
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Energy transfer and formation of long-lived 3MLCT states in multimetallic complexes with extended highly conjugated bis-terpyridyl ligands†
Maria
Wächtler
a,
Joachim
Kübel
ab,
Kevin
Barthelmes
cd,
Andreas
Winter
cd,
Alexander
Schmiedel
e,
Torbjörn
Pascher
f,
Christoph
Lambert
e,
Ulrich S.
Schubert
cd and
Benjamin
Dietzek
*abd
aLeibniz Institute of Photonic Technology e.V., Albert-Einstein-Straße 9, 07745 Jena, Germany. E-mail: benjamin.dietzek@ipht-jena.de; Fax: +49 (0)3641 206-390; Tel: +49 (0)3641 206-332
bInstitute of Physical Chemistry and Abbe Center of Photonics, Friedrich Schiller University Jena, Helmholtzweg 4, 07743 Jena, Germany
cLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstraße 10, 07743 Jena, Germany
dJena Center for Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany
eInstitut für Organische Chemie, Universität Würzburg, Wilhelm Conrad Röntgen Research Center for Complex Material Systems, Center for Nanosystems Chemistry, Am Hubland, 97074 Würzburg, Germany
fPascher Instruments AB, Stora Råbybyaväg 24, 22478 Lund, Sweden
First published on 21st September 2015
Abstract
Multimetallic complexes with extended and highly conjugated bis-2,2′:6′,2′′-terpyridyl bridging ligands, which present building blocks for coordination polymers, are investigated with respect to their ability to act as light-harvesting antennae. The investigated species combine Ru(II)- with Os(II)- and Fe(II)-terpyridyl chromophores, the latter acting as energy sinks. Due to the extended conjugated system the ligands are able to prolong the lifetime of the 3MLCT states compared to unsubstituted terpyridyl species by delocalization and energetic stabilization of the 3MLCT states. This concept is applied for the first time to Fe(II) terpyridyl species and results in an exceptionally long lifetime of 23 ps for the Fe(II) 3MLCT state. While partial energy (>80%) transfer is observed between the Ru(II) and Fe(II) centers with a time-constant of 15 ps, excitation energy is transferred completely from the Ru(II) to the Os(II) center within the first 200 fs after excitation.
Introduction
The design of artificial antennae systems for efficient collection of solar radiation, i.e., multicomponent systems in which several molecular components absorb the incident light and channel the energy to an acceptor unit, is an active field of research.1–8 The ultimate goal is to build systems, which are capable of harvesting light over a large part of the visible spectrum. In this respect a number of systems containing transition metal complexes have been prepared and studied.1,2,5,9–16 Multimetallic complexes containing several metal centers connected via large conjugated ligands can show photoinduced energy and/or electron transfer processes between the individual chromophoric centers.13,16,17 A challenge in the design of such systems is to control the direction of the energy/electron transfer, to transport the excitation over large distances, e.g., in wirelike structures.17–20 This can be achieved by structural and electronic variations of the bridging ligands and/or by changing metal centers and their coordination environment to create a gradient for energy or electron transport in the system.13 In this respect, hierarchically structured coordination polymers are interesting systems for the design of artificial light-harvesting antennas. Especially, low-spin d6 polypyridyl transition metal complexes play an important role in this field due to their strong absorption of light in the visible spectral range and the favorable and tunable photophysical properties of their charge-transfer states.13,17,21–23In this contribution bi- and trimetallic systems are investigated, which may serve as building blocks for coordination polymers. For this purpose a bis-2,2′:6′,2′′-terpyridyl ligand L bearing a conjugated spacer, which is closely related to the widely used poly[phenylene–vinylene] and poly[phenylene–ethynylene] conjugated polymers (Fig. 1), is used.24,25 The strong conjugation of the bridging ligand together with the tridentate coordination site enables to build linear, rodlike structures and no stereoisomer mixtures are formed upon complexation, as it is the case for bidentate ligands.13,17,20,26,27 The herein investigated species combine Ru(II) and Fe(II) (RuFeRu) or Ru(II) and Os(II) (RuOs) centers, which are coordinated by the terpyridyl units. This combination of metal centers in the supramolecular assembly is advantageous due to their complementary absorption features: both Fe(II) and Os(II) complexes absorb at wavelengths longer than the 1MLCT (metal-to-ligand charge transfer) absorption band of Ru(II)-terpyridyl centers: Fe(II) terpyridyl complexes possess a 1MLCT transition at ca. 560 nm28–30 and the Os(II) terpyridyl chromophore shows, besides a 1MLCT absorption band in the same wavelengths range as the Ru(II) 1MLCT transitions, a 3MLCT absorption band spanning the entire visible range of the absorption spectrum17 extending the overall absorption spectrum of the assembly far into the red up to 700 nm. In these assemblies the Ru(II) centers are expected to serve as excitation energy donor while Os(II) and Fe(II) centers possess lower lying excited states and serve as potential energy accepting units.13,15,17,18,28,31–33
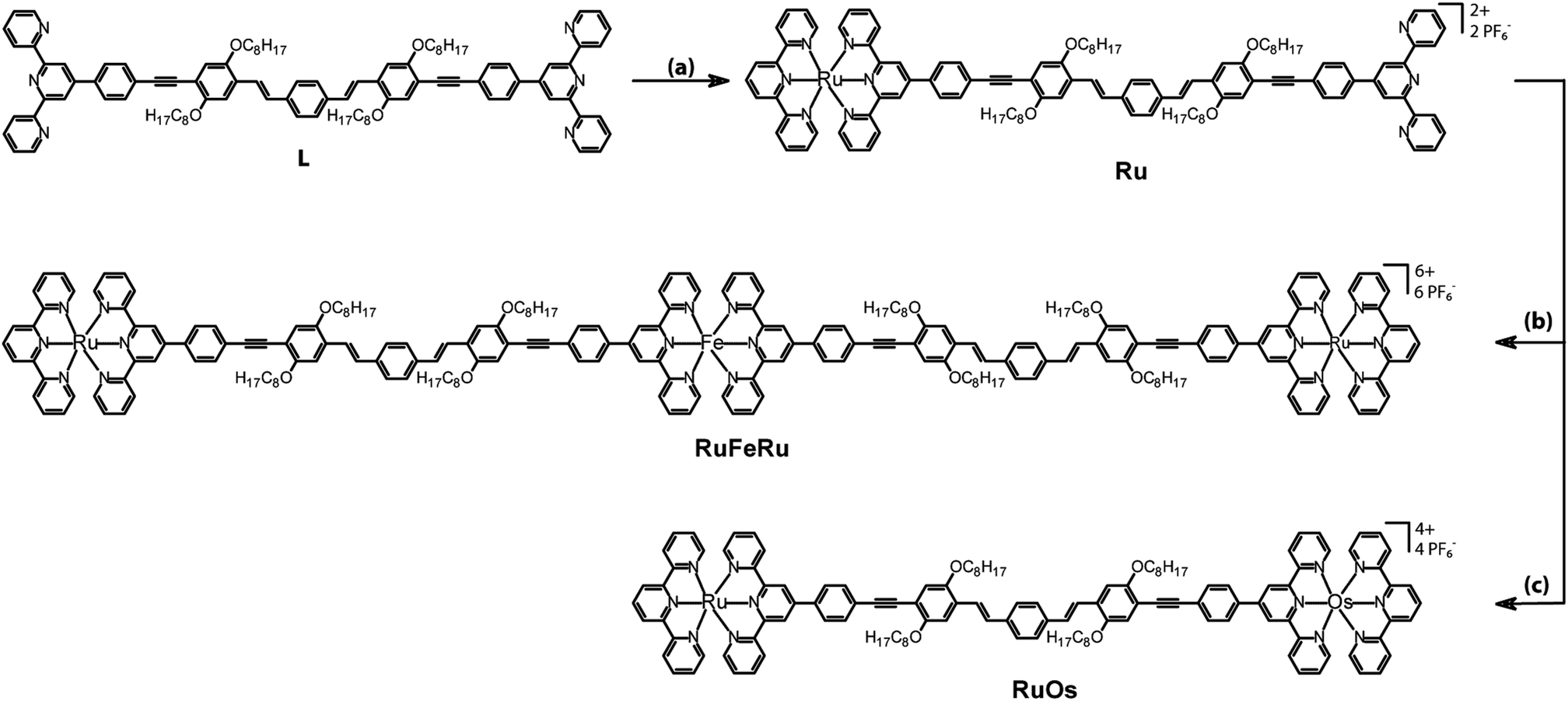 | ||
| Fig. 1 Structures and synthesis of the investigated polynuclear complexes RuFeRu and RuOs and the mononuclear reference complex Ru. Legend: (a) [Ru(tpy)(acetonitrile)3](PF6)2, DMF, 160 °C, 3 h; (b) FeSO4·7H2O, dichloromethane/methanol, room temperature, 2 h; (c) (i) Os(tpy)Cl3, AgBF4, acetone, 70 °C, 2 h, (ii) DMA/ethylene glycol, 160 °C, 24 h. | ||
