
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular tectonics: tetracarboxythiacalix[4]arene derivatives as tectons for the formation of hydrogen-bonded networks†
A. S.
Ovsyannikov
bc,
M. N.
Lang
a,
S.
Ferlay
 *a,
S. E.
Solovieva
*bc,
I. S.
Antipin
*bc,
A. I.
Konovalov
bc,
N.
Kyritsakas
a and
M. W.
Hosseini
*a
*a,
S. E.
Solovieva
*bc,
I. S.
Antipin
*bc,
A. I.
Konovalov
bc,
N.
Kyritsakas
a and
M. W.
Hosseini
*a
aMolecular Tectonics Laboratory, UMR UdS-CNRS 7140, University of Strasbourg, Institut Le Bel, 4, rue Blaise Pascal, F-67000 Strasbourg, France. E-mail: ferlay@unistra.fr; hosseini@unista.fr
bKazan Federal University, Kremlevskaya str. 18, Kazan 420008, Russian Federation
cA. E. Arbuzov Institute of Organic and Physical Chemistry, Russian Academy of Science, Arbuzov str. 8, Kazan 420088, Russian Federation
First published on 6th October 2016
Abstract
A series of thiacalix[4]arene derivatives blocked in the 1,3-alternate conformation and bearing four carboxylic acids have been designed and synthesized. These compounds, owing to the H-bond donor (OH moiety) and acceptor (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group) nature of the carboxylic acid moieties, behave as self-complementary tectons and lead to the formation of tubular 1D H-bonded networks in the crystalline phase. Upon deprotonation of the self-complementary neutral compounds, i.e. transformation of carboxylic acid moieties into carboxylates, anionic tectons are generated. Due to their propensity to form H-bonded networks in the presence of a dicationic H-bond donor tecton of the cyclic bis-amidinium type, designed to behave as a molecular staple interconnecting two carboxylates moieties, 1- and 2-D H-bonded networks are formed under self-assembly conditions.
O group) nature of the carboxylic acid moieties, behave as self-complementary tectons and lead to the formation of tubular 1D H-bonded networks in the crystalline phase. Upon deprotonation of the self-complementary neutral compounds, i.e. transformation of carboxylic acid moieties into carboxylates, anionic tectons are generated. Due to their propensity to form H-bonded networks in the presence of a dicationic H-bond donor tecton of the cyclic bis-amidinium type, designed to behave as a molecular staple interconnecting two carboxylates moieties, 1- and 2-D H-bonded networks are formed under self-assembly conditions.
