
Possible interstellar formation of glycine from the reaction of CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) NH, CO and H2O: catalysis by extra water molecules through the hydrogen relay transport†
NH, CO and H2O: catalysis by extra water molecules through the hydrogen relay transport†
Zanele P.
Nhlabatsi
,
Priya
Bhasi
and
Sanyasi
Sitha
*
Department of Chemistry, University of Johannesburg, P.O. Box 524, Auckland Park, Johannesburg, South Africa. E-mail: ssitha@uj.ac.za
First published on 12th November 2015
Abstract
“How the fundamental life elements are created in the interstellar medium (ISM)?” is one of the intriguing questions related to the genesis of life. Using computational calculations, we have discussed the reaction of CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH, CO and H2O for the formation of glycine, the simplest life element. This reaction proceeds through a concerted mechanism with reasonably large barriers for the cases with one and two water molecules as reactants. For the two water case we found that the extra water molecule exhibits some catalytic role through the hydrogen transport relay effect and the barrier height is reduced substantially compared to the case with one water molecule. These two cases can be treated as ideal cases for the hot-core formation of the interstellar glycine. With an increasing number of water molecules as the reactants, we found that when the numbers of water molecules are three or more than three, the barrier height reduced so drastically that the transition states were more stable than the reactants. Such a situation gives a clear indication that with excess water molecules as the reactants, this reaction will be feasible even under the low temperature conditions existing in the cold interstellar clouds and the exothermic nature of the reaction will be the driving force.
NH, CO and H2O for the formation of glycine, the simplest life element. This reaction proceeds through a concerted mechanism with reasonably large barriers for the cases with one and two water molecules as reactants. For the two water case we found that the extra water molecule exhibits some catalytic role through the hydrogen transport relay effect and the barrier height is reduced substantially compared to the case with one water molecule. These two cases can be treated as ideal cases for the hot-core formation of the interstellar glycine. With an increasing number of water molecules as the reactants, we found that when the numbers of water molecules are three or more than three, the barrier height reduced so drastically that the transition states were more stable than the reactants. Such a situation gives a clear indication that with excess water molecules as the reactants, this reaction will be feasible even under the low temperature conditions existing in the cold interstellar clouds and the exothermic nature of the reaction will be the driving force.
1. Introduction
There exists one fundamental question related to the origin of life and that is “How life is created?” or more technically “How the essential life elements, i.e., the amino acids are formed?”1,2 To find an answer to this question various possible angles have been tried by many chemists, biologists, astronomers and geologists. Among the various amino-acids, glycine, (H2N–CH2–COOH), is one of the simplest amino acids found in the earth. The era of the astronomical search for glycine begins with the availability of the laboratory spectra for it,3 but its presence in the interstellar medium (ISM) is still controversial.4–6 On the other side, many amino-acids including glycine have been found on meteorites.7–18 In a recent discovery glycine is being detected even in comets, as evidenced from the pristine cometary samples sent back by NASA STARDUST mission,18 which can be treated as a validation for its extraterrestrial origin. On the other hand our knowledge on its formation in the extraterrestrial space through various chemical pathways seems to be either incomplete or inconclusive.19 To imitate the synthesis in the laboratory, there are two widely exploited approaches that can be found in the literature. The first one is Strecker's synthesis,20 and the second one is the Miller experiment,21 where in both the cases it is believed that the key intermediate is HCHO (formaldehyde). Similar to formaldehyde, another important reactant, methanimine (CH2NH) which is isoelectronic with HCHO is also suggested as a potential prebiotic precursor for glycine.22–25 Starting with this methanimine, in this work using computational calculations, we have carried out a complete potential energy surface (PES) analysis of the CH2NH + CO + H2O → glycine reaction. A recent work, “Discovering chemistry with an ab initio nano reactor”26 by Wang et al. also indicates the possibility of such a reaction, but without any further discussions. With a detailed potential energy surface analysis, we have shown that the reaction suggested by Wang et al.26 proceeds through a concerted type of reaction mechanism from CH2NH, CO and H2O to give glycine as the end product. Also we have discussed about the cooperative and catalytic role of extra water molecules in this reaction through hydrogen relay transport phenomena. The role of water acting as a proton transfer helper was first described by Radom and co-workers.27–29 But, such phenomena are less investigated for the interstellar formation of Glycine.25,30–33 The recent work reported by Rimola et al.,33 where they have carried out computational studies on the formation of glycine from the reaction CH2NH and CO on radical surfaces of water-ice dust particles, indicates a clear catalytic role exhibited by the water molecules of the water-ice cluster. Moreover in the work of Rimola et al.,33 they have shown how the defects formed in the water-ices are capable of making the reaction feasible and the mechanism proposed is multi-stepped in nature. The work discussed here shows a concerted type of mechanism for the formation of glycine, also with discussions related to the catalytic role of extra water molecules, which is capable of bringing down the reaction barrier drastically.