A critical property for future applicability of such structures is the excited state lifetime of the acceptor centers, which can be a crucial efficiency determining factor for subsequent reaction steps, e.g., charge separation and generation of redox-equivalents. The 3MLCT states of Os(II)-bis(terpyridyl) chromophores possess sufficiently long lifetimes in the range of 200 ns in aerated solution at room temperature.17 Due to the high ligand-field strength in Os complexes the energy gap between 3MLCT states and metal-centered (MC) ligand field states, which offer an efficient route for radiationless deactivation, is large.17,28 This prevents deactivation of the 3MLCT state via thermal population of the MC states.34 In the order Os(II), Ru(II) and Fe(II) the ligand field strength decreases and, hence, the MC states are significantly closer in energy to the 3MLCT states in comparable Ru(II) and Fe(II) complexes. Furthermore, in tridentate terpyridyl complexes additional weakening of the ligand field is observed due to the unfortunate bite angle of the terpyridyl ligand, which causes a distortion of the octahedral coordination sphere.13,17 This further decreases the energetic separation between MC and MLCT states which is reflected in the short room-temperature lifetime of the 3MLCT excitation in Ru(II)-terpyridyl complexes ([Ru(tpy)2]2+τ = 124 ps)17,35 and Fe(II)-polypyridyl complexes in general ([Fe(tpy)2]2+, [Fe(bpy)3]2+τ ≤ 0.2 ps).36–43 This is also the reason why – in contrast to Ru(II) and Os(II)-polypyridyl centers – Fe(II)-polypyridiyl complexes are not established in light-harvesting applications yet, despite its high abundance and low cost of production. Only ultrafast charge separation processes on the sub 100 fs timescale are able to compete with the ultrafast deactivation to the high spin quintet state (which is probably mediated by ligand field triplet states) in Fe(II)-polypyridyl complexes,36–42e.g., ultrafast injection of electrons into TiO2.37,44,45 Hence, to boost the applicability of Fe(II)-polypyridyl dyes in light-harvesting applications one important goal is to prolong the 3MLCT lifetime.
One approach to prolong the lifetime of 3MLCT states is to increase the relative energy of the MC states via coordination of ligands with high ligand field strength, e.g., strong σ-donor ligands. Attractive candidates in this respect are cyclometallated ligands. In a first attempt 3MLCT lifetimes in Ru(II) complexes were extended by four orders of magnitude46–48 and in Fe(II) cyclometallated complexes 3MLCT lifetimes of 9 ps and even 13 ps were achieved, which corresponds to an extension of the 3MLCT lifetime by two orders of magnitude.49–51 A second possibility to increase the energy gap between 3MLCT and MC states is to coordinate stronger π-acceptor ligands, which also increases the ligand field strength, hence raises the energy of the MC states and further results in lower energies of the 3MLCT state. This can be achieved by substitution in the 4′-position of the terpyridyl ligand and extending the conjugated system of the terpyridyl coordination sphere. Applying this approach the lifetimes in Ru(II) complexes were extended to reach tens of ns.27 An additional prolongation of the lifetime of the 3MLCT state can be achieved by applying highly extended conjugated 4′-terpyridyl ligands. In such systems low lying 3LC (ligand-centered) states may form equilibria with the 3MLCT state.27,30,52,53 Though structures fulfilling these preconditions are known in literature,54–56 the Fe(II) 3MLCT lifetimes in such systems have not been addressed yet. A third approach was applied by Heinze et al.57 by coordinating ligands with larger bite angle than the classical terpyridyl units, which increases the relative energy of MC states due to less distortion of the coordination polyhedron resulting in a stronger ligand field.58–62 Additionally, ligands with strong electron withdrawing/electron donating character have been combined resulting in push–pull systems with low lying 3MLCT states.63–65 The first attempts following this approach for Fe(II) complexes, however, did not lead to formation of a long-lived CT (charge-transfer) states.57
In the here investigated systems the highly conjugated bis-2,2′:6′,2′′-terpyridine ligands offer the possibility to stabilize the 3MLCT states and additionally to form equilibria with low lying 3LC states as was previously reported for Ru(II) coordinated by related ligands.66 In the following the photoinduced processes in the multimetallic systems, especially with respect to possible energy transfer pathways between the specific metal centers, will be investigated by time-resolved spectroscopy. Special emphasis will be on the 3MLCT lifetime of the Fe(II) 3MLCT states in these systems.
Experimental section
For spectroscopic measurements all compounds were dissolved in aerated acetonitrile (spectrophotometric grade). Absorption spectra were recorded with a Lambda 750 (PerkinElmer) UV/VIS spectrometer in a cell with 1 cm pathlength. Emission spectra were recorded with a Jasco FP-6200 spectrofluorometer. For all time-resolved measurements the stability of the samples was verified by recording the absorption spectra before and after each measurement.For the ns time-resolved transient absorption (TA) measurements the samples were excited by 5 ns pulses at 520 nm with a repetition rate of 10 Hz. The probe light is delivered by a pulsed 75 W Xe arc lamp and detected on a PMT after passing a monochromator. By switching off the probe light emission decay can be detected with ns-temporal resolution with the same set-up.
The fs time-resolved transient absorption measurements were performed in a cuvette with 1 mm path length. Ru and RuFeRu were excited by 140 fs pulses at 520 nm and 520/575 nm, respectively. A white light continuum generated in a CaF2 crystal was used to probe the sample between 350 and 800 nm. The relative temporal delay between the pump and probe pulses was varied over a maximum range of 8 ns. The fs time-resolved measurements for RuOs were performed applying pump pulses centered at 520 and 670 nm with a duration of ∼80 fs and probed by a white-light continuum between 450 and 700 nm, which is produced in a sapphire crystal. The pump pulses are delayed with respect to the probe pulses by means of an optical delay stage over a maximum range of 2 ns. The mutual polarization of the pump and probe pulses was set to magic angle.
Prior to data analysis the experimental data from the fs time-resolved measurements were chirp corrected. To avoid prominent contributions from the coherent artifact the pulse overlap region (±200 fs) around time zero was excluded in the data fitting procedure. The data were fitted with a global fitting routine applying a sum of exponential functions for data analysis. Additionally, the data for RuFeRu upon excitation at 520 nm were numerically fitted with a home-written algorithm for non-sequential (e.g., including branching of the relaxation pathway) kinetic schemes.
