
Characterization of nitrogen species incorporated into graphite using low energy nitrogen ion sputtering†
Hisao
Kiuchi
a,
Takahiro
Kondo
b,
Masataka
Sakurai
b,
Donghui
Guo
b,
Junji
Nakamura
b,
Hideharu
Niwa
cd,
Jun
Miyawaki
cd,
Maki
Kawai
a,
Masaharu
Oshima
d and
Yoshihisa
Harada
*cd
aDepartment of Applied Chemistry, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, 113-8656, Japan
bFaculty of Pure and Applied Sciences, University of Tsukuba, Japan
cThe Institute for Solid State Physics (ISSP), University of Tokyo, Japan. E-mail: harada@issp.u-tokyo.ac.jp
dSynchrotron Radiation Research Organization, University of Tokyo, Japan
First published on 16th November 2015
Abstract
The electronic structures of nitrogen species incorporated into highly oriented pyrolytic graphite (HOPG), prepared by low energy (200 eV) nitrogen ion sputtering and subsequent annealing at 1000 K, were investigated by X-ray photoelectron spectroscopy (XPS), angle-dependent X-ray absorption spectroscopy (XAS), and Raman spectroscopy. An additional peak was observed at higher binding energy of 401.9 eV than 400.9 eV for graphitic1 N (graphitic N in the basal plane) in N 1s XPS, where graphitic2 N (graphitic N in the zigzag edge and/or vacancy sites) has been theoretically expected to appear. N 1s XPS showed that graphitic1 N and graphitic2 N were preferably incorporated under low nitrogen content doping conditions (8 × 1013 ions cm−2), while pyridinic N and graphitic1 N were dominantly observed under high nitrogen content doping conditions. In addition, angle-dependent N 1s XAS showed that the graphitic N and pyridinic N atoms were incorporated into the basal plane of HOPG and thus were highly oriented. Furthermore, Raman spectroscopy revealed that low energy sputtering resulted in almost no fraction of the disturbed graphite surface layers under the lowest nitrogen doping condition. The suitable nitrogen doping condition was discovered for realizing the well-controlled nitrogen doped HOPG. The electrochemical properties for the oxygen reduction reaction of these samples in acidic solution were examined and discussed.
1. Introduction
Nitrogen doping of carbon-based materials such as graphene, carbon nanotubes, and graphite is an effective way to tailor their electronic, chemical, optical, and magnetic properties. Recently, nitrogen-doped carbon materials have attracted an increasing amount of attention for use in electrodes for electric double-layer capacitors,1,2 anode materials of Li-ion batteries,3 catalysts for organic synthesis,4 and biosensing systems.5 In particular, carbon-based oxygen reduction catalysts are expected to be alternatives to high cost Pt-based cathode catalysts for polymer electrolyte fuel cells.6–13 In the last decade, the mechanism of their excellent oxygen reduction reaction (ORR) activity has been under extensive debate.6–20 There are various theoretical14–16 and experimental17–19 studies on the roles of pyridinic and graphitic nitrogen in ORR activity. In a diverse range of nitrogen-doping methods, including chemical vapor deposition,6,7 pyrolysis of nitrogen containing polymers,8–10 pyrolysis of the nitrogen source mixture,8–13,19 and nitrogen ion sputtering,20 a complicated relationship exists between nitrogen doping and ORR activity because many parameters, such as morphology, surface area, and crystallinity, are involved. It is therefore essential to prepare a well-defined reference system exhibiting not only high crystallinity and surface morphology, but also possessing select nitrogen species in order to determine the relationship between nitrogen configuration and ORR activity. In this respect, highly oriented pyrolytic graphite (HOPG) is expected to provide a well-defined surface for the ORR due to its ordered sp2-carbon network. Recently, Kondo et al. studied the local electronic structure of nitrogen incorporated into HOPG using a combination of low energy (200 eV) nitrogen ion sputtering (<0.2 × 1013 ions cm−2) and scanning tunneling microscopy (STM).21In this study, we report the low energy nitrogen ion sputtering (8–4000 × 1013 ions cm−2) of HOPG in order to realize effective nitrogen doping (N = 0.4–8.4 at%) while retaining the flatness of the surface sp2-carbon structure. The chemical states of the doped nitrogen will be controlled by altering the amount of impinging nitrogen ions and the nitrogen species will be clarified using a combination of Raman and X-ray spectroscopic analyses. The results will be compared with the resulting electrochemical properties in order to discuss the possible ORR active site.
2. Experimental
An HOPG substrate (PGCSTM, Panasonic Inc.) was cleaved in air using adhesive tape, and set in an ultra-high vacuum (UHV) chamber equipped with a molybdenum holder. The substrate was then annealed at 1000 K for 30 min, and sputtered with nitrogen ions at 300 K using an ion gun (OMI-0730, Omegatron Inc.). The acceleration voltage of the ion gun was set to 200 V. The nitrogen ion dose was estimated from the total ion current divided by the irradiated spot area. The amounts of dosed nitrogen ions were calculated to be 8 × 1013, 8 × 1014 and 4 × 1016 ions cm−2, equivalent to 2%, 21%, and 1030% nitrogen ions, respectively, with respect to surface carbon atoms. Finally, the samples were annealed at 1000 K for 1 h for surface cleaning. These samples will be referred to as N-HOPG hereafter.X-ray photoelectron spectroscopy (XPS) was performed at BL27SU in SPring-8 using a photoelectron analyzer (PHOIBOS 150, SPECS), with 850 eV incident photon energy. Backgrounds of the core level spectra were subtracted using the Shirley method. The N 1s XPS spectra were fitted with Voigt functions (0.98 eV Gaussian width and 0.25 eV Lorentzian width). The energy resolution of XPS at BL27SU was 170 meV. Conventional laboratory-based XPS was also performed using a monochromatic and a non-monochromatic Al-Kα source (JPS-9010, JEOL) with energy resolutions of 0.7–0.8 eV and 1.1–1.3 eV, respectively. X-ray absorption spectroscopy (XAS) was applied in the partial electron yield (PEY) mode in order to increase surface sensitivity. XAS spectra were obtained at BL27SU and BL07LSU in SPring-8 by setting θ to 0°, 45°, 60° or 70°. The angle, θ, is defined as the angle between the incident X-ray beam and the surface normal. The spectra were fitted with six Gaussian functions for the π* and σ* states, and two step functions convoluted by error function and exponential decay function for the continuum states in the π* and σ* regions.22 The energy resolution of XAS was greater than 100 meV. The samples were annealed at 900 K for 30 min to remove the initially adsorbed gas before XPS and XAS measurements were carried out. For each sample, the nitrogen content remained constant both before and after annealing, while the oxygen contamination percentage decreased dramatically (<0.15 at%) after annealing.