2. Computational methods
All the calculations have been carried out using the Gaussian software package.34 We have carried out calculations using various methods like, B3LYP, B3PW91, CBS-QB3, MP2, and also various composite methods which are known for accurate energy predictions, like, G3B3, G3MP2B3, G4 and G4MP2. For the B3LYP method we have also tested the effect of various basis sets. The true minima and the transition states were confirmed from the analysis of their frequencies by ensuring that all frequencies were positive for the minimum, there is only one imaginary frequency for the transition state. We have also carried out the analysis of the displacement vectors for the imaginary frequency to ascertain that the TS is a structurally true TS which is also confirmed by the IRC analysis. All the thermodynamic quantities were calculated from the zero point energy (ZPE) corrected energies of the stationary points in the PES. As all the methods predict a similar kind of trend, in the main text we have limited our discussion only to the B3LYP/6-31++G(3df,3pd) method only with very little discussions on effects of methodologies and basis sets.3. Results and discussion
3.1. PES for the CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) NH + CO + H2O → glycine reaction
NH + CO + H2O → glycine reaction
The PES of the termolecular reaction between CH2NH, CO and H2O to form glycine is shown in Fig. 1. The PES for the MP2 method is provided in the ESI.†
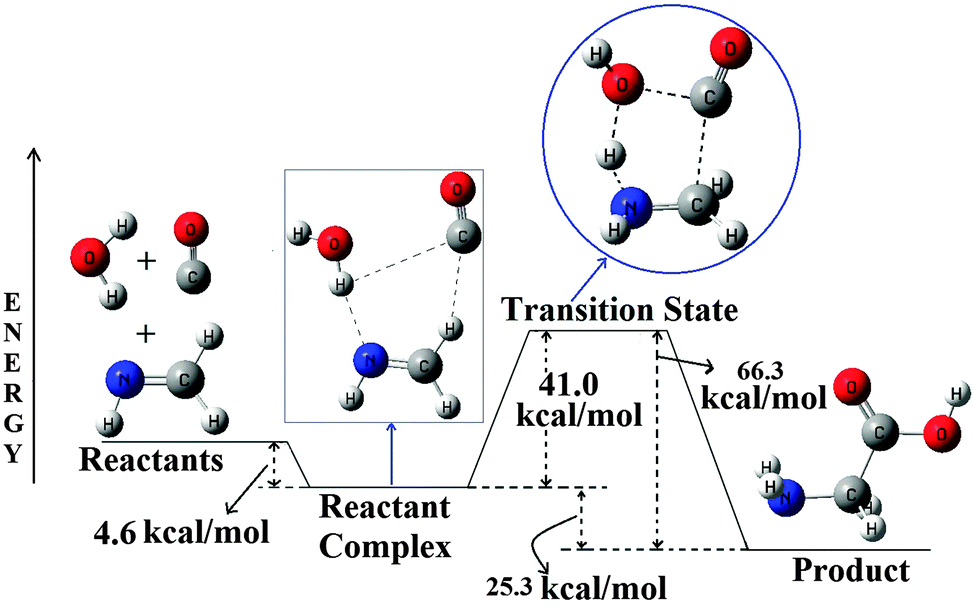 | ||
| Fig. 1 B3LYP/6-31++G(3df,2pd) optimized PES for the CH2NH + CO + H2O → Glycine, reaction. All the energies are in kcal mol−1 and the diagram is not to scale. | ||
Analysis of the PES shows that a well synchronized approach of the three reactants first leads to the formation of a stable hydrogen-bonded complex, which ultimately passes through a transition state to reach the product, glycine. Energetics of the PES shows that the complex is around 4.6 kcal mol−1 more stable than the corresponding reactants. The complex then passes through a transition state, where the TS is 41.0 kcal mol−1 higher in energy than the complex (36.4 kcal mol−1 is higher in energy compared to the reactants). Analysis of the product, glycine, shows that it is 25.3 kcal mol−1 more stable compared to the reactant complex or 29.9 kcal mol−1 more stable compared to the reactants. The PES for the MP2 method shows a similar kind of behavior to that of the B3LYP surface with a little difference in the energetics. The exothermic nature of this reaction is a clear indication for its thermochemical feasibility in the ISM, but the reasonably large barrier may act as a bottleneck to this reaction. We have also investigated the effect of various basis sets with the B3LYP method for the PES of this reaction and also the effect of various methodologies for the PES of this reaction. The detailed discussions related to these are provided in the ESI.† As the reactant complex and the transition state are vital to the feasibility of this reaction under the interstellar conditions, a concise discussion about their structures and nature of interactions is shown below.