For a more detailed description of the synthetic procedures, characterization, experimental set-ups and the fitting procedure the reader is referred to the ESI.†
Results and discussion
Synthesis
The synthesis of the bridging ligand L, which is used in this study, was reported recently.67 In order to obtain heterometallic oligonuclear complexes, a two-step assembly approach had to be followed (Fig. 1). The first step was the preparation of the mononuclear Ru(II) bisterpyridyl complex Ru by reaction of [Ru(tpy)(acetonitrile)3](PF6)268 with ligand L in DMF (tpy – 2,2′:6′,2′′-terpyridine).69 The trinuclear complex RuFeRu was prepared by addition of a methanolic Fe(II) sulphate solution to a solution of Ru in dichloromethane. The dinuclear complex RuOs was prepared by the reactions of Ru with silver-activated Os(tpy)Cl3 in a DMAc/ethylene glycol mixture.70Steady-state absorption and emission spectroscopy
The absorption spectra of Ru, RuFeRu and RuOs in acetonitrile are displayed in Fig. 2. The spectra show multiple electronic transitions in the visible and UV spectral region. ππ* transitions, which are located at the terpyridine sphere, dominate at wavelengths shorter than 350 nm. At longer wavelengths the spectra contain a superposition of ligand-centered ππ* transitions and transitions which appear upon coordination of the metal center and, according to literature, most likely are due to MLCT transitions.17,28–30,71 The ligand-centered ππ* transitions of the extended bridging ligand can be identified by the comparison with the absorption spectrum of the pure ligand L (absorption maximum at 429 nm). No significant shifts are observed in the ππ* transition upon coordination of one or two metal centers: for Ru the maximum of the ππ* transition is at 426 nm and for RuFeRu at 429 nm. For RuOs no separate maximum of the ππ* transition can be distinguished. The Ru(II) 1MLCT transitions in Ru and RuFeRu peak at 486 nm while in RuOs the maximum of the superimposed 1MLCT transitions of both the Ru(II) and Os(II) centers is at 490 nm. The 1MLCT transitions at the Ru(II) and Os(II) center present a mixture of transitions to the terminal tpy ligand and the 4′-substituted bridging ligand with the extended chromophore. The torsional angle between the tpy unit and the adjacent phenyl ring, which is typically in the range of 30°,72–74 reduces conjugation but does not inhibit it completely. Hence, the chromophore is delocalized over the tpy and the directly connected phenyl ring, but – on the other hand – does not extent over the ethynyl bond.72 When comparing the absorption spectrum of RuFeRu to the one of Ru an additional band at 574 nm is observed, which is assigned to the Fe(II) 1MLCT transition to the bridging ligand. Also the spectrum of RuOs is significantly broadened due to additional weak spin-forbidden 3MLCT transitions spanning the visible spectral region up to 700 nm with a maximum at 665 nm.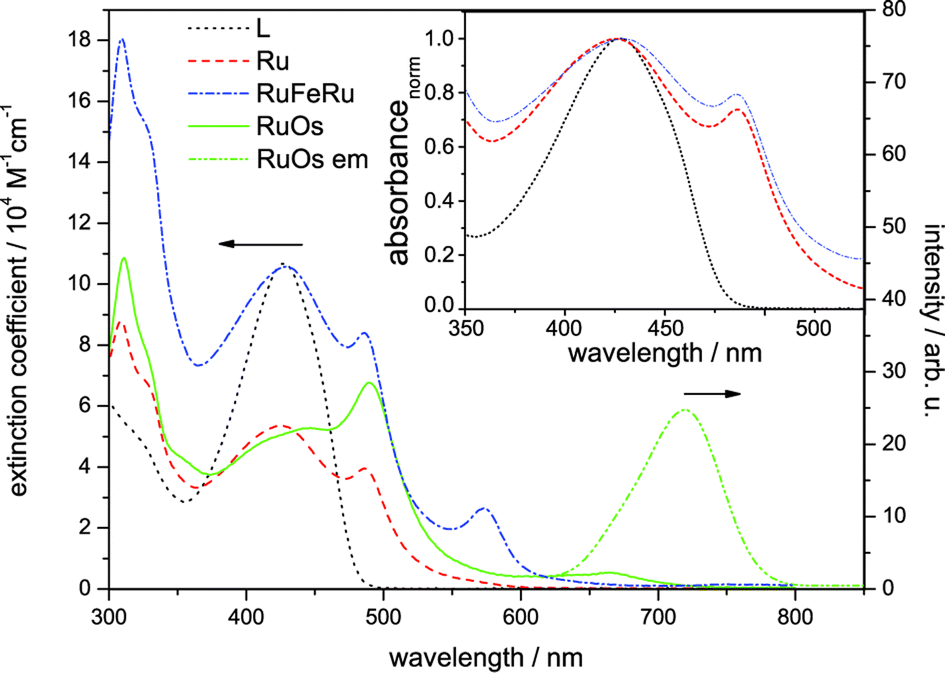 | ||
| Fig. 2 Steady-state absorption spectra of Ru, RuFeRu and RuOs and emission spectrum of RuOs after excitation at 488 nm in aerated acetonitrile. For comparison the absorption spectrum of the bridging ligand L in dichloromethane is given. The inset shows the normalized absorption spectra of L, Ru and RuFeRu for comparison of the position of the LC absorption band. | ||
Only for RuOs a weak triplet emission could be detected at room temperature in aerated solution. This emission peaks at 720 nm and is due to 3MLCT phosphorescence from the Os(II) center. In all three investigated species, even the mononuclear complex Ru, phosphorescence from the Ru(II) 3MLCT state is not detectable, hence no conclusions about energy transfer between the metal centers can be drawn from emission experiments. The missing emission could be an indication for a strong delocalization of the 3MLCT state over the extended bridge in these systems, decreasing the oscillator strength for the radiative return to the ground state and open the competition with more prone non-radiative deactivation processes. Besides its probably low quantum yield, unambiguous detection of the Ru(II) 3MLCT emission in Ru unfortunately is additionally hampered by residual fluorescence originating from LC states in the same spectral region where the emission of the 3MLCT is expected (for further details see ESI,† Fig. S1 and S2). Such LC emission was reported earlier for related 2,2′:6′:2′′-terpyridine complexes coordinating two highly conjugated 4′-(4-{[2,5-bis(octyloxy)-4-styrylphenyl]ethynyl}-phenyl)-2,2′:6′,2′′-terpyridine ligands.30,75 Emission originating from the Fe(II) 3MLCT in RuFeRu is not expected due to the generally observed fast deactivation of the 3MLCT states in Fe(II)-polypyridyl complexes either populating a non-luminescent high-spin quintet state36–43 or rapidly repopulating the ground state via3MC states.50,51
Lifetime of long-lived excited states
The lifetime of the Os(II) 3MLCT based phosphorescence of RuOs in aerated acetonitrile at room temperature was determined to 100 ns, which is in good agreement with the decay time of the excited-state absorption (ESA) signal in ns time-resolved transient absorption (TA) measurements (τ = 84 ns) (Fig. 3). | ||
| Fig. 3 (A) Transient absorption kinetics of RuOs at selected probe wavelengths: 450 nm (dash/black), 590 nm (solid/black) and emission decay at 750 nm (red solid). (B) Direct comparison of the decay kinetics at 600 nm probe wavelength for Ru (black) and RuFeRu (red). | ||
For Ru and RuFeRu, although no Ru(II) 3MLCT emission is detectable at room temperature, transient absorption spectroscopy with ns time-resolution reveals the presence of a long-lived excited state. The excited state lifetimes of Ru and RuFeRu are 132 and 129 ns, respectively (Fig. 3), which is in good agreement with the lifetime determined for the analogues homo-bimetallic Ru(II) complex (145 ns).66 The nearly identical values of their lifetimes and spectral shapes of the transient spectra of the long-lived excited state in Ru and RuFeRu (see ESI,† Fig. S8) point to a similar origin of the signals, i.e. an excited state connected with the Ru(II) center. Contributions of an Fe(II) excited state to the transient spectra are not expected on the timescale of the experiment, as Fe(II)-terpyridyl species show typically lifetimes <10 ns (see below).17,29,36,76–78 The observation of an excited Ru(II) state suggests that energy transfer between the Ru(II) and the Fe(II) metal centers in RuFeRu occurs with much less than 100% efficiency.
fs time-resolved spectroscopy
To identify and to follow possible energy transfer processes in the polynuclear systems RuFeRu and RuOs fs time-resolved TA spectroscopy was applied. To disentangle the contributions of the Ru(II) and Fe(II)/Os(II) centers to the observed photoinduced dynamics and to be able to identify possible interactions and transfer processes between the different metal centers, first the characteristic dynamics of each metal center is studied by selective excitation at carefully chosen excitation wavelengths. In contrast to the Os(II) and the Fe(II) centers, selective excitation of the Ru(II) chromophore is not possible in the polymetallic species, hence the monometallic compound Ru serves as reference model for the Ru(II) intrinsic spectral signatures and dynamics.Upon excitation at 520 nm, i.e., in the Ru(II) 1MLCT band of Ru, the transient spectra (Fig. 4) are dominated by excited-state absorption (ESA) at probe wavelengths longer than 520 nm with a maximum at 630 nm. Below 500 nm negative signal contributions due to ground-state bleach (GSB) dominate. Within the first 100 ps the signal intensity increases and the minimum in the GSB at 482 nm vanishes, while the second minimum shifts from 432 to 418 nm. After 1000 ps the overall intensity starts to decay (by approximately 8% up to 8 ns).