Introduction
Tubular architectures1–3 are of interest as they form channels that may lead to the transport of neutral or charged species. Such entities may be either discrete or infinite networks displaying translational symmetry. For the latter category, they may be formed by self-assembly processes upon interconnection of cyclic entities using either hydrogen or coordination bonds.4–7 Tubular architectures may also be helical assemblies.8 Organisation of cyclic units into tubular architectures using liquid crystalline phases9 or polymeric backbones10 has been also reported.Following the concepts developed in molecular tectonics11 for the design and formation by self-assembly processes of infinite molecular networks in the crystalline phase, we are interested in tubular architectures based on H-bonds between macrocyclic entities behaving as tectons.12 Owing to their cyclic nature, calix[4]arene13 and thiacalix[4]arene 1–314,15 (Fig. 1) in their 1,3-alternate conformation are interesting backbones for the design of such tectons. Indeed the 1,3-alternate conformation of these two platforms allows up to 4 interaction sites to be positioned in a divergent fashion, i.e. two above and two bellow the main plane of the calixarene. We have previously exploited this feature and designed metallatubulanes based on calix[4]arene derivatives16 or on [1]-metacyclophane in the 1,3-alternate fixed conformation, an analogous backbone to calix[4]arene.17
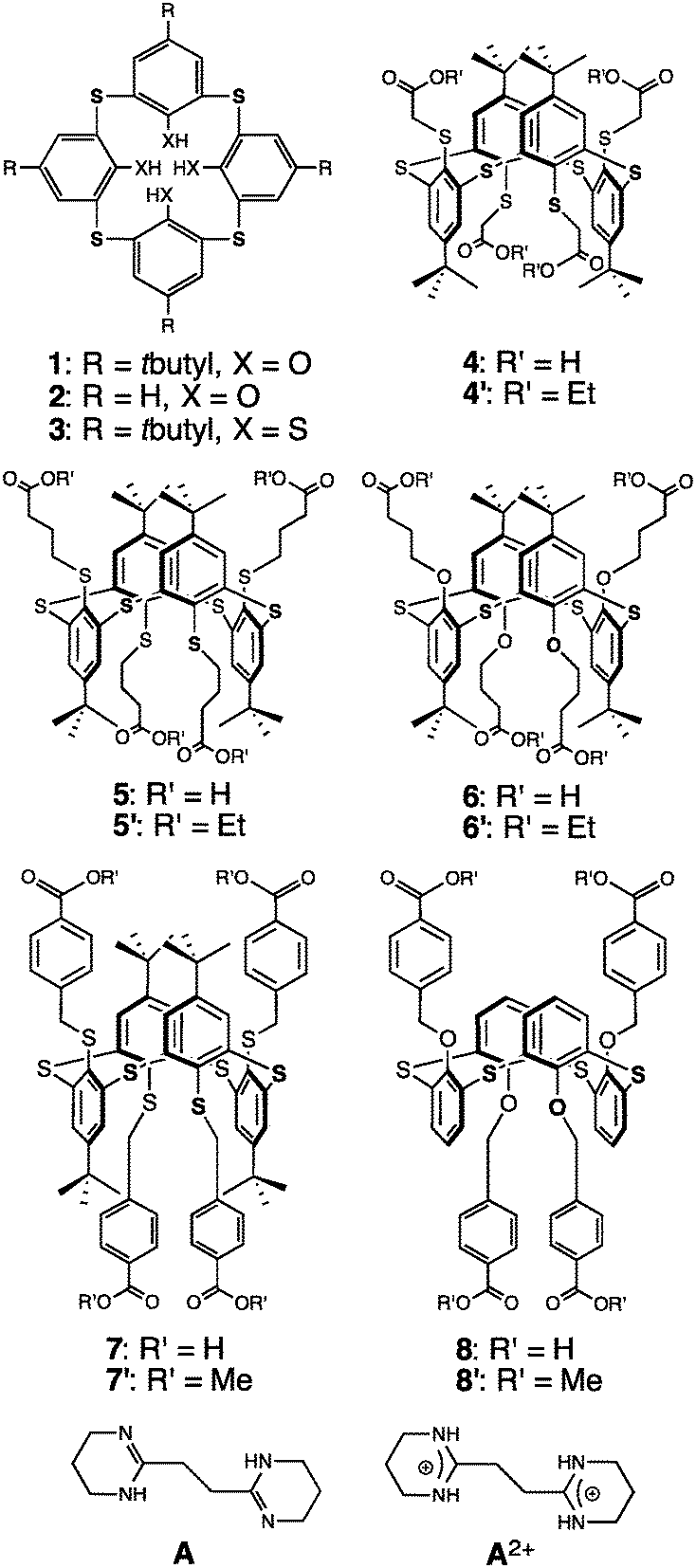 | ||
| Fig. 1 Tetrathia- and tetrathiatetramercaptocalix[4]arene precursors 1, 2 and 3, and tetrakis carboxyl derivatives 4–8 in the 1,3-A conformation. | ||
Here we report on the design, synthesis and characterization of thiacalixarene based derivatives 4–8, in their 1,3-alternate conformation, bearing four carboxylic acid groups and thus behaving as self-complementary tectons. We describe their self-assembly in the crystalline phase into 1D tubular H-bonded networks as well as combinations of their deprotonated derivatives as anionic tectons with a dicationic tecton A2+, behaving as a molecular staple, capable of bridging consecutive anionic units, leading to extended H-bonded networks.