NH; (1) above or below the plane of the molecule (perpendicular to the π-cloud of the molecule), (2) in-plane from one side of methanimine. The optimized structure of the reactant-complex is shown in Fig. 2(a). Analysis of the reactant-complex shows that the approach of the reactants towards CH2NH is almost in-plane and also is in the same side of the plane. The CO molecule is placed on the side of CH2 of methanimine and makes a hydrogen bonded interaction with one of the H-atoms of the CH2 unit (C–H–O hydrogen bonding. Hydrogen bond distance is 2.895 Å). On the other hand the H2O molecule is on the NH unit side of methanimine, where one of the H-atoms of the water molecule interacts with the N-atom of methanimine and the nature of interaction is hydrogen bonding (O–H–N hydrogen bonding. Hydrogen bond distance is 1.941 Å). Besides these two major interactions there exist so many van der Waals types of interactions in the reactant complex.
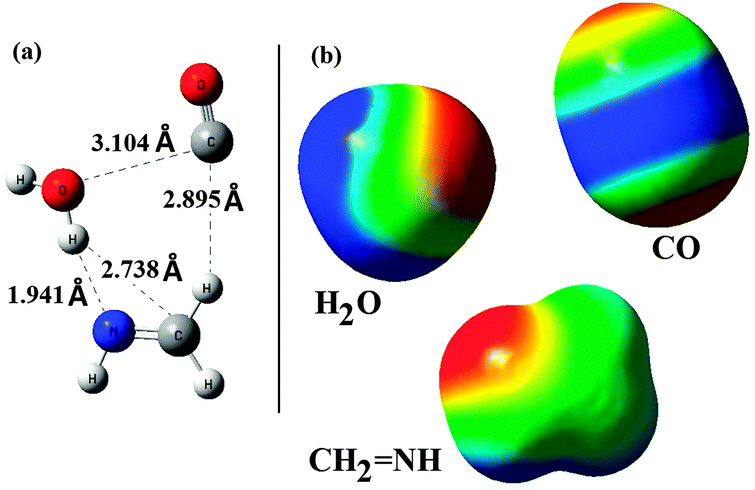 | ||
| Fig. 2 (a) B3LYP/6-31++G(3df,2pd) optimized structure of the reactant complex with important interaction distances. (b) Computed ESP maps of CH2NH, CO and H2O (oriented in the similar fashion to that of the reactant complex) calculated from the B3LYP/6-31++G (3df,2pd) method at 0.001 au electron density surfaces [ESP colour scheme: red (negative) – positive (blue)]. | ||
To study the nature of the interaction existing in the complex we have analyzed the electrostatic potential (ESP) maps of the reactants [Fig. 2(b)]. Analysis of the ESP maps shows that; (1) for CO: there is a maximum negative potential around the C-atom and a smaller negative potential around the O-atom, which are capable of interacting with positive potential regions of other reactants, (2) for H2O: two ends with H-atoms have positive potentials and the region around the central O-atom has the negative potential, (3) for CH2NH: except the region around the N-atom (showing negative potential) all the other regions have positive potentials. Analysis of the geometry of the reactant-complex shows a well synchronized approach of the reactants from the electrostatic potential point of view.
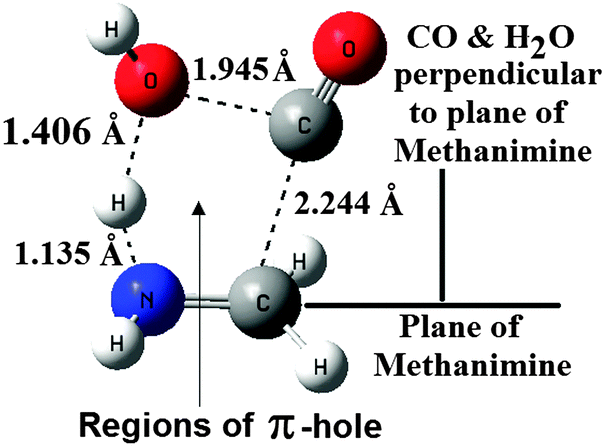 | ||
| Fig. 3 B3LYP/6-31++G(3df,2pd) optimized structure of the transition state with important interaction distances. | ||
The structure of the TS shows that the molecular structural characteristics of CO and CH2NH are almost retained in the TS, and at the same time one of the H–O bonds in H2O is largely elongated. Further analysis of the structure of the TS shows that the approach of CO + H2O towards CH2NH is perpendicular to the plane of CH2NH (perpendicular to the π-cloud of CH2NH). To know the nature of interaction existing at the TS, we have analyzed the ESPs of the reactants again. It can be seen that again H2O in the TS is on the side of the NH unit of CH2NH, with one of the H-atoms (with positive potential) of H2O sandwiched between the negative potentials of the N-atom of NH and the O-atom of H2O. On the other hand CO and OH of H2O interact with the π-hole regions35 of CH2NH rather than interacting with each other. In other words π-hole regions35 of CH2NH provide a well synchronized positioning of these two reactants in the TS.