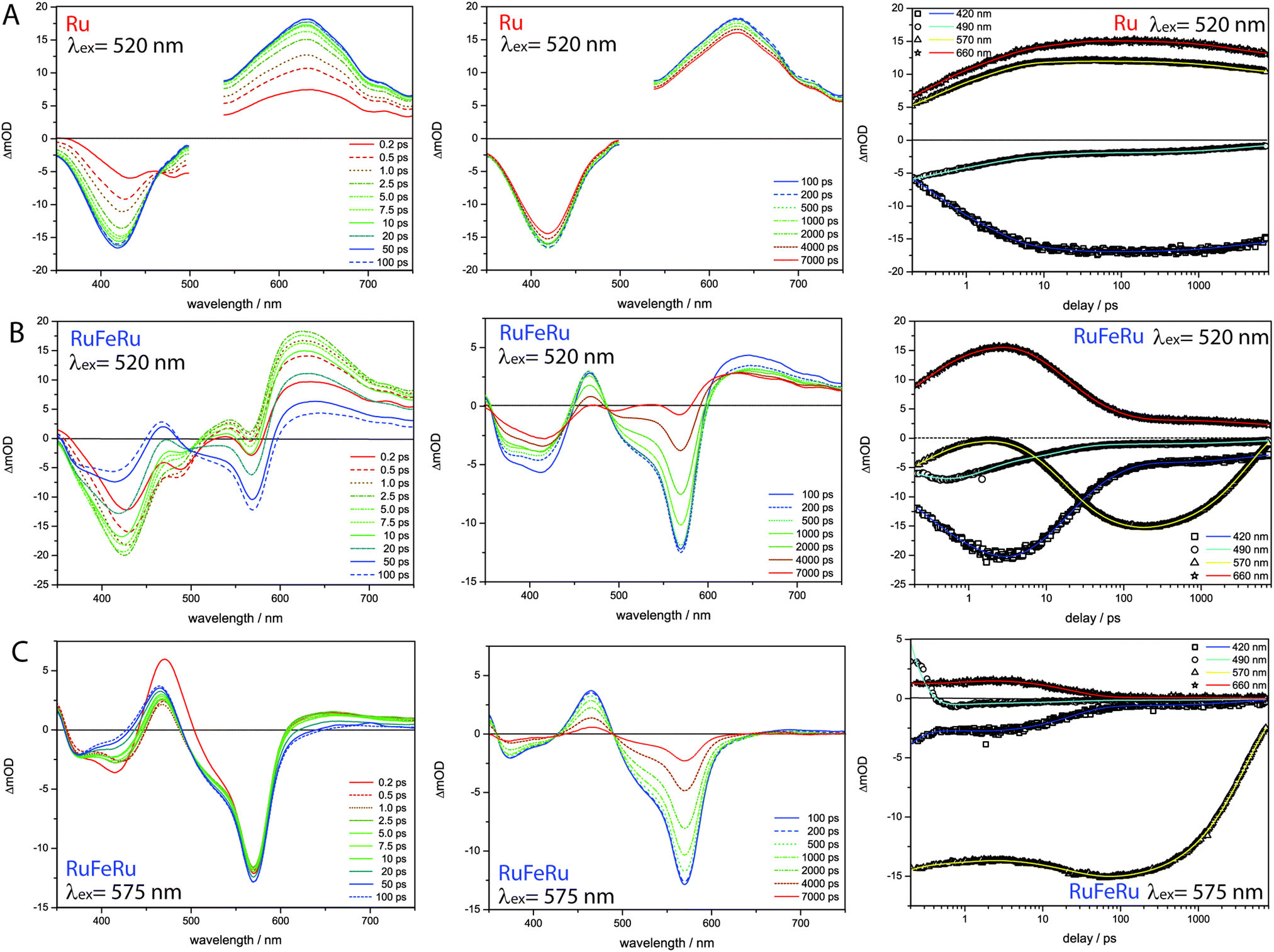 | ||
| Fig. 4 Transient absorption spectra at selected delay times (left and middle) for (A) Ru following excitation at 520 nm, (B) RuFeRu upon excitation at 520 nm and (C) RuFeRu upon excitation at 575 nm in acetonitrile and the respective kinetics at selected probe wavelengths (right). The pump region for Ru is neglected in the data evaluation due to scattered pump. | ||
The data was globally fitted with a sequential relaxation scheme. To describe the data four rates and an additional long-lived component, with a lifetime exceeding the observation window, had to be taken into account (Table 1). A feasible assignment of the processes and the species associated spectra (SAS) corresponding to the excited states contributing to the relaxation processes (Fig. 5) is displayed in Scheme 1. The sub-ps process can be related to internal vibrational energy redistribution (IVR) and intersystem crossing (ISC), which leads to the population of the manifold of vibrationally hot Ru(II) 3MLCT states within the first ∼200 fs with a quantum yield close to unity.53,79–83 The two components in the range of a few ps describe intramolecular relaxation processes: τ2 = 2 ps is assigned to vibrational cooling23,53,71,82–86 and interligand electron transfer (ILET)35 after which the lowest vibrationally relaxed Ru(II) 3MLCT state in the system is populated and τ3 = 15 ps describes further structural relaxation, i.e., planarization of the bridging ligand, and as a consequence delocalization of the Ru(II) 3MLCT state beyond the terpyridine sphere of the bisterpyridine bridging ligand.30,66 The slowest component τ5 responsible for the slight decay of the signal amplitude at later delay times can be identified as equilibration with a 3LC state (see ESI,† Fig. S10).66 This equilibration is found to be approached much slower in the monometallic complex Ru than in the related homobimetallic complex RuRu (Ruτ5 = 3100 ps, RuRuτ = 361 ps66). This could be an indication for changes in the relative energetic positions of the 3MLCT and 3LC states induced by the coordination of the second metal center leading to changes in the activation barriers impacting the equilibration process.
| λ ex/nm | k 1/ps−1 | τ 1/ps | k 2/ps−1 | τ 2/ps | k 3/ps−1 | τ 3/ps | k 4/ps−1 | τ 4/ps | k 5/ps−1 | τ 5/ps | k 6/ns−1 | τ 6/ns | k inf /ns−1 | τ inf /ns | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Infinite component in the fs time-resolved measurements, rate/lifetime determined by ns time-resolved TA and emission measurements. b Numerical fit applying model described in Scheme 1. | |||||||||||||||
| Ru | 520 | 3.1 | ≤0.3 | 0.48 | 2.0 | 0.066 | 15 | — | — | 0.00033 | 3100 | — | — | 0.0076 | 132 |
| RuFeRu modelb | 520 | 4.1 | ≤0.2 | 0.74 | 1.4 | 0.067 | 15 | 0.018 | 56 | — | — | 0.24 | 4.2 | 0.0077 | 129 |
| RuFeRu | 575 | 9.7 | ≤0.1 | 0.92 | 1.1 | — | — | 0.043 | 23 | — | — | 0.25 | 4.1 | — | — |
| RuOs | 670 | — | — | 1.3 | 0.8 | 0.11 | 9 | — | — | 0.0016 | 650 | — | — | 0.01 | 100 |
| RuOs | 520 | 4.8 | ≤0.2 | 0.63 | 1.6 | 0.090 | 11 | — | — | 0.0014 | 700 | — | — | ||
 | ||
| Fig. 5 SAS resulting from the global fit (A) Ru excitation 520 nm sequential model, (B) RuFeRu excitation 520 nm model with energy transfer according to Scheme 1, (C) RuFeRu excitation at 575 nm sequential model. | ||
 | ||
| Scheme 1 Proposed relaxation schemes for Ru and RuFeRu, solid lines define energy levels with defined energetic positions, while dashed lines define excited states, the energy of which can only be indirectly inferred or depends on the excitation wavelength, processes in grey are not directly observable in the data. | ||
Upon selective excitation of RuFeRu into the Fe(II) 1MLCT transition at 575 nm, the Fe(II) intrinsic dynamics is accessible (Fig. 4). Transient spectra show GSB at 570 nm corresponding to the maximum of the Fe(II) 1MLCT transition. Three regions dominated by ESA are observed: at wavelength longer than 605 nm, between 490 and 430 nm and in the UV below 360 nm. The ESA feature in the red spectral region decays within 100 ps. On the same timescale the second ESA feature gains intensity and broadens, extending further to the blue spectral range. After 100 ps a global decay of the overall signal intensity occurs. This development can quantitatively be described by a multi-exponential fit corresponding to the sequential relaxation scheme presented in Scheme 1 applying four exponentials (see Table 1 and Fig. 5 for SAS). The sub-ps and 1.1 ps processes describe – in analogy to the processes at the Ru(II) center – population of the Fe(II) 3MLCT manifold and vibrational cooling. Especially interesting is the process with a time constant of τ4 = 23 ps. This process is connected to the decay of the ESA feature in the red probe range (see also decay associated spectra (DAS) in the ESI,† Fig. S10), which is – according to literature – a characteristic feature of the Fe(II) 3MLCT state.37,38 Hence, the process associated with τ4 probably describes the depopulation of the Fe(II) 3MLCT state via3MC states resulting in population of the high-spin Fe(II) quintet state. From the decay of the overall signal representing repopulation of the ground state a Fe(II) quintet lifetime of τ6 = 4.1 ns is determined, which is comparable to lifetimes reported for the quintet state of [Fe(tpy)2]2+.17,29,36,76,77 Neither planarization nor formation of an equilibrium with an 3LC state is observed upon excitation of the Fe(II) center. The latter is probably due to the lower energetic position of the excited states of the Fe(II) center compared to the Ru(II) center, preventing a thermal population of the 3LC states, while the former might skip detection due to negligible spectral changes in the observation range. Nevertheless, from these results it can be concluded that the system at hand shows a lifetime of 23 ps for the Fe(II) 3MLCT state, which is the longest reported in literature.50,51 Hence, the strong conjugated and extended ligand system shows a similar stabilizing effect on the Fe(II) 3MLCT state as for the Ru(II) 3MLCT states.
