
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of environmentally persistent free radicals from the heterogeneous reaction of ozone and polycyclic aromatic compounds
Cuyler K.
Borrowman
,
Shouming
Zhou
,
Timothy E.
Burrow
and
Jonathan P. D.
Abbatt
*
Department of Chemistry, University of Toronto, Toronto, Canada M5S 3H6. E-mail: jabbatt@chem.utoronto.ca
First published on 25th November 2015
Abstract
In the 1980s long-lived radical species were identified in cigarette tar. Since then, environmentally persistent free radicals (EPFRs) have been observed in ambient particulate matter, and have been generated in particulate matter generated from internal combustion engines. For the first time, we measure in situ the formation and decay of EPFRs through the heterogeneous reaction of ozone and several polycyclic aromatic compounds (PAC). Solid anthracene (ANT), pyrene (PY), benzo[a]pyrene (BAP), benzo[ghi]perylene (BGHIP), 1,4-naphthoquinone (1,4NQ), and 9,10-anthraquinone (AQ) were reacted with gas-phase ozone in a flow system placed in the active cavity of an electron paramagnetic resonance (EPR) spectrometer, and the formation of radicals was measured on the timescale of tens of minutes at ambient levels of ozone down to 30 ppb. For most substrates the net radical production is initially rapid, slows at intermediate times, and is followed by a slow decay. For oxidized solid BAP, radical signal persists for many days in the absence of ozone. To evaluate the effect of substrate phase, the solid PAHs were also dissolved in squalane, an organic oil inert to ozone, which yielded a much higher maximum radical concentration and faster radical decay when exposed to ozone. With higher mobility, reactants were apparently able to more easily diffuse and react with each other, yielding the higher radical concentrations. The EPR spectra exhibit three radicals types, two of which have been assigned to semiquinone species and one to a PAH-derived, carbon-centered radical. Although our system uses levels of PAC not typically found in the environment it is worth noting that the amounts of radical formed, on the order of 1018 radicals per g, are comparable to those observed in ambient particulate matter.
Introduction
Due to the negative impacts to human health posed by particulate matter (PM),1–3 much research has been done in trying to identify the chemical culprits of this toxicity. Polycyclic aromatic hydrocarbons (PAH) are frequently found in ambient PM and have been shown to have toxic and mutagenic effects, as have their oxygenated derivatives.4–6 PAH plus their oxygenated derivatives will be referred to as polycyclic aromatic compounds, PAC, henceforth in this work. Long-lived radicals, termed environmentally persistent free radicals (EPFRs), have also been observed in PM7 and their ability to catalytically generate reactive oxygen species (ROS)8,9 in the human body enables the induction of oxidative stress.EPFRs were first detected in cigarette tar, and identified to be primarily semiquinone in nature.10 Additionally, EPFRs have been generated on transition metal-doped particles in post flame and cool zone regions of combustion systems, and have also been identified as being primarily semiquinone.11,12 These formation mechanisms are believed to rely on thermally-driven processes occurring in the combustion system. This work seeks to determine if EPFRs may also be formed under atmospherically relevant conditions via heterogeneous oxidation mechanisms.
PAH are commonly found within PM and can react heterogeneously with ozone13–18 to produce a variety of oxygenated functional groups including quinones and diol PAC17,19–23 In previous work done in our laboratory on soot particles, and PAH-coated soot particles, it was found that ozone exposure led to the decay of soot PAHs with two general trends, an initial fast decay followed by a secondary slower decay, resulting in increased redox activity of the particles. The increased redox activity was attributed to an increase of surface bound quinone-like structures, formed from PAH and graphitic carbon reacting with ozone.24 Quinones are well known electron shuttles, with more chemically reduced hydroquinones formed under reducing conditions and semiquinone radicals acting as intermediates (Fig. 1). A mixture of quinones, hydroquinones, and long-lived semiquinone radicals present on the surface would indeed enhance the redox cycling ability of primarily hydrocarbon and elemental carbon particles, though the semiquinone radical has yet to be observed in such systems.
 | ||
| Fig. 1 The interconversion between quinones, semiquinones, and hydroquinones, shown for benzo[a]pyrene-1,6-dione, with one resonance structure of the semiquinone radical showing that the radical can reside on either oxygen or carbon. | ||
It has been shown in work done by Shiraiwa et al. that in the heterogeneous reaction of PAH and ozone reactive oxygen intermediates (ROI), with life times on the order of 100 seconds, may be formed on the path to final oxygenated PAC products.25 It is possible that these ROI have radical character. In the same work by Shiraiwa et al.25 it is shown that NO2 uptake is enhanced in particles generated by the protein bovine serum albumin when they are pretreated with ozone. This enhanced uptake is indirectly attributed to the formation of long-lived ROI radicals on aromatic amino acids in later work by Shiraiwa et al.26
We seek to answer several questions in this study. Does the heterogeneous reaction of solid PAH and gas-phase ozone proceed through an EPFR intermediate? Is the formation of EPFRs exclusive to the reaction of ozone with PAH, or may other PAC also form EPFRs under atmospherically relevant conditions? Furthermore, if EPFRs form in this system how long lived are they, and how does the phase of PAH effect their behaviour? Finally, if long-lived EPFRs form on PAH is their longevity affected by the presence of other radicals, such as NO2? To answer these questions an experimental system was developed to carry out the reaction of PAH with ozone while simultaneously monitoring for radical formation.