3.2. Catalysis by an extra water molecule
Rimola et al. have carried out computational studies on the formation of glycine on radical surfaces of water-ice dust particles which involves CH2NH and CO as reactants.33 In their work they have used a cluster of water molecules where many water molecules are interlinked with each other.33 Though the mechanism predicted in their work is a step-wise mechanism, their work clearly indicates the prominent hydrogen transport catalytic effect exhibited by those interlinked water molecules. Similarly, Wang et al. in their recent work also indicated the possible catalytic effect for this reaction, but without any further details on the mechanistic aspect of this reaction.26 So in this work we have carried out calculations related to the PES of the reaction between CH2NH, CO and two molecules of H2O to study the mechanistic aspect of this reaction. In our study we have placed the two water molecules adjacent to each other and at the same time also interlinked with each other. This inter linked system also sometimes can be viewed as a H2O–H2O binary complex. We have compared our results of the reaction PES consisting of two individual molecules of water with that of the reaction PES of the H2O–H2O binary complex. The potential energy surface of the reaction between CH2NH, CO and two water molecules is shown in Fig. 4. The PESs related to the H2O–H2O binary complex for both the MP2 and B3LYP methods are provided in the ESI.†
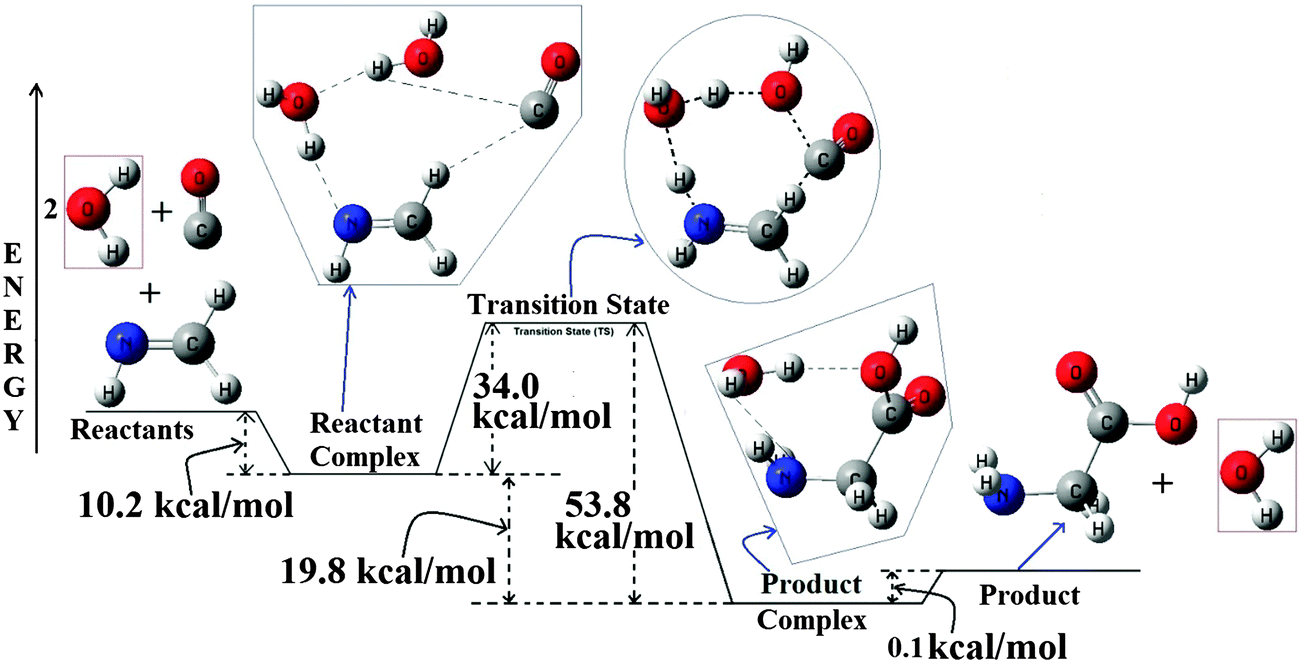 | ||
| Fig. 4 B3LYP/6-31++G(3df,2pd) optimized PES for the CH2NH + CO + 2H2O → Glycine, reaction. All the energies are in kcal mol−1 and the diagram is not to scale. | ||
Analysis of the potential energy surface shows that a well-coordinated approach of the four reactants leads to the formation of a stable hydrogen bonded reactant-complex (multiple hydrogen bonding situations can be seen in the complex) at the entry channel of this reaction. This complex ultimately passes through a transition state to reach a stable product-complex, an exit channel complex for this reaction. This exit channel complex then endothermically dissociates to give glycine and water as final products. Without any further discussion about the structures of the all the stationary points in the PES we have only discussed the energetics of this reaction PES to account for the extent of the catalytic effect by the extra water molecule as a reactant. More details about the optimized structures of these stationary points can be found in the ESI.† Analysis of the reactant complex shows that it is highly stable (around 10.2 kcal mol−1 more stable than the corresponding reactants). The extra stability of the reactant complex is coming from the more number of well synchronized H-bonded interactions existing in the complex than the previous case as discussed above for one water molecule. Instead of the two separate water molecules, we have extended our discussions for the H2O–H2O complex reacting with CH2NH and CO and this is based on the fact that the binary complex of H2O also being detected in the ISM.36 Now, with the H2O–H2O binary complex as one of the reactants, the reactant complex is 7.0 kcal mol−1 stable compared to the reactants. The loss in the stability is hidden in the H2O–H2O binary complexation energy.