Compounds 4–7, and 8 are based on p-tert-butylthiacalix[4]arene and on the H-thiacalix[4]arene backbone in its 1,3-alternate conformation, respectively (Fig. 1). They are analogous derivatives bearing four carboxylic acid moieties and differ by the nature of the spacer connecting the carboxyl groups to the backbone. They have been designed in order to investigate the role played by the spacer in their ability to form tubular H-bonded networks (Fig. 2a) based on the formation of H-bonded dimeric nodes (Fig. 2b) between carboxylic acids belonging to consecutive self-complementary tectons. Owing to the acidic nature of compounds 4–8, upon their deprotonation leading to anionic species, they offer another possibility for the design of tubular H-bonded networks. Indeed these anionic tectons bearing carboxylate moieties may be combined with the dicationic organic tecton A2+ (Fig. 1) designed to bridge two carboxylates, one on each of its faces, and thus behave as a molecular staple (Fig. 2c) connecting consecutive carboxylate-bearing components.18,19 Depending on the degree of deprotonation of neutral compounds 4–8 and the nature of the spacer connecting the calix backbone to the carboxylate units, one may expect the formation of 1D tubular H-bonded networks (Fig. 2a). The driving force for the formation of such extended architectures is the establishment of charge-assisted H-bonds.20
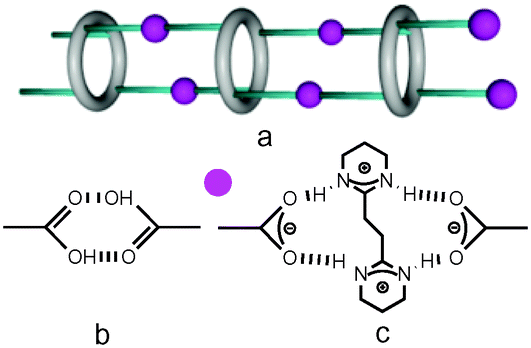 | ||
| Fig. 2 Recognition patterns through H bonds between tectons: a) schematic view of tetra-substituted macrocyclic species leading to 1D tubular systems, b) a dimeric node involving two carboxylic acid moieties and c) dihapto mode between carboxylate and amidinium moieties, behaving as molecular staples. | ||
Although few tetra-acid derivatives based on calix[4]arene21 (CA) and thiacalix[4]arene22 (TCA) backbones have been reported, to the best of our knowledge, no example of tetramercaptothiacalixarene (TMTCA) 3 (Fig. 1) based derivatives has been documented to date.
Few examples of H-bonded nanotubular assemblies based on p-tert-butylthiacalix[4]arene 3 bearing carboxylic acid and/or urea have been reported.23 An interesting investigation dealing with the photoisomerization of a H-bonded network based on a p-tert-butylthiacalix[4]arene derivative bearing carboxylic acid moieties combined with bipyridylethene has been also reported.24
Experimental
Characterization techniques
1H-NMR and 13C-NMR spectra were recorded at room temperature on a Bruker 300 MHz and 500 MHz.FT-IR spectra were recorded on a Perkin Elmer ATR spectrometer.
Mass spectra (MS (ES+)) were recorded on a Bruker MicroTOF spectrometer.
Melting points were measured in capillary tubes on a Stuart Scientific Melting Point SMP-1 apparatus.
Elemental analyses were performed by the Service de Microanalyses de la Fédération de Recherche Chimie of the Université de Strasbourg.
Single-crystal studies
Data were collected at 173(2) K on a Bruker APEX8 CCD diffractometer equipped with an Oxford Cryosystem liquid N2 device, using graphite-monochromated Mo-Kα (λ = 0.71073 Å) radiation. For all structures, diffraction data were corrected for absorption. Structures were solved using SHELXS-97 and refined by full matrix least-squares on F2 using SHELXL-97. The hydrogen atoms were introduced at calculated positions and not refined (riding model).25 CCDC: 1504484–1504490 and 1505426 (see Tables S4 and S5, ESI†).Powder X-ray diffraction
Powder diffraction (PXRD) diagrams were collected on a Bruker D8 diffractometer using monochromatic Cu-Kα radiation in a scanning range between 3.8 and 40° using a scan step size of 2° mn−1.As already demonstrated and currently admitted, for all the compounds, discrepancies in intensity between the observed and simulated patterns are due to the preferential orientations of the microcrystalline powders.