In the next step the photoinduced processes in RuFeRu upon excitation at 520 nm, where simultaneously excitation of Ru(II) 1MLCT and Fe(II) 1MLCT transitions occurs, are investigated. Immediately after excitation the transient spectra (Fig. 4) can be described as a superposition of the spectral characteristics of both centers at early delay times. During the first 100 ps the spectral features characteristic for the excited states of the Ru(II) center decay (e.g., the strong ESA signal in the red probe range) while the characteristic signatures of the Fe(II) excited states (e.g., bleach at 570 nm) gain in intensity. In the following the signal amplitude is decaying on a similar timescale as observed for the ground-state repopulation from the Fe(II) quintet state. Nonetheless, a residual amplitude remains beyond the timescale of the experiment (8 ns). This long-lived species shows positive and negative signal above and below 500 nm, respectively, and thus reflects the transient spectra of the photoexcited Ru(II)-subunit at long delay times (see ESI,† Fig. S14). A simple multi-exponential fit assuming a superposition of the characteristic Ru(II) and Fe(II) centered photoinduced dynamics was not sufficient to describe the observed temporal development of the signal (see ESI,† Table S1 and Fig. S9). Hence, changes of the values of the rates of the processes at the Ru(II) center due to coordination of the second metal center and additional processes like energy transfer between the Ru(II) and the Fe(II) centers need to be taken into account. A further complication is that by applying a sum of exponential functions to model the data only a strictly parallel or sequential relaxation scheme can be analytically described.87 Hence, to describe the data exactly a numerical fit based on the relaxation scheme in Scheme 1 was performed (Table 1): due to the very similar time constants and close resemblance of the spectral contributions of the sub-ps τ1 and cooling processes τ2 at both metal centers, the applied model regards the initial excited state as weighted mixture of Ru(II) and Fe(II) 1MLCT states. These states are decaying in parallel to the respective thermally relaxed 3MLCT states, described by a component τ1 ≤ 0.2 ps representing population of the respective triplet manifolds. The component τ2 mainly represents the vibrational cooling at the Ru(II) center, contributions from cooling at the Fe(II) center are neglected, this process probably is not detectable due only minor changes in the SAS (Fig. 5) between Fe(II) 3MLCThot and Fe(II) 3MLCTcold. From the development of the spectral signature with time (Fig. 4), i.e. decay of the Ru(II) features and increase of the Fe(II) signatures on a timescale up to 50 ps, it can be deduced that the energy transfer from the Ru(II) center to the Fe(II) center occurs in parallel to or even before further relaxation (planarization and equilibration) at the Ru(II) center. The time constant for the energy transfer τ3 is found to be with 15 ps nearly identical to that describing the planarization in Ru. Further, a fraction of excitation remains at the Ru(II) center, which is indicated by the remaining residual Ru(II) signature beyond 8 ns. This suggests that in this system energy transfer might occur from a non-planarized state in parallel to planarization and a fraction of the excited molecules remains trapped in the energetically low-lying planarized Ru(II) excited state. From the intensity ratio of the long-lived component in the data of Ru and RuFeRu upon excitation at 520 nm the efficiency of the energy transfer from the Ru(II) center to the Fe(II) center is determined to be at least 80%, assuming that neither the extinction coefficient in the ground nor the excited state changes significantly upon coordination of the Fe(II) center (details see ESI†). In the model applied, equilibration at the Ru(II) center is not explicitly included. This process is expected to be slower than the energy transfer and occurs only in the residual fraction of the excited molecules remaining in the Ru(II) excited state, resulting in very weak contributions to the overall signal development, too low to be determined properly (see ESI,† Fig. S12). The final equilibrated Ru(II) state is considered in the fit by incorporation of a constant component. Hence, the further development of the signal represents Fe(II) centered kinetics only. The process associated with τ4 probably is connected to the depopulation of the 3MLCT state to form the Fe(II) quintet state. The reason for the observed deceleration of this process upon excitation at 520 nm compared to direct excitation at 575 nm remains unclear at the moment and is subject to further investigations. τ6 describes ground-state repopulation originating from the quintet state. The description of the experimental data applying this model results in SAS for the involved excited states, which are in good agreement with the spectra determined for the respective states at the Ru(II) and Fe(II) center from the reference measurements described above (Fig. 5). This resemblance confirms the validity of the applied model.
In contrast to RuFeRu, RuOs shows only minor differences in the dynamic behavior upon direct excitation of the 3MLCT of the Os(II) center at 670 nm and upon excitation at 520 nm, where 1MLCT transitions of both the Ru(II) and the Os(II) center are excited (Fig. 6). Under both conditions the transient spectra show strong GSB below 510 nm and a broad ESA feature at wavelengths longer than 510 nm. The ESA intensity increases during the first 10 ps, while the GSB intensity remains constant. After 10 ps the overall signal amplitude decays reaching a plateau at 80% of the maximum value of signal amplitude. Assuming excitation of both Ru(II) and Os(II) 1MLCT states at 520 nm, the absence of signatures of Ru(II) excited states in the transient spectra suggests that the energy transfer from the Ru(II) to the Os(II) center occurs on an ultrafast timescale (<200 fs) with 100% efficiency and escapes detection due to the limited time-resolution of the experiment. Such highly efficient ultrafast energy transfer processes have been reported before for other multimetallic systems combining Ru(II) and Os(II) metal centers.15,18,28,31,56,88–90 This assumption is corroborated by the results of the multi-exponential fit (Table 1): independent of the excitation conditions vibrational cooling (∼1 ps), ligand planarization (∼10 ps) and equilibration with the 3LC state (∼600–700 ps) are observed. Further, the SAS (Fig. 7) for the involved excited states match each other very closely for varying excitation conditions and hence, the observed processes are assigned to occur in the Os(II) excited states manifold. The equilibration in RuOs between the Os(II) 3MLCT state and the 3LC state is slower than the equilibration between the Ru(II) 3MLCT states and the 3LC states in the homobimetallic complex RuRu,66 which is probably due to different relative energetic positions between the respective 3MLCT state and the 3LC state and altered activation barriers, the Os(II) 3MLCT states are typically 0.20–0.3 eV lower in energy than the Ru(II) 3MLCT states.17 The only difference in the pump-wavelength dependent data can be found on timescales <0.5 ps: here, an increase in GSB intensity and a slight blueshift in the maximum is present upon excitation at 520 nm (Fig. 6), which is described by the sub-ps component τ1 (Table 1). This component could be due to 3MLCT population processes from the excited 1MLCT states under these excitation conditions but could also show contributions of the energy transfer. However, to unambiguously assign these possible processes measurements with higher time resolution are necessary.