The complex then passes through a transition state and the analysis of the TS shows that it is a distorted cyclic 8-membered ring. At the TS all the four reactants are arranged in a nonplanar three dimensional way. Energetics of the TS shows that it is 34.0 kcal mol−1 higher in energy than the reactant complex (23.8 kcal mol−1 higher in energy compared to the reactants). This clearly shows a 7.0 kcal mol−1 of decrease in energy compared to the uncatalyzed case (∼12 kcal mol−1 compared to the reactants in the un-catalyzed case) and a direct indication of the prominent catalytic activity by the extra water molecule for this reaction.26 In the TS, the catalytic effect of the extra H2O molecule can be considered as being facilitated by the relay effect it is exhibiting in transporting the H-atom to the N-atom site of CH2NH.37 In the transition state the water molecule close to the N-atom of CH2NH acts as the communicator in relay transporting the H-atom from the other water molecule placed close to the CO molecule to the N-atom of methanimine. Though prominent hydrogen transport occurs in the transition state, the low value of imaginary frequency of the TS clearly gives an indication of negligible chances for the low temperature tunneling mechanism to occur for this reaction.38 While descending from the TS towards the product in the PES, we observed a product-complex formation (complex between Glycine and water). In the complex the hydrogen bonding complexation of H2O seems to be with both NH2 and COOH groups of glycine. Interestingly we found that the exit channel product complex to be marginally more stable (0.1 kcal mol−1) than that of glycine and water separated. Being a stable complex it might help to prevent the photochemical degradation of glycine, a phenomenon that needs separate and further investigations. At the end, under suitable conditions, the product-complex will dissociate endothermically to give glycine and water separately. We have also investigated the effect of various basis sets with the B3LYP method for the PES of this reaction and also the effect of various methodologies for the PES of this reaction. The detailed discussions related to these are provided in the ESI.†
3.3. Effect of excess water molecules
As we have seen in the earlier case of two water molecules, the catalytic effect exhibited by the extra water molecule is very significant in reducing the barrier by an amount of around 12.6 kcal mol−1. So to have some preliminary idea about the above mentioned catalytic effect when the water is in excess, we extended our investigation for three and four water molecules. Without going further detail into the PES, we tried to optimize the transition states for these two cases. The optimized structure of the transition states for three and four water molecules is shown in Fig. 5. With the limited computational resource available with us, first we optimized both the transition states using the B3LYP/6-31G method. Then to compare with our results of the other two PESs as discussed above, we did only the energy calculations at the B3LYP/6-31++G(3df,2pd) level, using those optimized geometries. Then the energies were corrected for ZPVE with the zero point energies obtained during the B3LYP/6-31G optimizations.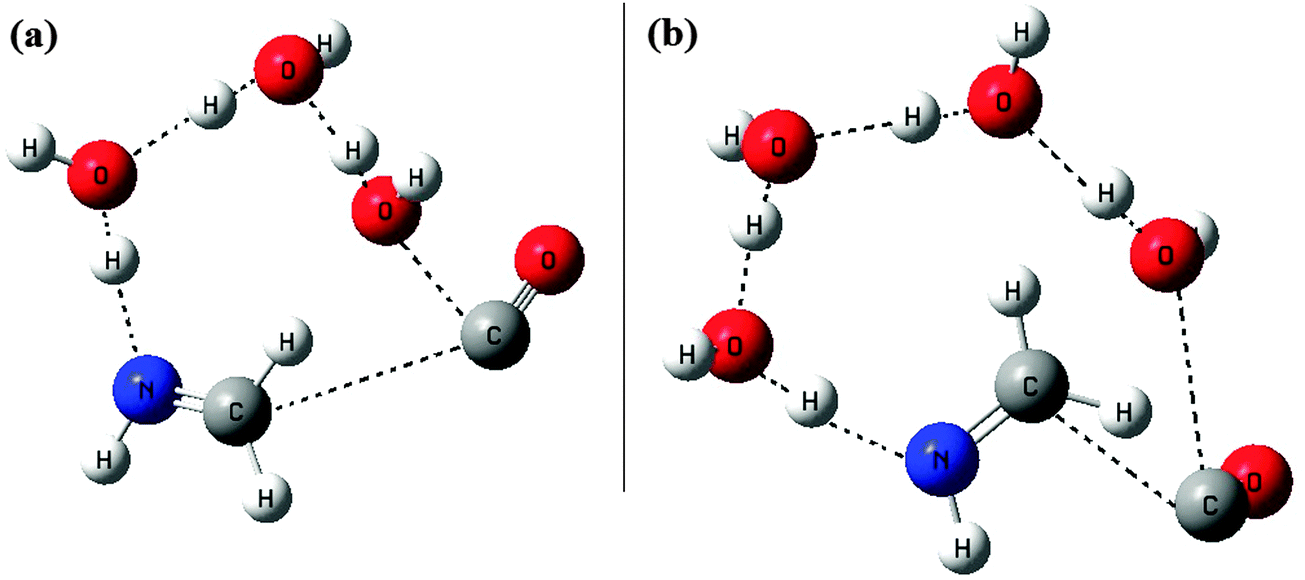 | ||
| Fig. 5 B3LYP/6-31G optimized geometries of the transition states, (a) with three water molecules as reactants and (b) with four water molecules as reactants. | ||
As it can be seen from the two transition state structures, by adding extra water molecules to the reaction, the relay transport of hydrogen occurs over longer and longer distances. Besides these the major differences observed are in the structure of the transition state, i.e. the positioning of the CO molecule. For the one H2O case, CO and H2O are in the same side of the plane, and for the two H2O case, though the CO position is shifted a little bit, the two H2O molecules and the CO are still in the same side of the plane (this is perpendicular to the molecular plane of CH2NH, or we can say, above the molecular plane of CH2NH). With three H2O and four H2O, the situations are quite similar in the sense that all H2O and CO are above the molecular plane of CH2NH. The major differences as stated earlier are with respect to the positions of CO molecules in these two cases. For 3H2O, the CO molecule is just above the C–N bond axis of