 2013, 15, 3320–3328 RSC.
- O. Werzer, E. D. Cranston, G. G. Warr, R. Atkin and M. W. Rutland, Phys. Chem. Chem. Phys., 2012, 14, 5147–5152 RSC.
- R. Álvarez Asencio, E. D. Cranston, R. Atkin and M. W. Rutland, Langmuir, 2012, 28, 9967–9976 CrossRef PubMed.
- S. Perkin, Phys. Chem. Chem. Phys., 2012, 14, 5052–5062 RSC.
- S. Perkin, T. Albrecht and J. Klein, Phys. Chem. Chem. Phys., 2010, 12, 1243–1247 RSC.
- A. M. Smith, K. R. J. Lovelock, N. N. Gosvami, T. Welton and S. Perkin, Phys. Chem. Chem. Phys., 2013, 15, 15317–15320 RSC.
- G. Straffelini, in Friction and Wear, Springer International Publishing, 2015, ch. 3, pp.61–84 DOI:10.1007/978-3-319-05894-8_3.
- R. G. Horn, D. F. Evans and B. W. Ninham, J. Phys. Chem., 1988, 92, 3531–3537 CrossRef CAS.
- R. Atkin and G. G. Warr, J. Phys. Chem. C, 2007, 111, 5162–5168 CAS.
- A. M. Smith, K. R. J. Lovelock, N. N. Gosvami, P. Licence, A. Dolan, T. Welton and S. Perkin, J. Phys. Chem. Lett., 2013, 4, 378–382 CrossRef CAS PubMed.
- R. M. Espinosa-Marzal, A. Arcifa, A. Rossi and N. D. Spencer, J. Phys. Chem. Lett., 2013, 5, 179–184 CrossRef PubMed.
- R. M. Espinosa-Marzal, A. Arcifa, A. Rossi and N. D. Spencer, J. Phys. Chem. C, 2014, 118, 6491–6503 CAS.
- M. A. Gebbie, M. Valtiner, X. Banquy, E. T. Fox, W. A. Henderson and J. N. Israelachvili, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 9674–9679 CrossRef CAS PubMed.
- R. Atkin, N. Borisenko, M. Druschler, S. Z. El Abedin, F. Endres, R. Hayes, B. Huber and B. Roling, Phys. Chem. Chem. Phys., 2011, 13, 6849–6857 RSC.
- R. Hayes, N. Borisenko, M. K. Tam, P. C. Howlett, F. Endres and R. Atkin, J. Phys. Chem. C, 2011, 115, 6855–6863 CAS.
- R. Atkin, S. Z. El Abedin, R. Hayes, L. H. S. Gasparotto, N. Borisenko and F. Endres, J. Phys. Chem. C, 2009, 113, 13266–13272 CAS.
- D. Wakeham, R. Hayes, G. G. Warr and R. Atkin, J. Phys. Chem. B, 2009, 113, 5961–5966 CrossRef CAS PubMed.
- A. Elbourne, J. Sweeney, G. B. Webber, E. J. Wanless, G. G. Warr, M. W. Rutland and R. Atkin, Chem. Commun., 2013, 49, 6797–6799 RSC.
- A. Elbourne, K. Voitchovsky, G. G. Warr and R. Atkin, Chem. Sci., 2015, 6, 527–536 RSC.
- M. Mezger, B. M. Ocko, H. Reichert and M. Deutsch, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3733–3737 CrossRef CAS PubMed.
- M. Mezger, S. Schramm, H. Schroder, H. Reichert, M. Deutsch, E. J. De Souza, J. S. Okasinski, B. M. Ocko, V. Honkimaki and H. Dosch, J. Chem. Phys., 2009, 131, 094701 CrossRef PubMed.
- M. Mezger, H. Schroder, H. Reichert, S. Schramm, J. S. Okasinski, S. Schoder, V. Honkimaki, M. Deutsch, B. M. Ocko, J. Ralston, M. Rohwerder, M. Stratmann and H. Dosch, Science, 2008, 322, 424–428 CrossRef CAS PubMed.
- D. Wakeham, A. Nelson, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2011, 13, 20828–20835 RSC.
- P. Niga, D. Wakeham, A. Nelson, G. G. Warr, M. Rutland and R. Atkin, Langmuir, 2010, 26, 8282–8288 CrossRef CAS PubMed.
- R. G. Horn and J. N. Israelachvili, J. Chem. Phys., 1981, 75, 1400–1411 CrossRef CAS.
- H. K. Christenson and J. N. Israelachvili, J. Chem. Phys., 1984, 80, 4566–4567 CrossRef CAS.