 | ||
| Fig. 6 Transient spectra and kinetic traces for RuOs upon excitation at (A) 670 nm and (B) 520 nm. The insets show a magnification of the ultrafast sub 0.5 ps development of the GSB region. | ||
 | ||
| Fig. 7 SAS for RuOs resulting from a fit with a sequential model according to Scheme 2 upon excitation at (A) 670 nm and (B) 520 nm. | ||
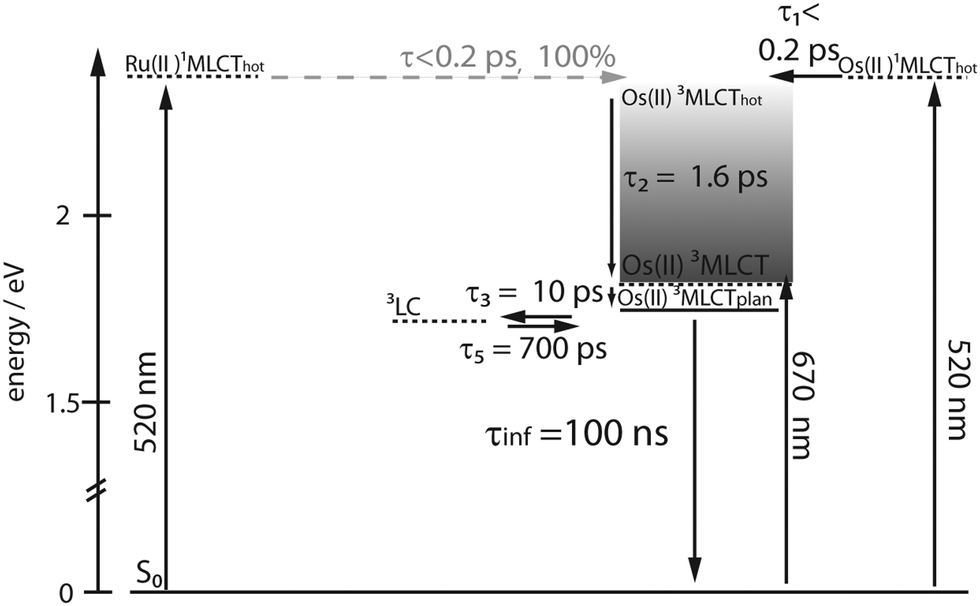 | ||
| Scheme 2 Proposed relaxation scheme for RuOs, solid lines define energy levels with defined energetic positions, while dashed lines define excited states, the energy of which can only be indirectly inferred or depends on the excitation wavelength, processes in grey are not directly observable in the data. | ||
The effect that energy transfer occurs on the ultrafast timescale and with 100% efficiency from Ru(II) to Os(II) centers while in comparable systems combining Ru(II) and Fe(II) centers energy transfer to the lower lying Fe(II) centered states is slower and less efficient, has been observed before but no explanation was delivered.56 In general, two mechanisms for energy transfer are distinguished: energy transfer via dipole–dipole interactions (Förster-type)91 and energy transfer via electron-exchange mechanism (Dexter-type).92 A precondition for the dipole mechanism to be efficient is that transitions with high oscillator strength at the donor and acceptor site are involved and spectral overlap between donor emission and acceptor absorption is necessary. Dexter transfer requires orbital overlap for efficient electron exchange and is usually discussed as short range mechanism, but the conjugated system of the ligand system can support long-range energy transfer of Dexter-type. The missing donor emission would rule out energy transfer by Förster-type in our systems, but the situation is more complicated, because the energy transfer originates from non-relaxed states. For the ultrafast energy transfer in RuOs two scenarios can be discussed:15,88–90 singlet–singlet transfer and triplet–triplet transfer. For the triplet transfer mechanism, Dexter transfer might be favored, due to the low oscillator strength of the contributing triplet transitions though contributions of Förster transfer cannot be ruled out completely with the available data. For the transfer between the singlet states both Förster and Dexter mechanism could contribute, as the singlet states at both centers possess substantial oscillator strengths. Similar conclusions can be reached for the transfer mechanism in RuFeRu. As the oscillator strength of the radiative transitions from the non-relaxed 3MLCT state is not assessable from the available data, it is not possible to completely rule out a transfer following Förster mechanism, but a number of reports in literature suggest Dexter-type energy transfer between Ru(II) and Fe(II)-polypyridyl centers to be the most probable pathway.93–97 Unfortunately, no further insight in the mechanism is possible with the available data, but the observed differences in the rates might be determined by which of the proposed routes is followed and simply be based on differences in the relative energy of donor and acceptor states.
Summary and conclusion
In this investigation polymetallic complexes are investigated, which combine Ru(II) and Fe(II) (RuFeRu) or Os(II) (RuOs) bisterpyridine chromophores by coordination to an extended highly conjugated bis-4′-terpyridine bridging ligand. Due to their broad absorption range these mixed metal systems represent interesting candidates for light-harvesting antennae systems. Time-resolved spectroscopy reveals that energy transfer to the metal center with the lowest excited states in the assembly occurs in both systems with high efficiencies. While in RuFeRu an energy transfer of 80% occurs with a time constant of 15 ps from the Ru(II) to the Fe(II) center, the transfer from the Ru(II) to the Os(II) center in RuOs occurs on the ultrafast timescale ≤200 fs with unity quantum yield. These high efficiencies make these structures feasible candidates for incorporation into larger assemblies for long-range energy transfer.The high conjugation of the bis-4′-terpyridyl ligand leads to a stabilization of the 3MLCT states resulting in a prolonged lifetime. This is of special importance with respect to the lifetime of the 3MLCT state at the Fe(II) center, which up to now limits the applicability of Fe(II) polypyridyl systems for collection of solar energy. The results presented in this study indicate that in these Fe(II)-terpyridyl structures with extended conjugated ligands an exceptional high 3MLCT lifetime of 23 ps is achieved, which is the longest reported in literature.
In summary, the investigations presented in this work demonstrate that the compounds employed may be suitable building blocks for light-harvesting antennas. Future work will focus on the transfer of these structural motives into oligomeric structures.
Acknowledgements
We acknowledge support from Deutsche Forschungsgemeinschaft (DFG, Grant No. SCHU1229-16/1 and DI1517-3/1), LaserLab Europe (LLC001917), the Fonds der Chemischen Industrie and COST Action CM1202 Perspect-H2O. The group at Würzburg acknowledges support by the SolTech Initiative of the Bavarian State Ministry of Science, Research and the Arts.References
- T. Zhang and W. B. Lin, Chem. Soc. Rev., 2014, 43, 5982–5993 RSC
.
- P. D. Frischmann, K. Mahata and F. Würthner, Chem. Soc. Rev., 2013, 42, 1847–1870 RSC
- B. Albinsson, J. K. Hannestad and K. Borjesson, Coord. Chem. Rev., 2012, 256, 2399–2413 CrossRef CAS
- J. Yang, M. C. Yoon, H. Yoo, P. Kim and D. Kim, Chem. Soc. Rev., 2012, 41, 4808–4826 RSC
- F. Puntoriero, A. Sartorel, M. Orlandi, G. La Ganga, S. Serroni, M. Bonchio, F. Scandola and S. Campagna, Coord. Chem. Rev., 2011, 255, 2594–2601 CrossRef CAS
- M. Kozaki and K. Okada, J. Synth. Org. Chem., Jpn., 2011, 69, 1145–1157 CrossRef CAS
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS PubMed
- F. Puntoriero, F. Nastasi, M. Cavazzini, S. Quici and S. Campagna, Coord. Chem. Rev., 2007, 251, 536–545 CrossRef CAS
- F. Puntoriero, S. Serroni, M. Galletta, A. Juris, A. Licciardello, C. Chiorboli, S. Carnpagna and F. Scandola, ChemPhysChem, 2005, 6, 129–138 CrossRef CAS PubMed
- G. Calzaferri and K. Lutkouskaya, Photochem. Photobiol. Sci., 2008, 7, 879–910 CAS
- F. Odobel and H. Zabri, Inorg. Chem., 2005, 44, 5600–5611 CrossRef CAS PubMed
- V. Balzani, A. Juris, M. Venturi, S. Campagna and S. Serroni, Chem. Rev., 1996, 96, 759–833 CrossRef CAS PubMed
- C. Chiorboli, C. A. Bignozzi, F. Scandola, E. Ishow, A. Gourdon and J. P. Launay, Inorg. Chem., 1999, 38, 2402–2410 CrossRef CAS
- J. Larsen, F. Puntoriero, T. Pascher, N. McClenaghan, S. Campagna, E. Åkesson and V. Sundström, ChemPhysChem, 2007, 8, 2643–2651 CrossRef CAS PubMed
- V. Balzani, G. Bergamini, F. Marchioni and P. Ceroni, Coord. Chem. Rev., 2006, 250, 1254–1266 CrossRef CAS
- J. P. Sauvage, J. P. Collin, J. C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. Decola and L. Flamigni, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS
- S. Welter, N. Salluce, P. Belser, M. Groeneveld and L. De Cola, Coord. Chem. Rev., 2005, 249, 1360–1371 CrossRef CAS
- S. Welter, N. Salluce, A. Benetti, N. Rot, P. Belser, P. Sonar, A. C. Grimsdale, K. Mullen, M. Lutz, A. L. Spek and L. De Cola, Inorg. Chem., 2005, 44, 4706–4718 CrossRef CAS PubMed
- A. Wild, A. Winter, F. Schlütter and U. S. Schubert, Chem. Soc. Rev., 2011, 40, 1459–1511 RSC
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Vonzelewsky, Coord.