Synthesis
General: all reagents were purchased from commercial sources and used without further purification. The synthesis of 1,14a2,26327 and 828 has been previously reported. The synthesis of 4–7 was adapted from previously reported procedures.22,29The bisamidinium compound A2+, used as its ditosylate salt (A2+, 2TsO−), has been prepared using an already reported procedure.18b
Mp: 310 °C (decomp.); MALDI TOF: m/z = 1129.3 [M]+ (calculated 1129.69); elemental analysis: % calculated: C, 59.54%; H, 6.42%; % found: C, 59.45%; H, 6.48%; 1H-NMR (CDCl3, 300 MHz, 25 °C): δ(ppm) = 1.18 (12H, t, –CH3), 1.26 (36H, s, tBu); 3.61 (8H, s, ArSCH2), 4.12 (8H, q, –C(O)CH2–), 7.76 (8H, s, Ar–H); 13C-NMR (CDCl3, 125 MHz, 25 °C): δ(ppm) = 14.4; 31.1; 34.8; 37.7; 61.5; 136.1; 139.9; 143.2; 150.9; 170.0.
Mp: 320 °C (decomp.) MALDI TOF: m/z: 1241.5 [M]+ (calculated 1241.88); elemental analysis: % calculated: C, 61.90%; H, 7.14%; % found: C, 61.68%; H, 7.10%; 1H-NMR (CDCl3, 400 MHz, 25 °C): δ(ppm) = 1,24 (12H, t, –CH3), 1,26 (36H, s, tBu); 1,75 (8H, p, –CH2CH2CH2–); 2,56 (8H, t, –CH2C(O)–); 2,93 (8H, t, –SCH2–); 4,14 (8H, q, –C(O)O–CH2–); 7,71 (8H, s, Ar–H).
Mp: 315–317 °C; MALDI TOF: m/z: 1176,85 [M]+ (calculated 1177,64); elemental analysis: % calculated: C, 65.28%; H, 7.53%; % found: C, 65.33%; H, 7.51%; 1H-NMR (CDCl3, 600 MHz, 25 °C): δ(ppm) = 1.25 (s, 36H, tBu), 1.25 (t, 12H, –CH3), 1.41 (m, 8H, –CH2CH2CH2–), 2.11 (t, 8H, –CH2C(O)–), 3.91 (t, 12H, –O–CH2–), 4.09 (q, 8H, –OCH2CH3), 7.36 (s, 8H, Ar–H); 13C-NMR (CDCl3, 125 MHz, 25 °C): δ ppm = 14.3, 24.3, 30.9, 31.2, 34.3, 60.1, 67.9, 127.8, 128.2, 145.9, 156.8, 172.8.
Mp: 300 °C (decomp.) MALDI TOF: m/z: 1377,15 [M]+ (calculated 1377,95); elemental analysis: % calculated: C, 66.25%; H, 5.85%; % found: C, 66.18%; H, 5.86%; 1H-NMR (CDCl3, 300 MHz, 25 °C): δ(ppm) = 1,06 (36H, s, tBu); 3.92 (12H, s, –C(O)O–CH3); 4,05 (8H, s, –SCH2–); 7,28 (8H, d, Ar–H); 7,61 (8H, s, Ar–H); 7,90 (8H, d, Ar–H); 13C-NMR (CDCl3, 125 MHz, 25 °C): δ(ppm) = 30.7; 34.4; 51.0; 52.2; 128.7; 129.1; 129.7; 134.9; 140.0; 143.4; 143.6; 150.4; 167.2.
1H-NMR (CDCl3, 300 MHz, 25 °C): δ(ppm) = 4.05 (12H, s, –C(O)O–CH3); 5.29 (8H, s, –OCH2–); 6.44 (4H, t, Ar–H); 7,01 (8H, d, Ar–H); 7,13 (8H, d, Ar–H); 8.1 (8H, d, Ar–H).