Raman spectroscopy was performed using a micro-Raman spectrometer (NR-1800, JASCO) with an excitation wavelength of 532 nm. The Raman spectral resolution was 2.6 cm−1. A Monte Carlo simulation tool, Stopping and Range of Ions in Matter (SRIM),23 was used to calculate the depth profile of doped nitrogen ions in HOPG in order to estimate the distribution of nitrogen species probed by XAS and XPS.
The ORR activity of N-HOPG was evaluated at room temperature in a 0.1 M H2SO4 electrolyte solution. N-HOPG electrodes were prepared by attaching an iron wire with a conductive dotite, pasting an inert polymer around the wire and the dotite, and subsequent drying in air. The working electrode, counter electrode, and reference electrode were N-HOPG, Pt wire, and a reversible hydrogen electrode, respectively. Linear sweep voltammograms (LSV) were recorded by sweeping the potential from 1.1 V to −0.05 V (vs. RHE) at 5 mV s−1 in a saturated solution of either N2 or O2. The ORR current was determined by subtracting the N2 data from the O2 data. Current density was obtained as measured current divided by the electrode HOPG surface area (e.g. ∼0.15 cm2) rather than actual surface area, as all surfaces of HOPG model samples were flat, with no clear difference being observed based on the atomic force microscopy (AFM) analysis (Fig. S1, ESI†).
3. Results and discussion
3.1 Chemical states of doped nitrogen in N-HOPG
Using the SRIM Monte Carlo simulation tool,23 we found that the doped nitrogen atoms were distributed near the surface region of HOPG as shown in Fig. S2 (ESI†) (projected range up to 2 nm). We observed that the accelerated nitrogen ions create a 2–3 nm thick nitrogen-doped region at the surface of the HOPG substrate. Since the nitrogen doped region is thinner than the probing depth of XPS and XAS with PEY, all doped nitrogen can be detected by both XPS and XAS. From the XPS spectra, the amount of surface nitrogen (X) was estimated to be equal to 0.4, 2.3, and 8.4 at% for N-HOPG samples prepared by doping with 2, 21, and 1030% nitrogen ions, respectively. Kondo et al. previously reported the N 1s XPS spectrum of N-HOPG produced by low energy (200 eV) nitrogen sputtering.21 In this case, a nitrogen content of 2.7 at% was reported, and the profile was similar to that of N-HOPG with X = 2.3 (at%) in this manuscript. The amount of dosed nitrogen ions in the previous work was not measured but thus can be estimated around 80 × 1013 ions cm−2. In this paper, we have for the first time reported the very low ion dose effects between 0.2 and 80 × 1013 ions cm−2; in particular emphasis is on 8 and 4000 × 1013 ions cm−2, which are highly below and above the nitrogen dose of the previous report.21Fig. 1 shows the N 1s XPS spectra of N-HOPG with different levels of doped nitrogen. In addition, the N 1s XPS spectra of all N-HOPG with low energy resolution (ΔE = 0.7–0.8 eV) are shown in Fig. S3 (ESI†). The N 1s XPS spectra at the low nitrogen content region with low energy resolution (ΔE = 1.1–1.3 eV) are also shown in Fig. S4 (ESI†). All spectra were normalized by the integrated intensity. As can be seen, the spectra are fitted with five Voigt functions, denoted as NP1 (398.0–398.5 eV), NP2 (399.9 eV), NP3 (400.9 eV), NP4 (401.9 eV), and NP5 (403.5 eV). The binding energy was treated as a fixed parameter except for NP1. The terminology of nitrogen components used hereafter is defined in Fig. 1. NP1 represents the pyridinic N where one nitrogen atom is connected with two carbon atoms. The peak positions at X = 0.4 and 2.3 are 398.0 eV and slightly lower than the reported energy (398.5–398.9 eV).7 Although the pyridinic N usually exists in the graphite edge, the majority of pyridinic N atoms in N-HOPG exist in the vicinity of in-plane defects at the beginning of nitrogen ion sputtering, as the relative amount of graphite edge in HOPG is small. The difference of the configuration is considered to induce the observed chemical shift. On the other hand, the peak position of pyridinic N at X = 8.4 is 398.5 eV and agrees well with the reported one. The agreement would be attributed to the dominant number of pyridinic N at the edge created by the large amount of sputtering. NP2 represents the cyanide N, where a triple bond is formed between a nitrogen and a carbon atom,24 or the pyrrolic N where one nitrogen atom is connected with two carbon atoms and one hydrogen atom in a five-membered ring.25 NP3 corresponds to the graphitic N in the basal plane,26 which substitutes the carbon site in the graphite plane, forming a nitrogen site bonded to three carbon atoms (denoted as graphitic1 N in Fig. 1). Furthermore, NP4 is separated from NP3 by 1.0 eV, which expects a different chemical environment. The peak at X = 0.4 was expected to be narrower than the peak at X = 2.3 since it would decrease the variation of nitrogen components due to decreased damage by nitrogen sputtering. On the contrary, the experimental XPS spectrum showed a broader profile at X = 0.4 and the more detailed doping dependence of XPS (Fig. S4, ESI†) confirmed NP4 at low nitrogen content. Thus, we assumed the existence of the other nitrogen components and examined the possibility of the graphitic2 N in Fig. 1 (graphitic N in the zigzag edge and/or vacancy sites).19,27 Casanovas et al. used cluster calculations to suggest that the peak position of graphitic N in the zigzag edge shifts to higher binding energy by approximately +1.0 eV compared to that of the in-plane graphitic N.26 Wang et al. also estimated the binding energy separation of the same chemical components to be approximately +0.6 eV.28 The presence of the zigzag edge in HOPG has been reported by scanning tunneling spectroscopy (STS),29 with step densities of 0.09–2.2% being recorded for a variety of grades using AFM.30 The nitrogen sputtering process not only causes nitrogen doping but also creates carbon vacancies. Such carbon vacancies can contribute to the formation of the pyridinic N and the graphitic2 N. For a graphene on Ir(111) with 25 eV N+ plasma treatment, Orlando et al. observed a peak around 402 eV and assigned it to graphitc2 N based on the XPD study and the theoretical expectation.31 Because graphene on a metal substrate forms a Moire structure by the interaction between graphene and the substrate, there was another possibility that the peak around 402 eV resulted from the chemical shift of graphitc1 N (401 eV). Orlando et al., however, excluded the possibility because the interaction between graphene and Ir(111) was weak,32 thus supporting our assignment. It must be nevertheless noted that there is not yet strong evidence to visualize the local atomic structure of the graphitic2 N using atomic scale microscopy such as STM and AFM and the assignment is not conclusive. From these theoretical and experimental results, it seems therefore most plausible to assign NP4 to graphitic2 N. Finally, NP5 represents oxide N, which forms direct bonds with one oxygen atom.