- T. Nakada, S. Miyashita, G. Sazaki, H. Komatsu and A. A. Chernov, Jpn. J. Appl. Phys., Part 2, 1996, 35, L52–L55 CrossRef CAS.
- L. T. W. Lim, A. T. S. Wee and S. J. O'Shea, J. Chem. Phys., 2009, 130, 134703 CrossRef CAS PubMed.
- K. Voitchovsky, J. J. Kuna, S. A. Contera, E. Tosatti and F. Stellacci, Nat. Nanotechnol., 2010, 5, 401–405 CrossRef CAS PubMed.
- Y. Wang and G. A. Voth, J. Am. Chem. Soc., 2005, 127, 12192–12193 CrossRef CAS PubMed.
- M. Wang, T. Zander, X. Y. Liu, C. Liu, A. Raj, D. C. F. Wieland, V. M. Garamus, R. Willumeit-Romer, P. M. Claesson and A. Dedinaite, J. Colloid Interface Sci., 2015, 445, 84–92 CrossRef CAS PubMed.
- A. Schirmeisen, L. Jansen, H. Holscher and H. Fuchs, Appl. Phys. Lett., 2006, 88, 123108 CrossRef.
- X. Y. Zhao, M. Hamilton, W. G. Sawyer and S. S. Perry, Tribol. Lett., 2007, 27, 113–117 CrossRef CAS.
- I. Barel, M. Urbakh, L. Jansen and A. Schirmeisen, Phys. Rev. Lett., 2010, 104, 066104 CrossRef PubMed.
- S. J. Manzi, W. T. Tysoe and O. J. Furlong, Tribol. Lett., 2014, 55, 363–369 CrossRef.
- W. A. Ducker, T. J. Senden and R. M. Pashley, Nature, 1991, 353, 239–241 CrossRef CAS.
- J. A. Smith, G. B. Webber, G. G. Warr and R. Atkin, J. Phys. Chem. B, 2013, 117, 13930–13935 CrossRef CAS PubMed.
- J. A. Smith, O. Werzer, G. B. Webber, G. G. Warr and R. Atkin, J. Phys. Chem. Lett., 2010, 1, 64–68 CrossRef CAS.
- S. B. Capelo, T. Mendez-Morales, J. Carrete, E. L. Lago, J. Vila, O. Cabeza, J. R. Rodriguez, M. Turmine and L. M. Varela, J. Phys. Chem. B, 2012, 116, 11302–11312 CrossRef CAS PubMed.
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Phys. Chem. Chem. Phys., 2011, 13, 3237–3247 RSC.
- R. Hayes, S. Imberti, G. G. Warr and R. Atkin, Angew. Chem., Int. Ed., 2012, 51, 7468–7471 CrossRef CAS PubMed.
- S. Wellens, B. Thijs and K. Binnemans, Green Chem., 2013, 15, 3484–3485 RSC.
- IoLiTec MSDS IL-0043 EAN, http://www.iolitec.de/MSDS-Ionic-Liquids/View-category.html?dir=ASC&limit=5&limitstart=35&order=name, accessed November 2015.
- V. N. Emel'yanenko, G. Boeck, S. P. Verevkin and R. Ludwig, Chem. – Eur. J., 2014, 20, 11640–11645 CrossRef PubMed.
- J. E. Sader, J. W. M. Chon and P. Mulvaney, Rev. Sci. Instrum., 1999, 70, 3967–3969 CrossRef CAS.
- R. Alvarez-Asencio, E. Thormann and M. W. Rutland, Rev. Sci. Instrum., 2013, 84, 096102 CrossRef CAS PubMed.
- J. Ralston, I. Larson, M. W. Rutland, A. A. Feiler and M. Kleijn, Pure Appl. Chem., 2005, 77, 2149–2170 CrossRef CAS.
- G. Amontons, De la résistance causée dans les Machines, Mémoires de l'Académie Royale A, 1699 Search PubMed.
- R. Zarrougui, M. Dhahbi and D. Lemordant, J. Solution Chem., 2015, 44, 686–702 CrossRef CAS.
- D. Y. C. Chan and R. G. Horn, J. Chem. Phys., 1985, 83, 5311–5324 CrossRef CAS.
- O. I. Vinogradova, Langmuir, 1995, 11, 2213–2220 CrossRef CAS.
- J. Sweeney, G. B. Webber and R. Atkin, Phys. Chem. Chem. Phys., 2015, 17, 26629–26637 RSC.
- J. Sweeney, G. B. Webber, M. W. Rutland and R. Atkin, Phys. Chem. Chem. Phys., 2014, 16, 16651–16658 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp05837f |
| This journal is © the Owner Societies 2016 |