Experimental
To measure radical production in real time a system was designed such that solid or dissolved PAC may react with gas-phase species within the active cavity of an electron paramagnetic resonance spectrometer (EPR). In solid-phase experiments, 1–2 mg of PAC were dissolved in 1 mL dichloromethane and the entire solution was transferred to a 10 mm outer diameter pyrex EPR tube from Wilmad. The solution was evaporated under a constant flow of nitrogen so that PAC were coated on the bottom two centimetres of the inside walls of the EPR tube. Dissolved-phase PAC substrates were prepared using the same method, but with squalane mixed into the dichloromethane–PAC solution at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio by mass with the PAC. Squalane was chosen to solvate PAC because it is inert to ozone and sufficiently viscous at room temperature to stick to the walls of a glass EPR tube.
:1 ratio by mass with the PAC. Squalane was chosen to solvate PAC because it is inert to ozone and sufficiently viscous at room temperature to stick to the walls of a glass EPR tube.
The coated EPR tube was connected to a flow system (see Fig. 2) using Teflon Swagelock fittings and an o-ring to ensure a tight seal. Gaseous species were introduced to the EPR tube via 1/8 inch Teflon tubing at a flow rate of 1.8 slpm of carrier gas. The 1/8 inch tubing ran through 1/4 inch Teflon tubing so that the PAC were exposed to a constant flow of ‘fresh’ gas; the residence time of gases in the tube was less than a second. After reaction, all excess ozone and any gas-phase products continued to flow out of the system to an ozone monitor (Thermo model 49c) and to an exhaust line. Ozone was generated by a home-built generator in which high purity air or pure oxygen flowed through a Pyrex glass tube and was irradiated by ultraviolet light from a mercury pen-ray lamp (UVP Inc.). All reactions were carried out at room temperature using nominally 100 ± 10 ppb ozone, except for experiments investigating the effect of ozone mixing ratio on radical production. In those experiments, ozone mixing ratios of approximately 30 ± 10 ppb and 900 ± 20 ppb were used.
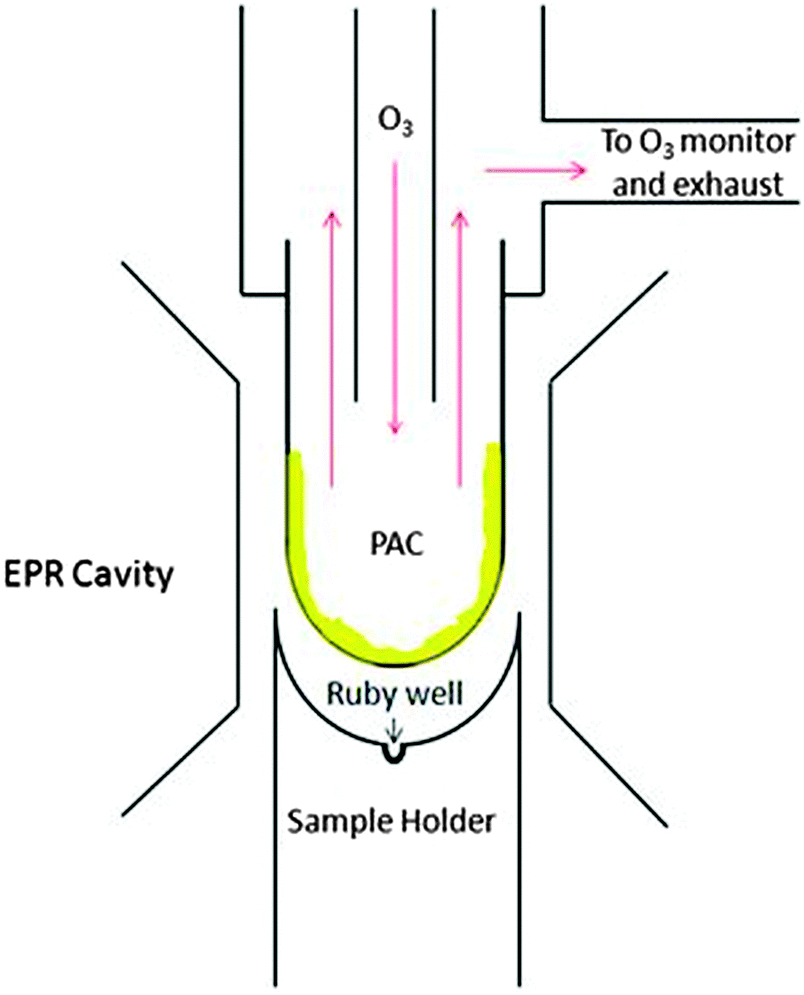 | ||
| Fig. 2 Experimental flow system schematic. A Teflon cylindrical rod, labelled ‘Sample Holder,’ was used to ensure the EPR tube was placed in the same spot in the EPR cavity between experiments. This also served to ensure that the ruby standard, located in the ruby well, was placed similarly between experiments. | ||
In experiments investigating the effects of gas-phase NO2 on PAC EPFR the same flow system was used, with the addition of a valve allowing NO2 and inert carrier gas to flow over the PAC after ozone exposure. The NO2 was supplied from a 2000 ppm NO2 cylinder and diluted in inert carrier gas to approximately 2 ppm.