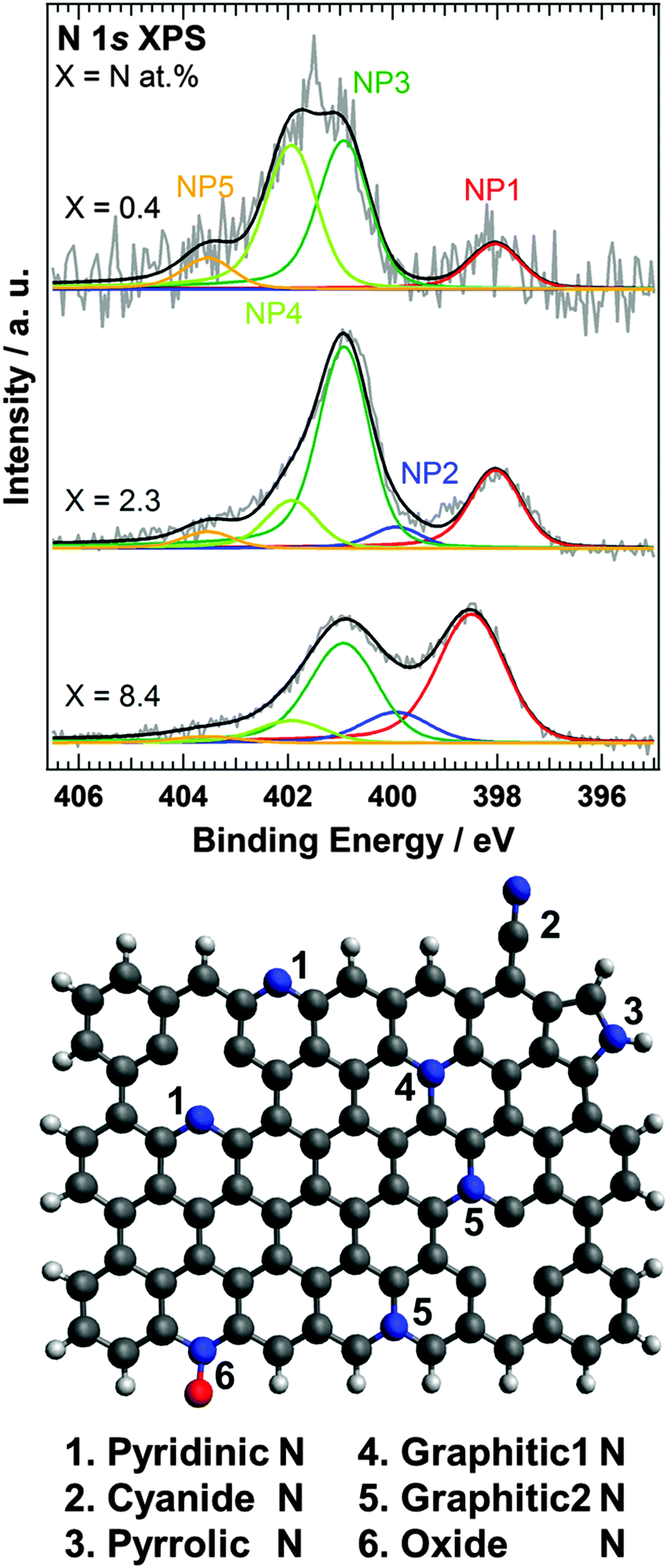 | ||
| Fig. 1 N 1s XPS spectra of N-HOPG (X = 0.4, 2.3, and 8.4). The energy resolution was 0.17 eV in N-HOPG with X = 0.4 and 2.3, and 0.7–0.8 eV in N-HOPG with X = 8.4. Red, blue, dark green, light green, and orange lines correspond to NP1 (pyridinic N), NP2 (cyanide N or pyrrole N), NP3 (graphitic1 N), NP4 (graphitic2 N), and NP5 (oxide N), respectively. Gray and black lines represent raw and fitted data, respectively. The atomic ratios of N-HOPG with X = 0.4, 2.3, and 8.4 are 0.04, 0.48, and 3.78 at% (NP1); 0.0, 0.13, and 0.91 at% (NP2); 0.14, 1.23, and 2.88 at% (NP3); 0.14, 0.29, and 0.65 at% (NP4); and 0.03, 0.10, and 0.17 at% (NP5), respectively. The lower panel shows a schematic representation of the nitrogen components discussed in the paper. Hydrogen, carbon, nitrogen and oxygen atoms correspond to white, black, blue and red balls, respectively. | ||
The graphitic1 N (NP3) and graphitic2 N (NP4) components were found to be dominant in N-HOPG with X = 0.4 and X = 2.3, which correspond to more than 70% of the total nitrogen content. The pyridinic N was the next common component. We found that the relative ratio of pyridinic N gradually increased with increasing total amount of doped nitrogen, i.e. 13% in N-HOPG with X = 0.4, 21% in N-HOPG with X = 2.3, and >40% in N-HOPG with X = 8.4. In graphene sputtered with nitrogen ions, a continuous increase in pyridinic N correlates to a greater amount of defects and some carbon loss.33 Thus, it was clear that the formation of the defects in graphite is suppressed by the low nitrogen concentration. In contrast, the relative ratio of graphtic2 N decreased from 39% in N-HOPG with X = 0.4 to 13% in N-HOPG with X = 2.3. This counterintuitive feature of graphitic2 N can probably be explained as follows. Graphite retains flat surface in the low nitrogen sputtering condition as later mentioned in Section 3.3. Raman spectra showed, however, that the carbon network was strongly disturbed above X = 2.3. At the low nitrogen doing region, the sputtering creates defects, like zigzag edges and point vacancies, whereas at the high nitrogen doping region, the sputtering creates more defects resulting in disorder of several carbon layers, that is, amorphous carbon as discussed in the previous study of Ar+ sputtering onto single- and few-layer graphene.34 The zigzag edges and point vacancies contribute to the formation of both pyridinic N and graphitic2 N. Therefore, at the low doping region, the absolute amount of pyridinic N and graphitic2 N increases in proportion to the total ion dose (Fig. S4(b), ESI†). At the high doping region, however, the evolution of the amorphous carbon (Fig. 3 and Fig. S6, ESI†) prevents the formation of graphitic2 N (Fig. 1 and Fig. S4(b), ESI†). It was amorphization of carbon that drastically reduced the proportion of graphitic2 N at the high doping region.