Chem. Rev., 1988, 84, 85–277 CrossRef CAS
-
V. Balzani, G. Bergamini, S. Campagna and F. Puntoriero, Photochemistry and Photophysics of Coordination Compounds I, 2007, vol. 280, pp. 1–36 Search PubMed
- A. Vlcek, Coord. Chem. Rev., 2000, 200, 933–977 CrossRef
- D. A. M. Egbe, B. Carbonnier, E. Birckner and U. W. Grummt, Prog. Polym. Sci., 2009, 34, 1023–1067 CrossRef CAS
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed
- R. Siebert, Y. X. Tian, R. Camacho, A. Winter, A. Wild, A. Krieg, U. S. Schubert, J. Popp, I. G. Scheblykin and B. Dietzek, J. Mater. Chem., 2012, 22, 16041–16050 RSC
- E. A. Medlycott and G. S. Hanan, Chem. Soc. Rev., 2005, 34, 133–142 RSC
- X. Y. Wang, A. Del Guerzo, S. Baitalik, G. Simon, G. B. Shaw, L. X. Chen and R. Schmehl, Photosynth. Res., 2006, 87, 83–103 CrossRef CAS PubMed
- H. Cho, M. L. Strader, K. Hong, L. Jamula, E. M. Gullikson, T. K. Kim, F. M. F. de Groot, J. K. McCusker, R. W. Schoenlein and N. Huse, Faraday Discuss., 2012, 157, 463–474 RSC
- R. Siebert, A. Winter, U. S. Schubert, B. Dietzek and J. Popp, Phys. Chem. Chem. Phys., 2011, 13, 1606–1617 RSC
-
C. Chiorboli, M. T. Indelli, F. Scandola, Molecular Wires: From Design to Properties, 2005, vol. 257, pp. 63–102 Search PubMed
- C. Chiorboli, M. A. J. Rodgers and F. Scandola, J. Am. Chem. Soc., 2003, 125, 483–491 CrossRef CAS PubMed
- L. Y. Zhu, A. Widom and P. M. Champion, J. Chem. Phys., 1997, 107, 2859–2871 CrossRef CAS
- P. S. Wagenknecht and P. C. Ford, Coord. Chem. Rev., 2011, 255, 591–616 CrossRef CAS
- J. T. Hewitt, P. J. Vallett and N. H. Damrauer, J. Phys. Chem. A, 2012, 116, 11536–11547 CrossRef CAS PubMed
- C. Creutz, M. Chou, T. L. Netzel, M. Okumura and N. Sutin, J. Am. Chem. Soc., 1980, 102, 1309–1319 CrossRef CAS
- J. E. Monat and J. K. McCusker, J. Am. Chem. Soc., 2000, 122, 4092–4097 CrossRef CAS
- W. Gawelda, A. Cannizzo, V. T. Pham, F. van Mourik, C. Bressler and M. Chergui, J. Am. Chem. Soc., 2007, 129, 8199–8206 CrossRef CAS PubMed
- A. L. Smeigh, M. Creelman, R. A. Mathies and J. K. McCusker, J. Am. Chem. Soc., 2008, 130, 14105–14107 CrossRef CAS PubMed
- C. Bressler, C. Milne, V. T. Pham, A. ElNahhas, R. M. van der Veen, W. Gawelda, S. Johnson, P. Beaud, D. Grolimund, M. Kaiser, C. N. Borca, G. Ingold, R. Abela and M. Chergui, Science, 2009, 323, 489–492 CrossRef CAS PubMed
- W. K. Zhang, R. Alonso-Mori, U. Bergmann, C. Bressler, M. Chollet, A. Galler, W. Gawelda, R. G. Hadt, R. W. Hartsock, T. Kroll, K. S. Kjaer, K. Kubicek, H. T. Lemke, H. Y. W. Liang, D. A. Meyer, M. M. Nielsen, C. Purser, J. S. Robinson, E. I. Solomon, Z. Sun, D. Sokaras, T. B. van Driel, G. Vanko, T. C. Weng, D. L. Zhu and K. J. Gaffney, Nature, 2014, 509, 345–348 CrossRef CAS PubMed
- C. Sousa, C. de Graaf, A. Rudavskyi, R. Broer, J. Tatchen, M. Etinski and C. M. Marian, Chem. – Eur. J., 2013, 19, 17541–17551 CrossRef CAS PubMed
- G. Auböck and M. Chergui, Nat. Chem., 2015, 7, 629–633 CrossRef PubMed
- S. Ferrere and B. A. Gregg, J. Am. Chem. Soc., 1998, 120, 843–844 CrossRef CAS
- D. N. Bowman, J. H. Blew, T. Tsuchiya and E. Jakubikova, Inorg. Chem., 2013, 52, 8621–8628 CrossRef CAS PubMed
- D. G. Brown, N. Sanguantrakun, B. Schulze, U. S. Schubert and C. P. Berlinguette, J. Am. Chem. Soc., 2012, 134, 12354–12357 CrossRef CAS PubMed
- S. U. Son, K. H. Park, Y. S. Lee, B. Y. Kim, C. H. Choi, M. S. Lah, Y. H. Jang, D. J. Jang and Y. K. Chung, Inorg. Chem., 2004, 43, 6896–6898 CrossRef CAS PubMed
- L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903–1912 RSC
- Y. Z. Liu, T. Harlang, S. E. Canton, P. Chabera, K. Suarez-Alcantara, A. Fleckhaus, D. A. Vithanage, E. Goransson, A. Corani, R. Lomoth, V. Sundström and K. Wärnmark, Chem. Commun., 2013, 49, 6412–6414 RSC
- L. A. Fredin, M. Papai, E. Rozsalyi, G. Vanko, K. Warnmark, V. Sundstrom and P. Persson, J. Phys. Chem. Lett., 2014, 5, 2066–2071 CrossRef CAS PubMed
- Y. Liu, K. S. Kjær, L. A. Fredin, P. Chábera, T. Harlang, S. E. Canton, S. Lidin, J. Zhang, R. Lomoth, K.-E. Bergquist, P. Persson, K. Wärnmark and V. Sundström, Chem. – Eur. J., 2015, 21, 3628–3639 CrossRef CAS PubMed
- P. P. Laine, S. Campagna and F. Loiseau, Coord. Chem. Rev., 2008, 252, 2552–2571 CrossRef CAS
- X. Y. Wang, A. Del Guerzo and R. H. Schmehl, J. Photochem. Photobiol., C, 2004, 5, 55–77 CrossRef CAS
- S. Baitalik, X. Y. Wang and R. H. Schmehl, J. Am. Chem. Soc., 2004, 126, 16304–16305 CrossRef CAS PubMed
- D. Maity, S. Mardanya, S. Karmakar and S. Baitalik, Dalton Trans., 2015, 44, 10048–10059 RSC
- D. Maity, C. Bhaumik, S. Mardanya, S. Karmakar and S. Baitalik, Chem. – Eur. J., 2014, 20, 13242–13252 CrossRef CAS PubMed
- A. K. C. Mengel, C. Forster, A. Breivogel, K. Mack, J. R. Ochsmann, F. Laquai, V. Ksenofontov and K. Heinze, Chem. – Eur. J., 2015, 21, 704–714 CrossRef CAS PubMed
- L. Hammarström and O. Johansson, Coord. Chem. Rev., 2010, 254, 2546–2559 CrossRef
- M. Abrahamsson, M. Jager, T. Osterman, L. Eriksson, P. Persson, H. C. Becker, O. Johansson and L. Hammarstrom, J. Am. Chem. Soc., 2006, 128, 12616–12617 CrossRef CAS PubMed
- M. Abrahamsson, M. Jager, R. J. Kumar, T. Osterman, P. Persson, H. C. Becker, O. Johansson and L. Hammarstrom, J. Am. Chem. Soc., 2008, 130, 15533–15542 CrossRef CAS PubMed