A method was developed for quantification of radical numbers formed based on Singer's work.27 In particular, spectra were collected in the presence of a ruby sphere (Alfa Aesar, 0.5 mm diameter, grade 25). This was used as a secondary standard because typical primary standards utilize organic radicals, whose signal overlaps with our signal of interest. Ruby has three EPR signals, all of which do not overlap with our signal of interest. The ruby radical content was first calibrated to a known amount of DPPH (2,2-diphenyl-1-picrylhydrazyl), a stable organic radical whose lone EPR signal directly overlaps with our signals of interest. The ruby was then placed in the ruby well on the Teflon sample holder, and used to calibrate the number of radicals formed during the PAC–ozone reaction. This method is convenient because it allows us to monitor instrumental drift, and to calibrate the g-factor as well as the absolute number of radicals formed. In these experiments DPPH was coated over the bottom 2 cm of an EPR tube using the same method used during PAC sample preparation in order to ensure similar instrument response between the calibration and different experiments. The amount of DPPH used in the calibration experiments was selected so that the number of radicals present would be on the same order as the number formed during the PAC–O3 reaction because single point calibrations were performed to quantify the amount of radicals formed and to ensure that the EPR would respond similarly to both the DPPH standard and PAC radicals formed during the experiment.
All EPR measurements were performed at room temperature using a Bruker ECS-EMX X-band EPR spectrometer equipped with an ER4119HS cavity. Typical operating parameters were as follows: microwave frequency 9.61 GHz, microwave power 54 mW, modulation amplitude 4 G, sweep width 205 G, time constant 0.01 ms, total sweep time 120 s. Quantification experiments were run under the same conditions except the sweep width was increased to 3301 G.
Spectra were acquired in ‘Field Delay’ mode. In this mode, spectra were acquired as subsequent slices utilizing the following parameters: four 30 s scans were averaged together, followed by a time delay which was typically 60 s for a total acquisition time of 180 s per slice. During the first slice of each experiment PAC were exposed to air only. The ozone generator was then turned on, and ozone exposure began during the delay time after the first spectrum was collected. By double integrating the peak area of the radical signal (Fig. 3) the radical growth relative to initial conditions may be determined. The absolute number of radicals formed may be obtained when spectra are acquired in the presence of a reference standard (ruby).
 | ||
| Fig. 3 The EPR signals from the reactions of solid and dissolved BAP and solid AQ with ozone. Solid benzo[a]pyrene (BAP, blue trace) radicals form a single, broad unstructured peak; solid anthraquinone (AQ, green trace) radicals form two peaks, and dissolved BAP (red trace) radicals form three separate peaks. The signal from solid BAP displayed is from approximately 12 hours of ozone exposure, the signal from solid AQ is from about 20 minutes of ozone exposure, and the signal from dissolved BAP is from 30 minutes of ozone exposure. An ozone mixing ratio of 100 ppb was used in each reaction. The AQ two peak signal is representative of all solid PAC studied other than BAP. | ||
Anthracene (ANT), pyrene (PY), benzo[a]pyrene (BAP), benzo[ghi]perylene (BGHIP), 9,10-anthraquinone (AQ), and 1,4-napthoquinone (1,4NQ) were all reacted with ozone (Table 1). Synthetic air (grade 0.1), used to generate ozone, as well as nitrogen (grade 5.0) used to flush over the sample were obtained from Linde. In some experiments pure oxygen (grade 2.6) was used to generate ozone instead of air.
| Compound | Abbreviation | Structure |
|---|---|---|
| 1,4-Napthoquinone | 1,4NQ |

|
| Anthraquinone | AQ |

|
| Anthracene | ANT |

|
| Pyrene | PY |

|
| Benzo[a]pyrene | BAP |

|
| Benzo[ghi]perylene | BGHIP |

|
Results and discussion