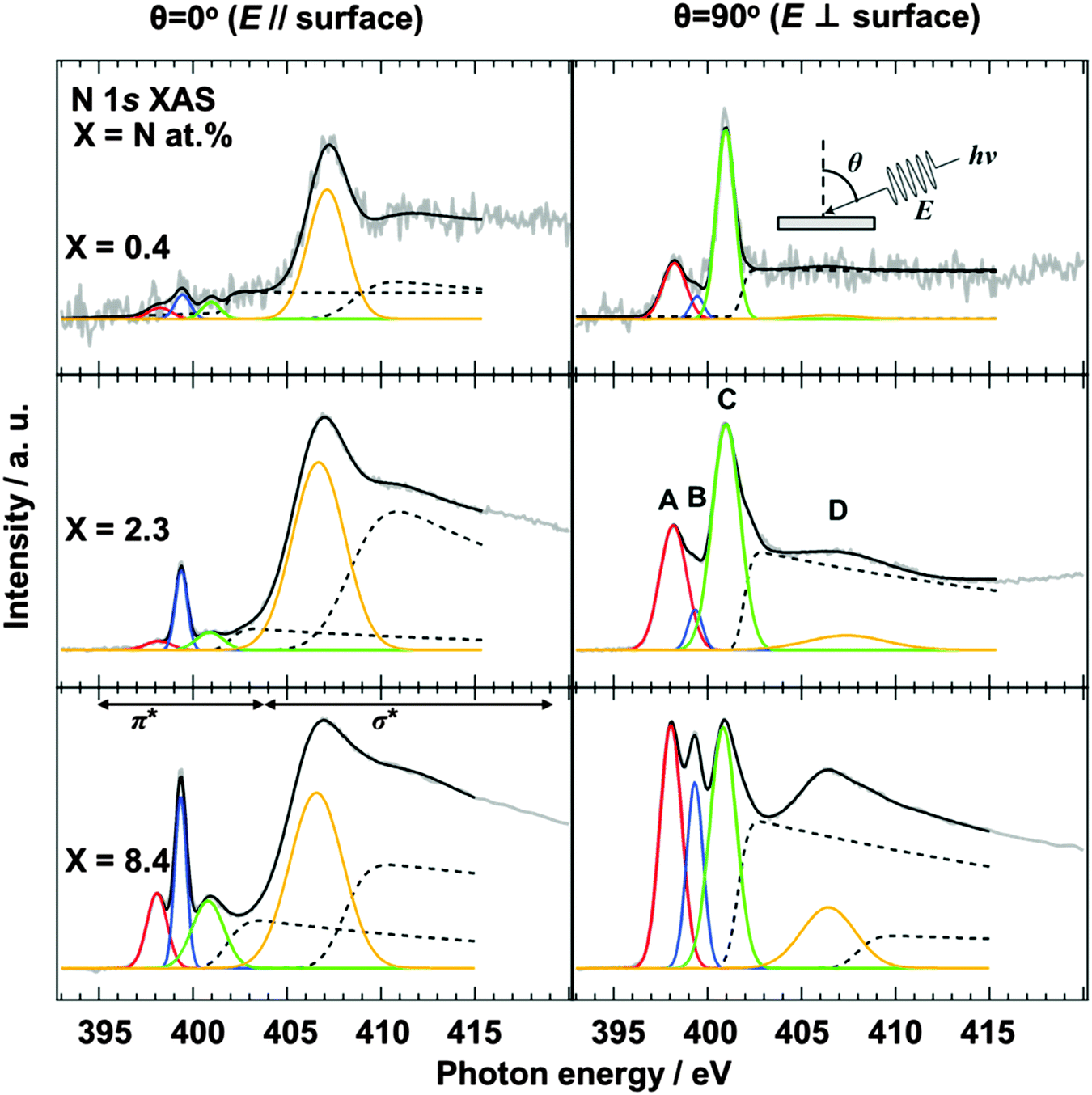 | ||
| Fig. 2 N 1s XAS spectra of N-HOPG (X = 0.4, 2.3 and 8.4). The spectra at θ = 90° and 0° are shown on the left and right hand panels, respectively. Red, blue, and green lines correspond to pyridinic N (A), cyanide N (B), and graphitic N (C) in the π* region, respectively. The orange line exhibits the lowest energy σ* peak (D). An additional Gaussian function used for the higher energy σ* component is not shown here. Dashed lines represent step functions which correspond to continuum states of π* and σ* regions. Gray and black lines represent raw and fitted data, respectively. | ||
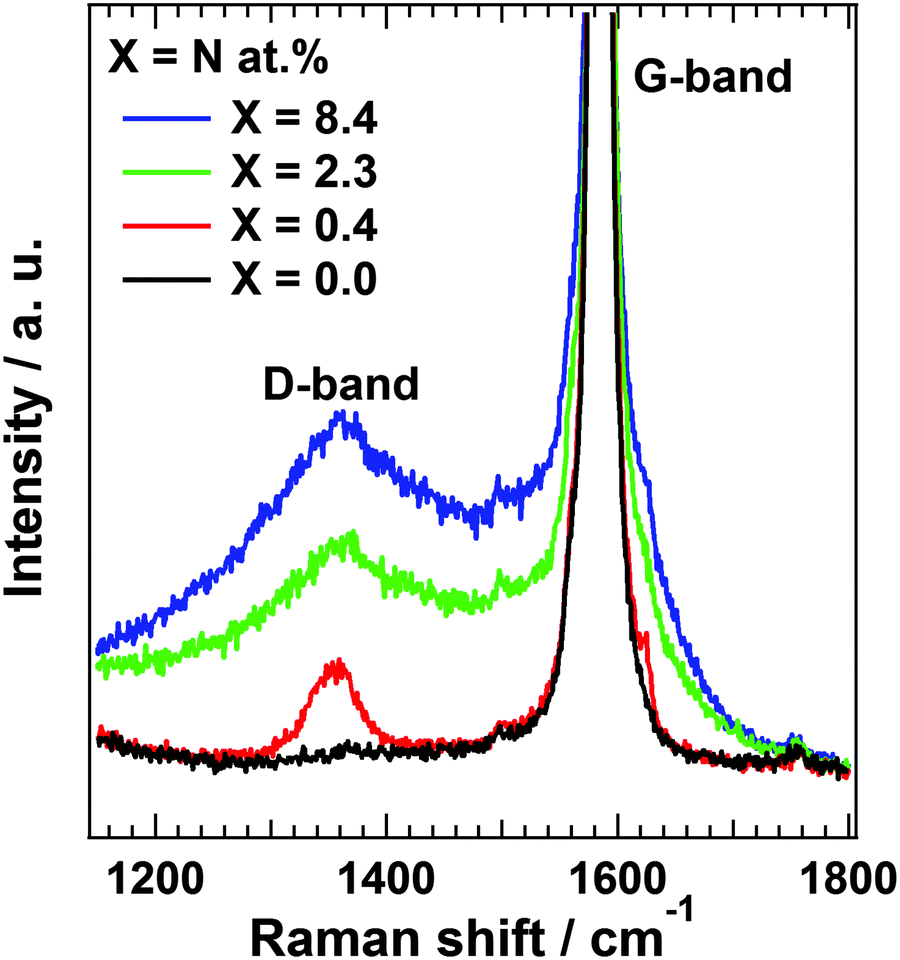 | ||
| Fig. 3 Raman spectra of each N-HOPG (X = 0.4, 2.3 and 8.4) and clean HOPG (X = 0.0). | ||
3.2 Configuration of doped nitrogen in N-HOPG
The configuration of doped nitrogen in N-HOPG was analyzed by angle-dependent N 1s XAS. The spectra were normalized according to intensity at 430 eV, taking an average between 390 and 395 eV as background. Out-of-plane (θ = 90°) unoccupied states of nitrogen were extracted by taking into account their contribution to the obtained (θ = 0°, 45°, 60°, or 70°) spectra (Fig. S5, ESI†). Fig. 2 shows the in-plane (θ = 0°) and the extracted out-of-plane (θ = 90°) N 1s XAS spectra. Three sharp peaks in the π* region can be assigned to (A) pyridinic N, (B) cyanide N, and (C) graphitic N sites,35–37 respectively, where the presence of pyrrolic N could be excluded by XAS rather than XPS because the peak position of the pyrrolic N was calculated as ∼402 eV.38 In addition, peak D corresponds to the lowest energy σ* states. The intensities of pyridinic N (A) and graphitic N (C) for the low nitrogen concentration samples (N-HOPG with X = 0.4 and 2.3) strongly depend on the X-ray incident angle. Since a similar angle dependence is observed in the carbon π* state of HOPG (C 1s XAS),39 those nitrogen species are incorporated into the graphite basal plane to give a planar N-HOPG structure under low nitrogen doping conditions. The simulation of both in-plane and out-of-plane XAS spectra has been reported by Hou et al. using density functional theory (DFT) calculations.40 They reported that both a graphitic N in perfect graphene and a pyridinic N with a monovacancy show perfect polarization dependence for π* and σ* components. Therefore, graphitic N and the pyridinic N are doped into the planar graphene lattice via substitution. Two main factors are known to facilitate the formation of the planar structure in this N-HOPG preparation method. Firstly, the low acceleration voltage (200 V) maintains the nitrogen ion at the surface of the graphite layer and reduces the collapse of the graphite basal plane by the recoil effect. Secondly, the