the CH2NH molecule, whereas for 4H2O, the CO molecule is away from the C–N bond axis of the CH2NH molecule. For the 4H2O case, 4H2O are on one side and CO is on the opposite side with respect to the C–N bond axis of the CH2NH molecule. Thus it can be imagined that this drastic shift in the positions of the CO molecules in these two cases might have some significant effect on the energetics of these two transition states. After comparing the energetics of these two transition states, we found that the results were quite surprising and indicate that both the transition states are now more stable than the reactants. For the case of 3H2O as a reactant, the transition state [Fig. 5(a)] is around 10.0 kcal mol−1 below the reactants, and for 4H2O, the transition state [Fig. 5(b)] is around 13.8 kcal mol−1 below the reactants. Such a situation gives a clear indication that with excess water molecules as the reactants, they will definitely have a significant catalytic effect in making the reaction feasible under the low temperature conditions.
3.4. Possible interstellar applications
Though the interstellar time scale is large, still these kinds of reactions are kinetically less probable owing to the large number of molecules involved in the reaction process. As the reaction proceeds through the formation of a stable complex (a super molecule), the kinetic behavior of the reaction can be expected to be unimolecular. The only problem arises here is the probability of the formation of this super molecule or the stable complex and this largely depends on the molecular composition of the ISM. It is well known that CO and H2O are among the most abundant molecules in the ISM (in fact it is believed that CO is the second most abundant molecule in the ISM) besides hydrogen.39–41 Based on this, along with the large concentrations of H2O available in the ISM, the formation of this complex is fairly probable. Once the complex is formed the reaction will proceed towards a unimolecular kinetics pathway to give glycine as an end product.As extreme temperature conditions are prevailing in the ISM, we tried to explore the possibility of this reaction in both hot-cores and cold interstellar clouds. As part of this we first tried to find whether the reactants are available in those hot-cores and cold interstellar clouds, or not? Methanimine (CH2NH) was first detected in Sgr B2 by Godfrey et al.,22 then subsequently has been found in many other hot core sources,23 and also recently Salter et al. detected it in the ultraluminous infrared galaxy (ULIRG) Arp 200 which is 250 million light years away, using an Arecibo radio telescope; certainly a remarkable discovery.24 Interestingly, besides the availability in hot cores, CH2NH was also observed to be present in the quiescent gas at the so-called “radical-ion peak” along the Orion ridge, where the temperature is about 20 K and also expected to be present even at regions of 10 K or low temperatures.23 Availability of CO and H2O in the ISM is well known.39–41 In the interstellar medium (ISM), hot cores are the dense and warm regions consisting of gases and dusts, which are rich in exotic gas chemistries.40,41 High temperature in those hot cores (around 200–1000 K) is capable of facilitating most of the high barrier reactions.40 Analysis of the PES shows that there is a reasonably large barrier for this reaction for the cases of one and two water molecules. We believe that under the existing condition in the hot-cores of the ISM and where water is not in very large excess the reaction might proceed through the paths shown in Fig. 1 and 4 and the large reaction barriers will not be able to act as a bottleneck to this reaction to occur. On the other hand the feasibility of this reaction in the cold interstellar clouds, where a large amount of water exists as water-ice and the temperature is extremely low, is affirmative only if there are three or more water molecules involved in the reaction. Our preliminary results on the transition state analysis for three water molecules involved in the reaction process show that, the transition state is even more stable than the reactants. This makes the entire process thermodynamically favourable to occur under the extreme low temperature conditions existing in the cold interstellar clouds. Under the low temperature conditions, these transition states being lower in energies compared to the reactants, the exothermic nature of the reaction will drive the reaction. Moreover though probability of tunnelling can be expected to be extremely low,38 but owing to the exhibited prominent proton dynamics, such a phenomenon can not be completely ruled out.37