Radical growth was observed for all solid PAC studied except PY, with an initial fast radical growth step which typically lasted for the first 10–20 minutes of ozone exposure (see Fig. 4a). This may be attributed to the reaction of ozone with molecules on the surface of the deposited PAC. With the exception of BAP, there was radical decay after the initial fast production for all PAC (Fig. 4a) with the decay much slower than the fast initial growth. For BAP, after the initial fast radical growth there was a second slower radical growth regime (see Fig. 5). It is possible that after all the available surface PAC have reacted with ozone any further radical production is limited by the diffusion of ‘fresh’ PAC from the bulk to the surface, allowing for the reaction of additional PAC with ozone. This is a manifestation of the ‘shielding’ effect, where non-volatile surficial species may protect buried species from oxidation.28 Simultaneously, radical loss processes compete, thus resulting in the net radical decay behaviour at long times. In the case of BAP, the radical species present may have a higher stability than the other PAC, thus leading to the net radical gain after the first initial production. In work done by Antiñolo et al. in which the decay of particle-bound PAH was measured, a similar pattern of fast initial decay followed by a secondary slower decay was observed, and the behaviour was attributed to an initial surface reaction followed by a diffusion limited reaction.24 Our results are analogous with these findings. | ||
| Fig. 4 Peak area of radical signal is plotted vs. reaction time. Each data point is obtained from the double integration of a single slice. Ozone exposure began after time zero and remained for the duration of the experiments. Polycyclic aromatic compounds (PAC) began with no radical character and radical growth was only observed after ozone exposure began. Panel A presents data for the reaction of ozone with solid PAC and Panel B is the reaction of ozone with PAC dissolved in squalane. Fits are included to guide the eye only. Note the different Y-axis ranges. | ||
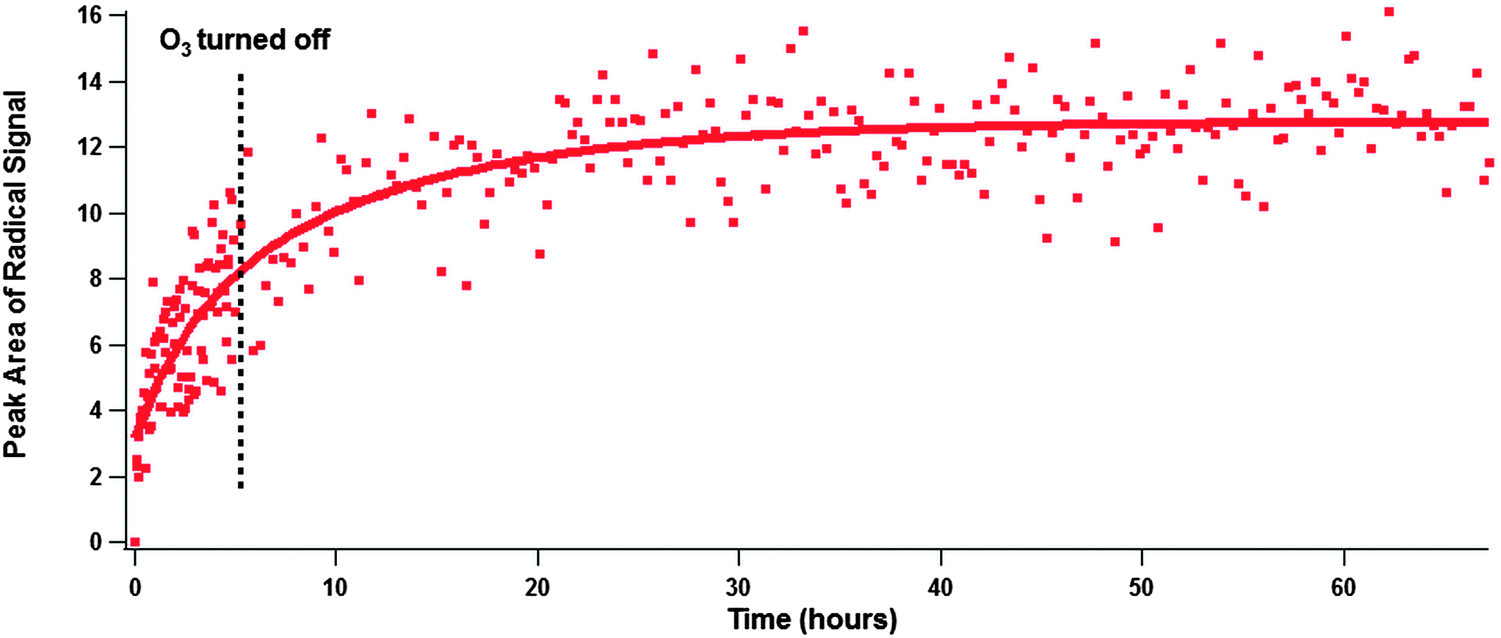 | ||
| Fig. 5 Radical formation over the course of 65 hours from the reaction of solid benzo[a]pyrene (BAP) and ozone. Ozone exposure began after the slice at time = 0 hours and spectra were acquired every 3 minutes. Ozone exposure ended after 5 hours and at that point the system was flushed with pure air, and subsequent spectra were acquired every 20 minutes. These data were fit with a double exponential function (red trace) to help guide the eye. | ||
To investigate the effect of the PAC phase on radical behaviour, the PAC were mixed with squalane at a 1:1 ratio by mass and were oxidized by ozone. In the dissolved-phase experiments radical growth was seen in all PAC studied, including PY (unlike in the solid phase experiments). The initial fast radical production step is still present, but a much higher total radical concentration is reached. Additionally, radical decay is enhanced with all PAC, except ANT, when dissolved. The squalane allows for better mobility of both PAC and ozone, on the surface and potentially in the bulk, thus leading to the higher total radical concentration. The increased mobility also allows for radical loss processes to occur more efficiently, without the hindrance arising of radicals locked into a solid matrix. Possible loss processes include dimerization26 of radical PAC or termination reactions with other radicals.
It should be mentioned that the apparent morphology of the deposited PAC had an important effect on radical behaviour. Most PAC, with the exception of PY and to some degree 1,4NQ, appeared amorphous. PY and 1,4NQ appeared to have both amorphous and crystalline morphologies. It is possible that the increased rigidity of a crystalline structure inhibits the formation of radicals. Additionally, ANT mixed poorly with squalane, and as a result ANT's reactivity is similar between solid and dissolved phase experiments. It is clear that the phase and morphology to the PAC plays an important role in EPFR formation and behaviour.