post-annealing step at 1000 K reduces defects and removes excess nitrogen from the system.However, in highly nitrogen-doped systems (N-HOPG with X = 8.4), this polarization dependence is less distinct when the same preparation method is used. The decrease in polarization dependence is clearly shown in Table 1, where the degree of the orientation of each nitrogen component is expressed by the intensity ratio [π*/σ* (at θ = 90°)]/[π*/σ* (at θ = 0°)], where π* and σ* represent the integrated intensity of the Gaussian functions for the π* (A, B, C) and σ* (D) components, respectively. In the case of peak A, the intensity ratios were >100 for X = 0.4 and 2.3, while a smaller ratio of ∼10 was observed for X = 8.4. Similarly, the intensity ratio of peak C decreases with increasing amount of nitrogen, i.e., 290 for X = 0.4, 110 for X = 2.3, and 9 for X = 8.4. Thus, by increasing the amount of doped nitrogen, the number of defects may increase, with nitrogen atoms thought to be incorporated in such defective and non-planar structures. This results in a lesser polarization dependence in the N 1s XAS spectra. The orientation of nitrogen sites is therefore gradually randomized from N-HOPG with X = 0.4 to N-HOPG with X = 8.4. It has also been reported by simulations that excessively high nitrogen content in the graphite lattice (X > 20 at%) can result in loss of the planar structure.41 However, a small polarization dependence is still present, even in the doped nitrogen saturated N-HOPG where X = 8.4, suggesting that each doped nitrogen may be incorporated into the relatively flat N-HOPG surface. In contrast to peaks A and C, peak B shows less polarization dependence with all N-HOPG samples, since it is not implemented in the graphite basal plane.35
| Nitrogen content (X = N at%) | θ = 90° (π*/σ*)/θ = 0° (π*/σ*) | ||
|---|---|---|---|
| A | B | C | |
| X = 0.4 | 130 | 21 | 292 |
| X = 2.3 | 120 | 5 | 112 |
| X = 8.4 | 10 | 4 | 9 |
In the XAS spectra at θ = 90°, the intensity ratio of pyridinic N (A) to graphitic N (C) increased from 0.37 (X = 0.4) to 0.53 (X = 2.3), which is the same trend as the intensity ratio of pyridinic N (NP1) to graphitic1 N and graphitic2 N (NP3 + NP4), with an increase from 0.16 (X = 0.4) to 0.32 (X = 2.3) in the XPS spectra. However, it should be noted that the graphitic2 N peak could not be identified in the XAS spectra, possibly due to overlap with the graphitic1 N peak, while in the XPS spectra a different peak position allowed the two environments to be distinguished. As discussed for the pyrrolic N in this manuscript, the chemical shifts in XAS and XPS are not necessarily the same.38 The assignment of the graphitic2 N species in the XAS spectra will therefore be investigated in a later study.