4. Conclusions
We have discussed a reaction for the possible interstellar formation of glycine from CH2NH, CO and H2O using computation calculations. We have carried out the complete PES analysis for this reaction using various methodologies. Our calculations show that the reaction is having a large barrier height, indicating that such a reaction is only feasible in the hot core regions of the ISM. We have also shown the catalytic role of an extra water molecule in reducing the barrier height, which can further enhance the rate and thus a clear indication that excess water is going to assist the reaction strongly in the hot core regions of the ISM. The catalytic role of the extra water molecule is explained by the relay hydrogen transport effect of the water. With the expectations that more number of water molecules as reactants (more than two water molecules) will be able to reduce the barrier significantly to a very low value, we carried out some preliminary investigation for the cases with three and four water molecules as reactants. Our preliminary studies related to finding the transition states for these two cases indicate that the respective transition states were found to be more stable than the reactants. Now with the transition states being lower in energy compared to the reactant, we can predict that this reaction will also be feasible in the cold interstellar clouds, and the exothermic nature of the reaction will be the driving force for this reaction to occur under such extreme low temperature conditions. A more detailed PES analysis of the reactions related to three and four water molecules needs further investigations to have an idea about the complete reaction profile. We hope that this work will be able to contribute to our future understanding of the formation of glycine in the ISM as well as its synthesis in the laboratory.
Acknowledgements
The authors like to thank the University of Johannesburg for the support. SS thanks the NRF, South Africa for incentive funding. ZPN thanks NRF, South Africa for PhD bursary. PB thanks the University of Johannesburg for the PhD bursary.References
- K. Ruiz-Mirazo, C. Briones and A. de la Escosura, Chem. Rev., 2014, 114, 285–366 CrossRef CAS PubMed.
- E. Herbst, Chem. Soc. Rev., 2001, 30, 168–176 RSC.
- R. D. Brown, P. D. Godfrey, J. W. V. Storey and M. P. Bassez, J. Chem. Soc., Chem. Commun., 1978, 547–548 RSC.
- Y.-J. Kuan, S. B. Charnley, H.-C. Huang, W.-L. Tseng and Z. Kisiel, Astrophys. J., 2003, 593, 848–867 CrossRef CAS.
- J. M. Hollis, J. A. Pedelty, L. E. Snyder, P. R. Jewell, F. J. Lovas, P. Palmer and S.-Y. Liu, Astrophys. J., 2003, 588, 353–359 CrossRef CAS.
- L. E. Snyder, F. J. Lovas, J. M. Hollis, D. N. Friedel, P. R. Jewell, A. Remijan, V. V. Ilyushin, E. A. Alekseev and S. F. Dyubko, Astrophys. J., 2005, 619, 914–930 CrossRef CAS.
- S. Pizzarello, Acc. Chem. Res., 2006, 39, 231–237 CrossRef CAS PubMed.
- A. S. Burton, J. C. Stern, J. E. Elsila, D. P. Galvin and J. P. Dworkin, Chem. Soc. Rev., 2012, 41, 5459–5472 RSC.
- P. Schmitt-Kopplin, Z. Gabelica, R. D. Gougeon, A. Fekete, B. Kanawati, M. Harir, I. Gebefuegi, G. Eckel and N. Hertkorn, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2763–2768 CrossRef CAS PubMed.
- J. R. Cronin and S. Pizarello, Science, 1997, 275, 951–955 CrossRef CAS PubMed.
- M. H. Engel and B. Nagy, Nature, 1982, 296, 837–840 CrossRef CAS.
- K. Kvenvolden, J. Lawless, K. Pering, E. Peterson, J. Flores, C. Ponnamperuma, I. R. Kaplan and C. Moore, Nature, 1970, 228, 923–926 CrossRef CAS PubMed.
- M. H. Engel, S. A. Macko and J. A. Silfer, Nature, 1990, 348, 47–49 CrossRef CAS PubMed.
- M. P. Callahan, A. S. Burton, J. E. Elsila, E. M. Baker, K. E. Smith, D. P. Glavin and J. P. Dworkin, Meteorit. Planet. Sci., 2013, 48, 786–795 CrossRef CAS.
- P. G. Brown, A. R. Hildebrand, M. F. Zolensky, M. Grady, R. N. Clayton, T. K. Mayeda, E. Tagliaferri, R. Spalding, N. D. MacRae, E. L. Hoffman, D. W. Mittlefehldt, J. F. Wacker, J. Andrew Bird, M. D. Campbell, R. Carpenter, H. Gingerich, M. Glatiotis, E. Greiner, M. J. Mazur, P. J. A. McCausland, H. Plotkin and T. R. Mazur, Science, 2000, 290, 320–325 CrossRef CAS PubMed.