When dissolved, the EPR signal changes from one peak in the case of solid BAP or two peaks in the case of the rest of the PAC, to 3 peaks (Fig. 3). The conventional interpretation of the triple peak pattern is a coupling of the radical to a nitrogen nucleus;29 however, there is no reactive nitrogen in the system. To rule out the possibility of any reactive nitrogen species being generated or somehow entering the system, several additional experiments were conducted. First, a NOx monitor (Thermo model 42i) was connected downstream from the ozone generator to determine if any NOx was being generated, and it was found that NOx was below the detection limit (0.4 ppb). The possibility exists that NOx is being generated below the detection limit of the monitor, so an experiment (results not shown) was run using pure oxygen to generate ozone instead of pure air, which has a substantial amount of N2. There was no significant difference between BAP experiments using either pure oxygen or pure air to generate ozone. Taken together, the spectrum in Fig. 3 indicates 3 radical species being formed in the dissolved-phase experiments. The EPR signals observed for solid PAC are likely an unresolved superposition of multiple radical signals.
Multiple long-lived organic radical species have been observed previously, for instance in cigarette tar10,30 and in Fe2(III)O3-doped combustion generated particles.12 In their work on cigarette tar, Pryor and his colleagues postulate that the long-lived paramagnetism is due to the formation of low molecular weight quinone–semiquinone–hydroquinone polymers, and that rapid interconversion between the species by hydrogen atom exchange gives rise to a single, broad, unstructured peak.30 Our work here is consistent with this concept. In a solid matrix such a polymer may or may not be formed, but in either case hydrogen atom exchange may occur thus causing the observed single, broad, unstructured peak. When in a dissolved matrix molecules are more mobile and are not locked in place with respect to each other. It is possible that in this dissolved phase, the hydrogen atom exchange is inhibited and three separate peaks are resolved.
The g-factor of a radical is a property of an electron in a specific environment (analogous to the chemical shift of a nucleus in NMR spectroscopy). Organic radicals typically have g-factors ranging from 1.99–2.01, with carbon centred radicals' g-factor < 2.003, and oxygen centred radicals' g-factor > 2.004.11 The presence of other nuclei can shift the g-factor: oxygen or halogens increase the g-factor. Knowing this, we attribute the radical with a g-factor of 2.0006 to a carbon centred PAH radical. The radical with a g-factor of 2.0056 may be attributed to an oxygen centred semiquinone radical.7,11 Organic radicals with g-factors >2.01 have been observed previously in the presence of halogenated solvents.31 Experiments were prepared using dichloromethane, so to rule out the possibility of trapped solvent interacting with the radicals, an experiment was performed (results not shown) using benzene as a solvent, thus removing any chance of halogens entering the system, and no differences were found between experiments with different solvents. Therefore the radical with a g-factor of 2.0116 is likely also an oxygen centred radical on highly oxidized PAC (Table 2).
| Radical | g-Factor | Assignment | % of radical population | |
|---|---|---|---|---|
| BAP in squalane | Rad 1 | 2.0116 | Semiquinone | 30.6 |
| Rad 2 | 2.0056 | Semiquinone | 32.6 | |
| Rad 3 | 2.0006 | PAH | 36.8 | |
| Solid BAP | 2.0036 | — | 100 | |
To gain information on the longevity of the EPFRs an experiment was conducted in which solid BAP was exposed to ozone for 5 hours and then flushed with pure air and allowed to sit over the course of a weekend while acquiring spectra (Fig. 5). Again, an initial fast radical growth was observed, and the second slower growth regime was present. After ozone exposure ended a small amount of radical growth was measured, but over the course of the weekend that radical growth stopped and the radical signal became steady. This indicates that under the right conditions PAC EPFRs formed heterogeneously may persist for an extended period of time, possibly indefinitely. The lifetime of EPFRs could be comparable to, or even longer than the lifetime of PM in the environment (typically several days to a few weeks).
Quantification of the radical signal through the use of a ruby as a secondary standard was performed in duplicate for the reaction of BAP mixed in squalane at an ozone mixing ratio of 100 ppb. It was found that the maximum concentration of radicals formed was very high, between 0.7–2.1 × 1018 radicals per g. To put this in perspective, assuming the radicals formed in the BAP experiments have a molecular weight of roughly 300 Da (i.e. a few oxygen atoms have been added to the BAP molecular backbone), this indicates that a lower limit to the molar yield for radical production in the reaction is 0.04–0.1%. That is, for every mole of parent PAC approximately 0.04–0.1% forms an EPFR. This is a lower limit given that radical decay is occurring at the same time as radical formation. To place this in context, we note that this value of radical content is comparable to values reported by Gehling and Dellinger who measured EPFR concentrations in ambient particulate matter, and found between 2.02 × 1016–3.48 × 1018 radicals per g.7 EPFR concentrations on diesel exhaust particles have also been previously reported to be in this range by Lim et al. who found 1.23 × 1018 radicals per g.32 Radical concentrations on the order of 1018 have also been reported for n-hexane soot particles.33 It is possible that each study is measuring roughly the same levels of radicals because they undergo self-reaction at higher levels, thus limiting the maximum concentration.