3.3 Surface morphology and defect sites
Raman scattering measurements provide additional information regarding the disorder in the carbon structure. Fig. 3 shows Raman spectra of N-HOPG along with that of clean HOPG (X = 0.0) as reference. The Raman spectra at the low nitrogen content region are also shown in Fig. S6 (ESI†). The spectra were normalized according to peak height at 1585 cm−1. The G-band at approximately 1585 cm−1 is derived from the graphite basal plane, while the D-band at ∼1360 cm−1 corresponds to defect sites in the graphite plane.34,42,43 A wide profile across the G- and D-bands appears at higher disorder levels corresponding to the graphite phonon density of states (PDOS-like background).44 The presence of this peak indicates the appearance of amorphization in the graphite layers.43 However, separation of the PDOS-like background from other bands is problematic due to its rather broad profile. In addition, a large G-band is always observed due to the presence of pristine graphite layers below the nitrogen ion sputtered surface.The sharp D-band in N-HOPG with X = 0.4 corresponds to the formation of defects with or without doped nitrogen. The absence of the PDOS-like background indicates almost no disturbance of the surface graphite layers by nitrogen doping. The broad D-band and PDOS-like background were found to increase with increasing amount of doped nitrogen (N-HOPG with X = 2.3, 8.4), indicating the increase in amorphization of graphite due to the collapse of nitrogen ions partially penetrating into several graphite layers from the surface. These results suggest that the controlled nitrogen doping of HOPG with minimal disturbance to the graphite layer orientation can only be realized under appropriate conditions, such as 200 eV nitrogen ions and 8 × 1013 ions cm−2. The Raman scattering results are not necessarily consistent with the XAS results, which demonstrated the high orientation of the doped nitrogen structures even at N-HOPG with X = 2.3. While N 1s XAS only probes the doped nitrogen, the Raman scattering measurements evaluate not only carbon sites close to the doped nitrogen, but also amorphous carbon and bulk graphite regions. From the high polarization dependence of graphitic N and pyridinic N in the N 1s XAS results, these nitrogen species could be preferably doped to a flat graphite surface region rather than an amorphous carbon region, while the absence of polarization dependence of cyanide N cannot exclude the possibility that cyanide N exists in the amorphous carbon.
3.4 Discussion for the nitrogen doping mechanism and electrochemical properties of N-HOPG
From the XAS, XPS, and Raman scattering results, the HOPG doping mechanism of nitrogen ions can be summarized by nitrogen being preferably doped in the form of graphitic1 N and graphitic2 N in N-HOPG with X = 0.4. In N-HOPG with X = 2.3, the defective and amorphous regions increase in size. However, pyridinic N and graphitic1 N are likely to be doped into the flat in-plane graphite region, rather than into the amorphous region. As the amount of doped nitrogen increases in N-HOPG with X = 8.4, the majority of flat graphite layers convert into defective and amorphous regions, resulting in reduced orientation of the doped nitrogen in these regions.The ORR activity measurements of N-HOPG were performed to discuss the possible contribution of nitrogen to ORR (Fig. S7, ESI†). The N-HOPG with X = 0.4 at the lowest nitrogen doping level showed the highest ORR activity (EO2 = 0.36 V at −2 μA cm−2) among the three N-HOPGs. However, it is difficult to clearly determine the relationship between ORR activity and each nitrogen configuration due to the presence of amorphous regions that significantly reduce the electron conductivity.