- R. J. Kumar, S. Karlsson, D. Streich, A. R. Jensen, M. Jager, H. C. Becker, J. Bergquist, O. Johansson and L. Hammarstrom, Chem. – Eur. J., 2010, 16, 2830–2842 CrossRef CAS PubMed
- F. Schramm, V. Meded, H. Fliegl, K. Fink, O. Fuhr, Z. R. Qu, W. Klopper, S. Finn, T. E. Keyes and M. Ruben, Inorg. Chem., 2009, 48, 5677–5684 CrossRef CAS PubMed
- A. Breivogel, C. Forster and K. Heinze, Inorg. Chem., 2010, 49, 7052–7056 CrossRef CAS PubMed
- A. Breivogel, C. Kreitner and K. Heinze, Eur. J. Inorg. Chem., 2014, 5468–5490 CrossRef CAS
- A. Breivogel, M. Meister, C. Forster, F. Laquai and K. Heinze, Chem. – Eur. J., 2013, 19, 13745–13760 CrossRef CAS PubMed
- R. Siebert, C. Hunger, J. Guthmuller, F. Schlütter, A. Winter, U. S. Schubert, L. Gonzalez, B. Dietzek and J. Popp, J. Phys. Chem. C, 2011, 115, 12677–12688 CAS
- A. Winter, C. Friebe, M. D. Hager and U. S. Schubert, Eur. J. Org. Chem., 2009, 801–809 CrossRef CAS
- B. Schulze, D. Escudero, C. Friebe, R. Siebert, H. Görls, S. Sinn, M. Thomas, S. Mai, J. Popp, B. Dietzek, L. Gonzalez and U. S. Schubert, Chem. – Eur. J., 2012, 18, 4010–4025 CrossRef CAS PubMed
- K. Barthelmes, J. Kübel, A. Winter, M. Wächtler, C. Friebe, B. Dietzek and U. S. Schubert, Inorg. Chem., 2015, 54, 3159–3171 CrossRef CAS PubMed
- F. Barigelletti, L. Flamigni, V. Balzani, J. P. Collin, J. P. Sauvage, A. Sour, E. C. Constable and A. M. W. C. Thompson, J. Am. Chem. Soc., 1994, 116, 7692–7699 CrossRef CAS
-
S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Photochemisty and Photophysics of Coordination Compounds I: Ruthenium, Springer, Berlin, 2007 Search PubMed
- R. Siebert, F. Schlutter, A. Winter, M. Presselt, H. Gorls, U. S. Schubert, B. Dietzek and J. Popp, Cent. Eur. J. Chem., 2011, 9, 990–999 CrossRef CAS
- M. Presselt, B. Dietzek, M. Schmitt, S. Rau, A. Winter, M. Jager, U. S. Schubert and J. Popp, J. Phys. Chem. A, 2010, 114, 13163–13174 CrossRef CAS PubMed
- M. Presselt, B. Dietzek, M. Schmitt, J. Popp, A. Winter, M. Chiper, C. Friebe and U. S. Schubert, J. Phys. Chem. C, 2008, 112, 18651–18660 CAS
- R. Siebert, A. Winter, B. Dietzek, U. S. Schubert and J. Popp, Macromol. Rapid Commun., 2010, 31, 883–888 CrossRef CAS PubMed
- J. K. Mccusker, K. N. Walda, R. C. Dunn, J. D. Simon, D. Magde and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 298–307 CrossRef CAS
- A. Hauser, C. Enachescu, M. L. Daku, A. Vargas and N. Amstutz, Coord. Chem. Rev., 2006, 250, 1642–1652 CrossRef CAS
- G. Vankó, A. Bordage, M. Pápai, K. Haldrup, P. Glatzel, A. M. March, G. Doumy, A. Britz, A. Galler, T. Assefa, D. Cabaret, A. Juhin, T. B. van Driel, K. S. Kjær, A. Dohn, K. B. Møller, H. T. Lemke, E. Gallo, M. Rovezzi, Z. Németh, E. Rozsályi, T. Rozgonyi, J. Uhlig, V. Sundström, M. M. Nielsen, L. Young, S. H. Southworth, C. Bressler and W. Gawelda, J. Phys. Chem. C, 2015, 119, 5888–5902 Search PubMed
- J. N. Demas and D. G. Taylor, Inorg. Chem., 1979, 18, 3177–3179 CrossRef CAS
- J. N. Demas and G. A. Crosby, J. Am. Chem. Soc., 1971, 93, 2841–2847 CrossRef
- A. I. Baba, J. R. Shaw, J. A. Simon, R. P. Thummel and R. H. Schmehl, Coord. Chem. Rev., 1998, 171, 43–59 CrossRef CAS
- N. H. Damrauer, G. Cerullo, A. Yeh, T. R. Boussie, C. V. Shank and J. K. McCusker, Science, 1997, 275, 54–57 CrossRef CAS PubMed
- J. K. McCusker, Acc. Chem. Res., 2003, 36, 876–887 CrossRef CAS PubMed
- M. Chergui, Dalton Trans., 2012, 41, 13022–13029 RSC
- A. C. Bhasikuttan, M. Suzuki, S. Nakashima and T. Okada, J. Am. Chem. Soc., 2002, 124, 8398–8405 CrossRef CAS PubMed
- W. R. Browne, W. Henry, C. G. Coates, C. Brady, K. L. Ronayne, P. Matousek, M. Towrie, S. W. Botchway, A. W. Parker, J. G. Vos and J. J. McGarvey, J. Phys. Chem. A, 2008, 112, 4537–4544 CrossRef PubMed
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, 2004, 1658, 262 CrossRef CAS
- H. B. Baudin, J. Davidsson, S. Serroni, A. Juris, V. Balzani, S. Campagna and L. Hammarström, J. Phys. Chem. A, 2002, 106, 4312–4319 CrossRef CAS
- J. Andersson, F. Puntoriero, S. Serroni, A. Yartsev, T. Pascher, T. Polivka, S. Campagna and V. Sundstrom, Faraday Discuss., 2004, 127, 295–305 RSC
- J. Andersson, F. Puntoriero, S. Serroni, A. Yartsev, T. Pascher, T. Polivka, S. Campagna and V. Sundstrom, Chem. Phys. Lett., 2004, 386, 336–341 CrossRef CAS
- T. Förster, Discuss. Faraday Soc., 1959, 7–17 RSC
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS
- J. Lombard, J. C. Lepretre, J. Chauvin, M. N. Collomb and A. Deronzier, Dalton Trans., 2008, 658–666 RSC
- J. Lombard, S. Romain, S. Dumas, J. Chauvin, M. N. Collomb, D. Daveloose, A. Deronzier and J. C. Lepretre, Eur. J. Inorg. Chem., 2005, 3320–3330 CrossRef CAS
- R. H. Schmehl, R. A. Auerbach, W. F. Wacholtz, C. M. Elliott, R. A. Freitag and J. W. Merkert, Inorg. Chem., 1986, 25, 2440–2445 CrossRef CAS
- S. L. Larson, S. M. Hendrickson, S. Ferrere, D. L. Derr and C. M. Elliott, J. Am. Chem. Soc., 1995, 117, 5881–5882 CrossRef CAS
- F. Lafolet, J. Chauvin, M. Collomb, A. Deronzier, H. Laguitton-Pasquier, J. C. Lepretre, J. C. Vial and B. Brasme, Phys. Chem. Chem. Phys., 2003, 5, 2520–2527 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp04447b |
| This journal is © the Owner Societies 2016 |