The effects of ozone mixing ratio on radical formation were also investigated (Fig. 6) by reacting BAP in squalane with a variety of different ozone mixing ratios.
 | ||
| Fig. 6 Peak area of radical signal normalized to mass of benzo[a]pyrene (BAP) is plotted vs. time. Experiments using ozone mixing ratios of 900 ppb (red plusses), 100 ppb (purple circles), and 30 ppb (blue squares) are plotted. Fits are included to guide the eye. | ||
Radicals form faster at higher ozone mixing ratios, and reach higher instantaneous concentrations. This is because radical loss processes occur slower than radical formation. If we assume that the reaction of PAH and ozone proceeds through a Langmuir–Hinshelwood mechanism (ozone first must sorb to the surface before reaction may occur, and therefore a finite number of sorption sites must exist), of which there is ample evidence,13,15,17,20,34 then the interpretation of the above data becomes clear. At higher ozone concentrations surface sites are occupied quickly and the formation of radicals is fastest, and unreacted PAH are consumed most quickly. Formation quickly slows because of lack of unreacted PAH, and net radical decay is observed. At lower ozone concentrations surface sites are not filled as quickly, and radical production occurs at a steadier rate that decreases as unreacted PAH dwindle.
If the data in Fig. 6 is broken into two regimes, a formation and a decay regime (though during either regime both formation and decay would be taking place), rate constants may be determined for each regime. The formation regime is defined as the time at which ozone exposure begins until the time net formation of radicals ends. Ozone exposure begins directly after time zero and net radical formation typically ends after 10–20 minutes of exposure. The rate constants (±one standard deviation) in the formation regime for experiments using 900 ppb O3, 100 ppb O3, and 30 ppb O3 are 0.085 ± 0.014 s−1, 0.040 ± 0.003 s−1, and 0.011 ± 0.001 s−1 respectively. The rate constant increases linearly between the 30 ppb and 100 ppb experiments with respect to O3 mixing ratio, but does not increase linearly between the 100 ppb and 900 ppb ozone mixing ratio. This indicates that at some O3 mixing ratio between 100 ppb and 900 ppb saturation behaviour is reached i.e. all available adsorption sites are occupied by O3, which is indicative of Langmuir–Hinshelwood kinetics. The rate constants in the decay regime may also be determined, but because there are many factors affecting the decay behaviour (simultaneous radical formation, uptake of ozone, diffusion of ozone and PAC, radical dimerization, other radical loss processes, secondary reactions of ozone) assigning a physical meaning to this decay constant is very difficult. Qualitatively speaking, the decay in the 900 ppb experiment occurs at roughly twice the rate during the 100 ppb experiment, probably because of faster radical–radical loss processes.
Finally, the effect of NO2 on PAC EPFR behaviour was investigated (Fig. 7) by reacting BAP in squalane first with ozone, and then with NO2. As soon as NO2 exposure began, radical loss was greatly enhanced (red plusses). This is an indication that NO2 radicals react efficiently with the PAC and semiquinone radicals, thus forming closed-shell products and eliminating radical character in the dissolved-phase PAC. In the absence of gas-phase radicals (blue squares) the EPFR signal persists much longer though still shows radical loss. As mentioned in the Introduction, the presence of an ROI formed when ozone is exposed to PAHs or albumin protein was inferred by Shiraiwa et al.,25 who showed that NO2 uptake was enhanced after earlier ozone exposure. While these earlier experiments are suggestive of radical character for the ROI, our observations directly demonstrate that some or all of the ROI formed during PAH heterogeneous oxidation have radical character.