Comparison of our graphite model with current catalysts synthesized from organic precursors is also of interest. In a standard synthetic protocol, such catalysts are subjected to heat treatment that converts an amorphous carbon structure into an ordered graphite structure. Above a critical temperature, their ORR activity dramatically improves, mainly due to a sharp increase in electron conductivity.45 In addition, at higher pyrolysis temperatures, the ordered carbon structure excludes impurities such as nitrogen atoms, and the remaining nitrogen species dominate as in-plane graphitic N and/or pyridinic N.46,47 In the study reported herein, N-HOPG with X = 0.4 under the lowest nitrogen doping conditions corresponds to this high temperature pyrolysis region of the current catalysts in terms of catalytic activity and electron conductivity. This N-HOPG preferably contains the graphitic1 N and graphitic2 N, due to the optimized low energy nitrogen ion sputtering and annealing conditions. The higher nitrogen doping thus produces an amorphous carbon structure on the surface of HOPG containing a variety of nitrogen species, comparable to that observed below the critical pyrolysis temperature of the current catalysts with high nitrogen concentrations. Thus, a detailed study on the properties of model N-HOPG catalysts under similar electron conductivity conditions (e.g., similar density of amorphous carbon) will provide further insight into the ORR active site, which accurately models the current catalysts. The highly oriented N-HOPG is expected to be a reference system for studying the orientation of nitrogen sites and the role of oxygen adsorption as the first step in the ORR48 in carbon-based fuel cell cathode catalysts. The adsorbed O2 configuration is expected to be critical with regard to the recently debated 2-electron or 4-electron mechanisms for the ORR.49,50
4. Conclusion
The characteristics of nitrogen doped highly ordered pyrolytic graphite (N-HOPG) using low energy nitrogen ion sputtering were investigated by X-ray photoelectron spectroscopy (XPS), X-ray absorption spectroscopy (XAS), and Raman spectroscopy in combination with electrochemical measurements. It was found that the relative amount of pyridinic N increased with an increase in doped nitrogen, indicating an increase in the number of defect sites on the HOPG surface. This is also reflected in the orientation of each nitrogen site. Under low nitrogen doping conditions (N-HOPG with X = 0.4), the doped nitrogen is well oriented in the graphite basal plane, substituting the graphite lattice to form the graphitic N and pyridinic N sites. On increasing the nitrogen dose the in-plane orientation of the incorporated nitrogen species becomes less remarkable, and under high nitrogen doping conditions (N-HOPG with X = 8.4) defective and non-planar structures in N-HOPG become dominant. Raman spectra exhibit a similar trend. However, in the case of N-HOPG with X = 2.3, XAS revealed a highly oriented doped nitrogen character, while an amorphous-like signature was observed in the Raman spectrum. It is therefore thought that nitrogen in N-HOPG with X = 2.3 is likely to be doped into the flat graphite region rather than into the amorphous region. Moreover, an additional peak was observed at higher binding energy of 401.9 eV than 400.9 eV for graphitic1 N (graphitic N in the basal plane) in N 1s XPS. According to the previous theoretical and experimental reports, it seems most plausible to assign the peak to graphitic2 N (graphitic N in the zigzag edge and/or vacancy sites). For the conclusive assignment, however, we should visualize the local carbon structure with graphitic2 N by microscopic measurements in future studies. The graphitic1 N and graphitic2 N were preferably formed when the amount of doped nitrogen was small (N-HOPG with X = 0.4), while the graphitic1 N was more abundant at higher nitrogen doping levels (N-HOPG with X = 2.3). The ORR activity measurements of N-HOPG were performed and the N-HOPG with X = 0.4 at the lowest nitrogen doping level showed the highest ORR activity among the three N-HOPGs. Applying our preparation and characterization methods of model N-HOPG catalysts is a novel and promising way to reveal the nitrogen doped carbon-based catalysts.Acknowledgements
This work was financially supported by the New Energy and Industrial Technology Development Organization (NEDO), Japan. The authors thank T. Muro and Y. Tamenori for their technical support in the BL27SU experiments (Proposal No. 2012B1027, 2013A1538). The XAS measurements at BL07LSU were performed jointly in the Synchrotron Radiation Research Organization and Institute for Solid State Physics, at the University of Tokyo, Japan (Proposal No. 2013A7403).References
- H. M. Jeong, J. W. Lee, W. H. Shin, Y. J. Choi, H. J. Shin, J. K. Kang and J. W. Choi, Nano Lett., 2011, 11, 2472–2477 CrossRef CAS PubMed.
- W. Li, H.-Y. Lü, X.-L. Wu, H. Guan, Y.-Y. Wang, F. Wan, G. Wang, L.-Q. Yan, H.-M. Xie and R.-S. Wang, RSC Adv., 2015, 5, 12583–12591 RSC.
- A. L. M. Reddy, A. Srivastava, S. R. Gowda, H. Gullapalli, M. Dubey and P. M. Ajayan, ACS Nano, 2010, 4, 6337–6342 CrossRef CAS PubMed.
- J. Long, X. Xie, J. Xu, Q. Gu, L. Chen and X. Wang, ACS Catal., 2012, 2, 622–631 CrossRef CAS.
- R. Lv, Q. Li, A. R. Botello-Méndez, T. Hayashi, B. Wang, A. Berkdemir, Q. Hao, A. L. Elías, R. Cruz-Silva, H. R. Gutiérrez, Y. A. Kim, H. Muramatsu, J. Zhu, M. Endo, H. Terrones, J.-C. Charlier, M. Pan and M. Terrones, Sci. Rep., 2012, 2, 586 Search PubMed.
- L. Qu, Y. Liu, J.-B. Baek and L. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- P. Matter, L. Zhang and U. Ozkan, J. Catal., 2006, 239, 83–96 CrossRef CAS.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed.
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed.
- E. Proietti, F. Jaouen, M. Lefèvre, N. Larouche, J. Tian, J. Herranz and J.-P. Dodelet, Nat. Commun., 2011, 2, 416 CrossRef PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- Y. Nabae, M. Sonoda, C. Yamauchi, Y. Hosaka, A. Isoda and T. Aoki, Catal. Sci. Technol., 2014, 4, 1400–1406 CAS.
- Y. Nabae, Y. Kuang, M. Chokai, T. Ichihara, A. Isoda, T. Hayakawa and T. Aoki, J. Mater. Chem. A, 2014, 11561–11564 CAS.
- T. Ikeda, M. Boero, S. F. Huang, K. Terakura, M. Oshima and J. I. Ozaki, J. Phys. Chem. C, 2008, 112, 14706–14709 CAS.
- X. Bao, X. Nie, D. Von Deak, E. J. Biddinger, W. Luo, A. Asthagiri, U. S. Ozkan and C. M. Hadad, Top. Catal., 2013, 56, 1623–1633 CrossRef CAS.
- H. Kim, K. Lee, S. I. Woo and Y. Jung, Phys. Chem. Chem. Phys., 2011, 13, 17505–17510 RSC.
- C. H. Choi, H.-K. Lim, M. W. Chung, J. C. Park, H. Shin, H. Kim and S. I. Woo, J. Am. Chem. Soc., 2014, 136, 9070–9077 CrossRef CAS PubMed.
- H. Niwa, M. Kobayashi, K. Horiba, Y. Harada, M. Oshima, K. Terakura, T. Ikeda, Y. Koshigoe, J. I. Ozaki, S. Miyata, S. Ueda, Y. Yamashita, H. Yoshikawa and K. Kobayashi, J. Power Sources, 2011, 196, 1006–1011 CrossRef CAS.
- W. Ouyang, D. Zeng, X. Yu, F. Xie, W. Zhang, J. Chen, J. Yan, F. Xie, L. Wang, H. Meng and D. Yuan, Int. J. Hydrogen Energy, 2014, 39, 15996–16005 CrossRef CAS.