- T. Hiroi, M. E. Zolensky and C. M. Pieters, Science, 2001, 293, 2234–2236 CrossRef CAS PubMed.
- J. G. Lawless, K. A. Kvenvolden, E. Peterson, C. Ponnamperuma and C. Moore, Science, 1971, 173, 626–627 CAS.
- J. E. Elsila, D. Glavin and J. P. Dworkin, Meteorit. Planet. Sci., 2009, 44, 1323–1330 CrossRef CAS.
- K. Ruiz-Mirazo, C. Briones and A. de la Escosura, Chem. Rev., 2014, 114, 285–366 CrossRef CAS PubMed.
- A. Strecker, Annalen der Chemie und Pharmazie, 1850, 75, 27–45 Search PubMed.
- S. L. Miller, Science, 1953, 117, 528–529 CAS.
- P. D. Godfrey, R. D. Brown, B. J. Robinson and M. W. Sinclair, Astrophys. Lett., 1973, 13, 119–121 CAS.
- J. E. Dickens, W. M. Irvine, C. H. DeVries and M. Ohishi, Astrophys. J., 1997, 479, 307–312 CrossRef CAS.
- C. J. Salter, T. Ghosh, B. Catinella, M. Lebron, M. S. Lerner, R. Minchin and E. Momjian, Astron. J., 2008, 136, 389–399 CrossRef CAS.
- D. M. Koch, C. Toubin, G. H. Peslherbe and J. T. Hynes, J. Phys. Chem. C, 2008, 112, 2972–2980 CAS.
- L.-P. Wang, A. Titov, R. McGibbon, F. Liu, V. S. Pande and T. J. Martínez, Nat. Chem., 2014, 6, 1044–1048 CrossRef CAS PubMed.
- J. W. Gauld, H. Audier, J. Fossey and L. Radom, J. Am. Chem. Soc., 1996, 118, 6299 CrossRef CAS.
- A. J. Chalk and L. Radom, J. Am. Chem. Soc., 1997, 119, 7573 CrossRef CAS.
- J. W. Gauld and L. Radom, J. Am. Chem. Soc., 1997, 119, 9831 CrossRef CAS.
- D. E. Woon, J. Phys. Chem. A, 2001, 105, 9478–9481 CrossRef CAS.
- A. Rimola, L. Rodríguez-Santiago, P. Ugliengo and M. Sodupe, J. Phys. Chem. B, 2007, 111, 5740 CrossRef CAS PubMed.
- A. Rimola, M. Sodupe and P. Ugliengo, Phys. Chem. Chem. Phys., 2010, 12, 5285–5294 RSC.
- A. Rimola, M. Sodupe and P. Ugliengo, Astrophys. J., 2012, 754, 24 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- A. Bauza, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496–2517 CrossRef CAS PubMed.
- M. Scherer, M. Havenith, R. Mauersberger and T. L. Wilson, Astron. Astrophys., 1998, 335, 1070–1076 CAS.
- X. Meng, J. Guo, J. Peng, J. Chen, Z. Wang, J.-R. Shi, X.-Z. Li, E.-G. Wang and Y. Jiang, Nat. Phys., 2015, 11, 235–239 CrossRef CAS.
- R. J. Shannon, M. A. Blitz, A. Goddard and D. E. Heard, Nat. Chem., 2013, 5, 745–749 CrossRef CAS PubMed.
- J. E. Dyson and D. A. Williams, The Physics of the Interstellar Medium. Series in Astronomy and Astrophysics, Institute of Physics, Bristol, 2nd edn, 1997 Search PubMed.
- R. T. Garrod and S. L. W. Weaver, Chem. Rev., 2013, 113, 8939–8960 CrossRef CAS PubMed.
- E. F. van Dishoeck and G. A. Blake, Annu. Rev. Astron. Astrophys., 1998, 36, 317–368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Potential energy surfaces for the MP2 method of the reactions, CH2NH + CO + 1H2O and CH2NH + CO + 2H2O, are shown in Fig. S1 and S2 respectively. Similarly, for the reaction CH2NH + CO + H2O–H2O (binary complex) the PESs for B3LYP and MP2 methods are shown in Fig. S3 and S4 respectively. Optimized structures with important geometric parameters for MP2 as well as B3LYP methods are shown in Fig. S5–S8. Table S1 summarizes the optimized geometries of the TS for the CH2NH + CO + 1H2O reaction for various methodologies and Table S2 summarizes the optimized geometries of the TS for the CH2NH + CO + 2H2O reaction case for various methodologies. Tables S3 and S4 are related to the effect of various basis sets and methodologies respectively for the reaction of CH2NH, CO with one H2O. Similarly, Tables S5 and S6 are related to the effect of various basis sets and methodologies respectively for the reaction of CH2NH, CO with two H2O. See DOI: 10.1039/c5cp04987c |
| This journal is © the Owner Societies 2016 |