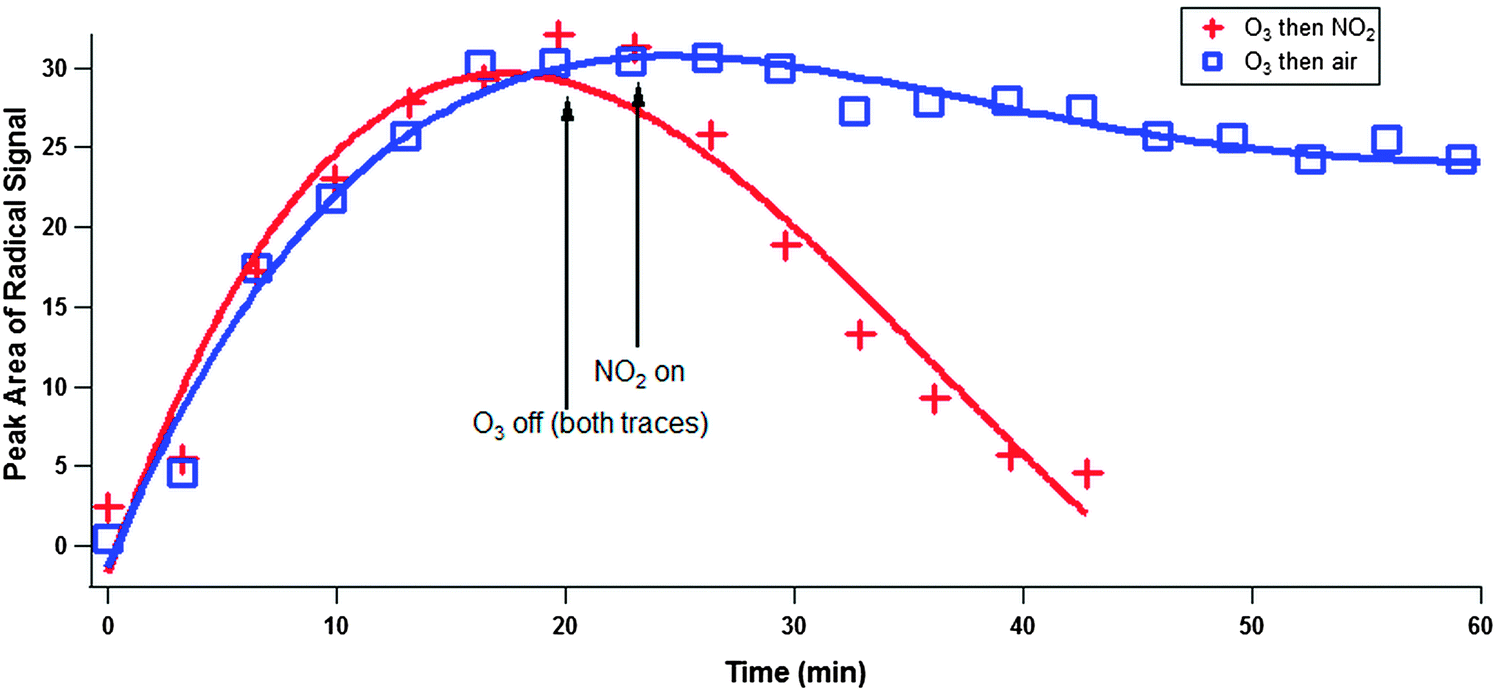 | ||
| Fig. 7 Peak area of radical signal vs. time is plotted. Benzo[a]pyrene (BAP) mixed with squalane was reacted with 100 ppb ozone for 20 minutes, then flushed with air for 3 minutes (indefinitely in the control experiment, blue squares), and finally exposed to 2 ppm of NO2 (red plusses). Fits are added to guide the eye only. | ||
We note that coordination of PAC to transition metals is described in the literature as a factor leading to enhanced stability of EPFRs. While our system did not have any transition metals added, it will be interesting to determine whether the presence of transition metal ions would alter any of the conclusions reached in this work.
Conclusions
EPFRs form from the heterogeneous reaction of PAH and quinones with atmospherically relevant levels of gas-phase ozone. Radicals are initially formed rapidly, which is attributed to the reaction of surficial PAC. After the initial rapid radical formation a slow radical decay was observed in the case of most PAC (BAP being the sole exception), likely due to radical–radical recombination processes. The longevity of PAC-derived EPFRs was investigated, and it was demonstrated that the radicals may persist for extended periods of time, comparable to the lifetime of PM for solid substrates. One mechanism to shorten the lifetime of the EPFRs is exposure to other radical species, in this case demonstrated NO2. The experiments were performed with ppm NO2 mixing ratios which are much higher than those experienced in ambient settings. Further experiments with other potential deactivating species, such as NO, HO2 and RO2 radicals, as well as with lower mixing ratios of NO2 will better evaluate the lifetime of these species under ambient conditions.When dissolved in a liquid oil the increased mobility of PAC allowed for a significantly higher maximum radical concentration to be achieved, 0.7–2.1 × 1018 in the case of BAP. This represents at least 0.04–0.1% molar yield, indicating a high conversion of reactants to long-lived radical intermediates. As well, this quantity is similar to that observed in ambient particulate matter.7 Dissolved-phase experiments revealed that three different radical structures form, two likely semiquinone in nature and the third a carbon-centered, PAH-based species. As well, the rate of radical loss was also increased with dissolved reactants relative to solids, likely arising from faster self-reaction of radical species.
Given that EPFRs have been identified in ambient PM and in combustion sources, the question now arises as to whether this radical signal all arises from PAC-derived EPFRs. As well, the balance between primary, combustion-derived sources and those arising from heterogeneous oxidation mechanisms needs to be established.
Acknowledgements
This work was supported by NSERC and the Sloan Foundation. We thank D. J. Donaldson for the suggestion of performing the NO2 experiment.References
- F. Lu, D. Xu, Y. Cheng, S. Dong, C. Guo, X. Jiang and X. Zheng, Environ. Res., 2015, 136, 196–204 CrossRef CAS PubMed.
- P. E. Schwarze, J. Ovrevik, M. Låg, M. Refsnes, P. Nafstad, R. B. Hetland and E. Dybing, Hum. Exp. Toxicol., 2006, 25, 559–579 CAS.
- L. W. Stanek, J. D. Sacks, S. J. Dutton and J. J. B. Dubois, Atmos. Environ., 2011, 45, 5655–5663 CrossRef CAS.
- J. N. Pitts, D. M. Lokensgard, P. S. Ripley, K. A. Van Cauwenberghe, L. Van Vaeck, S. D. Shaffer, A. J. Thill and W. L. Belser, Science, 1980, 210, 1347–1349 CAS.
- S. Hayano, L. Jang-Ho, K. Furuya, T. Kikuchi, T. Someya, C. Oikawa, Y. Iida, H. Matsushita, T. Kinouchi, Y. Manabe and Y. Ohnishi, Atmos. Environ., 1984, 19, 1009–1015 CrossRef.