- M. Favaro, L. Perini, S. Agnoli, C. Durante, G. Granozzi and A. Gennaro, Electrochim. Acta, 2013, 88, 477–487 CrossRef CAS.
- T. Kondo, S. Casolo, T. Suzuki, T. Shikano, M. Sakurai, Y. Harada, M. Saito, M. Oshima, M. I. Trioni, G. F. Tantardini and J. Nakamura, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 035436 CrossRef.
- J. Stöhr, NEXAFS Spectroscopy, Springer, 1992 Search PubMed.
- J. F. Ziegler, M. D. Ziegler and J. P. Biersack, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 1818–1823 CrossRef CAS.
- R. Jansen and H. Van Bekkum, Carbon, 1995, 33, 1021 CrossRef CAS.
- J. Pels, F. Kapteijn, J. Moulijn, Q. Zhu and K. Thomas, Carbon, 1995, 33, 1641–1653 CrossRef CAS.
- J. Casanovas, J. M. Ricart, J. Rubio, F. Illas and J. M. Jiménez-Mateos, J. Am. Chem. Soc., 1996, 118, 8071–8076 CrossRef CAS.
- T. Sharifi, G. Hu, X. Jia and T. Wågberg, ACS Nano, 2012, 6, 8904–8912 CrossRef CAS PubMed.
- X. Wang, Z. Hou, T. Ikeda, S.-F. Huang, K. Terakura, M. Boero, M. Oshima, M. Kakimoto and S. Miyata, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 245434 CrossRef.
- Y. Niimi, T. Matsui, H. Kambara, K. Tagami, M. Tsukada and H. Fukuyama, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 085421 CrossRef.
- A. N. Patel, M. G. Collignon, M. A. O’Connell, W. O. Y. Hung, K. McKelvey, J. V. Macpherson and P. R. Unwin, J. Am. Chem. Soc., 2012, 134, 20117–20130 CrossRef CAS PubMed.
- F. Orlando, P. Lacovig, M. Dalmiglio, A. Baraldi, R. Larciprete and S. Lizzit, Surf. Sci., 2015 DOI:10.1016/j.susc.2015.06.017.
- M. Batzill, Surf. Sci. Rep., 2012, 67, 83–115 CrossRef CAS.
- W. Zhao, O. Höfert, K. Gotterbarm, J. F. Zhu, C. Papp and H.-P. Steinrück, J. Phys. Chem. C, 2012, 116, 5062–5066 CAS.
- E. H. Martins Ferreira, M. V. O. Moutinho, F. Stavale, M. M. Lucchese, R. B. Capaz, C. A. Achete and A. Jorio, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125429 CrossRef.
- I. Shimoyama, G. Wu, T. Sekiguchi and Y. Baba, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, R6053–R6056 CrossRef CAS.
- T. Schiros, D. Nordlund, L. Pálová, D. Prezzi, L. Zhao, K. S. Kim, U. Wurstbauer, C. Gutiérrez, D. Delongchamp, C. Jaye, D. Fischer, H. Ogasawara, L. G. M. Pettersson, D. R. Reichman, P. Kim, M. S. Hybertsen and A. N. Pasupathy, Nano Lett., 2012, 12, 4025–4031 CrossRef CAS PubMed.
- H. Niwa, K. Horiba, Y. Harada, M. Oshima, T. Ikeda, K. Terakura, J. Ozaki and S. Miyata, J. Power Sources, 2009, 187, 93–97 CrossRef CAS.
- X. Wang, Z. Hou, T. Ikeda, M. Oshima, M. Kakimoto and K. Terakura, J. Phys. Chem. A, 2013, 117, 579–589 CrossRef CAS PubMed.
- F. Atamny, J. Blöcker, B. Henschke, R. Schlögl, T. Schedelniedrig, M. Keil and A. M. Bradshaw, J. Phys. Chem., 1992, 96, 4522–4526 CrossRef CAS.
- Z. Hou, X. Wang, T. Ikeda, K. Terakura, M. Oshima and M. A. Kakimoto, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 165401 CrossRef.
- M. dos Santos and F. Alvarez, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 13918–13924 CrossRef CAS.
- A. Cuesta, P. Dhamelincourt, J. Laureyns, A. Martínez-Alonso and J. M. D. Tascón, Carbon, 1994, 32, 1523–1532 CrossRef CAS.
- A. Jorio, M. M. Lucchese, F. Stavale, E. H. M. Ferreira, M. V. O. Moutinho, R. B. Capaz and C. A. Achete, J. Phys.: Condens. Matter, 2010, 22, 334204 CrossRef PubMed.
- A. Jorio, M. M. Lucchese, F. Stavale and C. A. Achete, Phys. Status Solidi B, 2009, 246, 2689–2692 CrossRef CAS.
- H. Niwa, M. Saito, M. Kobayashi, Y. Harada, M. Oshima, S. Moriya, K. Matsubayashi, Y. Nabae, S. Kuroki, T. Ikeda, K. Terakura, J. I. Ozaki and S. Miyata, J. Power Sources, 2013, 223, 30–35 CrossRef CAS.
- K. Kamiya, H. Koshikawa, H. Kiuchi, Y. Harada, M. Oshima, K. Hashimoto and S. Nakanishi, ChemElectroChem, 2014, 1, 877–884 CrossRef CAS.
- X. Li, G. Liu and B. N. Popov, J. Power Sources, 2010, 195, 6373–6378 CrossRef CAS.
- H. Kiuchi, H. Niwa, M. Kobayashi, Y. Harada and M. Oshima, Electrochim. Acta, 2012, 82, 291–295 CrossRef CAS.
- U. Tylus, Q. Jia, K. Strickland, N. Ramaswamy, A. Serov, P. Atanassov and S. Mukerjee, J. Phys. Chem. C, 2014, 118, 8999–9008 CAS.
- P. S. Miedema, M. M. van Schooneveld, R. Bogerd, T. C. R. Rocha, M. Hävecker, A. Knop-Gericke and F. M. F. de Groot, J. Phys. Chem. C, 2011, 115, 25422–25428 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp02305j |
| This journal is © the Owner Societies 2016 |