- E. B. Gehly, J. R. Landolph, C. Heidelberger, C. H. Clone, M. Fibroblasts and H. Nagasawa, Cancer Res., 1982, 42, 1866–1875 CAS.
- W. Gehling and B. Dellinger, Environ. Sci. Technol., 2013, 47, 8172–8178 CAS.
- W. Gehling, L. Khachatryan and B. Dellinger, Environ. Sci. Technol., 2014, 48, 4266–4272 CrossRef CAS PubMed.
- G. L. Squadrito, R. Cueto, B. Dellinger and W. A. Pryor, Free Radical Biol. Med., 2001, 31, 1132–1138 CrossRef CAS PubMed.
- W. A. Pryor, B. J. Hales, P. I. Premovic and D. F. Church, Science, 1982, 220, 425–427 Search PubMed.
- B. Dellinger, S. Lomnicki, L. Khachatryan, Z. Maskos, R. W. Hall, J. Adounkpe, C. McFerrin and H. Truong, Proc. Combust. Inst., 2007, 31, 521–528 CrossRef PubMed.
- E. Vejerano, S. Lomnicki and B. Dellinger, Environ. Sci. Technol., 2011, 45, 589–594 CrossRef CAS PubMed.
- B. T. Mmereki and D. J. Donaldson, J. Phys. Chem. A, 2003, 107, 11038–11042 CrossRef CAS.
- M. Cazaunau, K. Le Ménach, H. Budzinski and E. Villenave, Z. Phys. Chem., 2010, 224, 1151–1170 CrossRef CAS.
- S. N. Chu, S. Sands, M. R. Tomasik, P. S. Lee and V. F. Mcneill, J. Am. Chem. Soc., 2010, 132, 15968–15975 CrossRef CAS PubMed.
- N.-O. A. Kwamena, M. G. Staikova, D. J. Donaldson, I. J. George and J. P. D. Abbatt, J. Phys. Chem. A, 2007, 111, 11050–11058 CrossRef CAS PubMed.
- N. O. A. Kwamena, M. E. Earp, C. J. Young and J. P. D. Abbatt, J. Phys. Chem. A, 2006, 110, 3638–3646 CrossRef CAS PubMed.
- U. Poschl, T. Letzel, C. Schauer and R. Niessner, J. Phys. Chem. A, 2001, 105, 4029–4041 CrossRef.
- J.-J. Yao, H. Zhi-Heng and M. Susan J, Water Res., 1998, 32, 3235–3244 CrossRef CAS.
- J. Najera, R. Wamsley, D. Last, K. Leather, C. Percival and A. Horn, Int. J. Chem. Kinet., 2011, 44, 257–276 Search PubMed.
- Y. Zhang, J. Shu, Y. Zhang and B. Yang, J. Environ. Sci., 2013, 25, 1817–1823 CrossRef CAS.
- R. Koeber, J. M. Bayona and R. Niessner, Int. J. Environ. Anal. Chem., 1997, 66, 313–325 CrossRef CAS.
- T. Letzel, E. Rosenberg, R. Wissiack, M. Grasserbauer and R. Niessner, J. Chromatogr. A, 1999, 855, 501–514 CrossRef CAS PubMed.
- M. Antiñolo, M. D. Willis, S. Zhou and J. P. D. Abbatt, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- M. Shiraiwa, Y. Sosedova, A. Rouvière, H. Yang, Y. Zhang, J. P. D. Abbatt, M. Ammann and U. Pöschl, Nat. Chem., 2011, 3, 291–295 CrossRef CAS PubMed.
- M. Shiraiwa, K. Selzle, H. Yang, Y. Sosedova, M. Ammann and U. Pöschl, Environ. Sci. Technol., 2012, 46, 6672–6680 CrossRef CAS PubMed.
- L. S. Singer, J. Appl. Phys., 1959, 30, 1463–1464 CrossRef CAS.
- S. Zhou, A. K. Y. Lee, R. D. Mcwhinney and J. P. D. Abbatt, J. Phys. Chem. A, 2012, 116, 7050–7056 CrossRef CAS PubMed.
- Y. Lion, M. Delmelle and A. Van de Vorst, Nature, 1976, 263, 442–443 CrossRef CAS PubMed.
- D. F. Church and W. a. Pryor, Environ. Health Perspect., 1985, 64, 111–126 CrossRef CAS PubMed.
- E. P. Talzi, A. G. Stepanov, V. P. Babenko, V. M. Nekipelov, K. I. Zamaraev and S. Branch, Inst. Inorg. Chem., Sib. Branch, Acad. Sci. USSR, 1987, 30, 66–71 Search PubMed.
- J. Lim, L. E. Yu, Y. Y. Kostetski, C. Lim, J. Ryu and J. Kim, J. Air Waste Manage. Assoc., 2008, 58, 1077–1085 CAS.
- a. R. Chughtai, M. M. O. Atteya, J. Kim, B. K. Konowalchuk and D. M. Smith, Carbon, 1998, 36, 1573–1589 CrossRef CAS.
- J. McCabe and J. P. D. Abbatt, J. Phys. Chem. C, 2009, 113, 2120–2127 CAS.
| This journal is © the Owner Societies 2016 |