Mp: 312 °C (decomp.) MALDI TOF: m/z: 1039.19 [M + Na]+ (calculated 1040.47); elemental analysis: % calculated: C, 56.66%; H, 5.55%; % found: C, 56.40%; H, 5.50%; 1H-NMR (DMSO-d6, 300 MHz, 25 °C): δ(ppm) = 1.21 (s, 36H, tBu); 3.56 (s, 8H, ArSCH2), 7.76 (s, 8H, Ar–H); 13C-NMR (DMSO-d6, 125 MHz, 25 °C): δ(ppm) = 30.3; 34.1; 37.7; 135.7; 140.2; 141.9; 149.8; 170.5.
Mp: 315 °C (decomp.) MALDI TOF: m/z = 1167.31 [M + K]+ (calculated 1168.76); elemental analysis: % calculated: C, 59.54%; H, 6.42%; % found: C, 59.40%; H, 6.46%; 1H-NMR (DMSO-d6, 300 MHz, 25 °C): δ(ppm) = 1,23 (36H, s, tBu); 1,60 (8H, m, –CH2CH2CH2–); 2,46 (8H, t, –SCH2–); 2,88 (8H, t, –CH2–C(O)–); 7,71 (8H, s, Ar–H); 13C-NMR (DMSO-d6, 125 MHz, 25 °C): δ(ppm) = 24.2; 30.5; 32.6; 34.0; 34.6; 133.9; 140.0; 141.9; 149.4; 174.1.
Mp: 330 °C (decomp.) MALDI TOF: m/z = 1087.41 [M + Na]+ (calculated 1088.41); elemental analysis: % calculated: C, 61.09%; H, 6.90%; % found: C, 61.25%; H, 6.83%; 1H-NMR (DMSO-d6, 300 MHz, 25 °C): δ(ppm) = 1,21 (36H, s, tBu); 1,21 (8H, m, –CH2CH2CH2–); 1.99 (8H, t, –CH2–C(O)–); 3.80 (8H, t, –OCH2–); 7,34 (8H, s, Ar–H); 11.92 (4H, s, –COOH); 13C-NMR (DMSO-d6, 125 MHz, 25 °C): δ(ppm) = 23.7; 30.0; 30.9; 34.0; 67.3; 127.0; 127.3; 145.6; 156.2; 174.0.
Mp: 330 °C (decomp.) MALDI TOF: m/z = 1343.47 [M + Na]+ (calculated 1344.83); elemental analysis: % calculated: C, 65.42%; H, 5.49%; % found: C, 65.38%; H, 5.52%; 1H-NMR (DMSO-d6, 300 MHz, 25 °C): δ(ppm) = 0.98 (36H, s, tBu); 4,07 (8H, s, –SCH2–); 7.37 (8H, d, Ar–H); 7,65 (8H, s, Ar–H); 7.81 (8H, d, Ar–H); 13C-NMR (DMSO-d6, 125 MHz, 25 °C): δ(ppm) = 30.1; 33.9; 129.0; 129.2; 134.9; 140.3; 142.7; 142.7; 167.1.
1H-NMR (DMSO-d6, 300 MHz, 25 °C): δ(ppm) = 5.20 (8H, s, –OCH2–); 6.44 (4H, t, Ar–H); 6.98 (8H, d, Ar–H); 7,15 (8H, d, Ar–H); 7.95 (8H, d, Ar–H).
Crystallisation conditions
Results and discussion
Synthesis and solid state characterization of the tectons
The synthesis of thiacalix[4]arene derivatives 4–8 was achieved using reported procedures (see the Experimental section). Compounds 4–8 bearing four carboxylic acid moieties were obtained in 46–95% yield upon hydrolysis of the tetraester derivatives 4′–8′ (see ESI,† Table S1).In solution, all four compounds 4–8 were characterized by both 1H- and 13C-NMR spectroscopy which showed sharp signals indicating the presence of conformationally blocked 1,3-alternate isomers (see the Experimental section). Furthermore, among the five new compounds 4–8, four (compounds 4–7) have been also characterized in the solid state by X-ray diffraction methods on single crystals obtained upon slow diffusion or slow evaporation techniques (see the Experimental part and Table S4, ESI†). As expected, all compounds adopt the 1,3-A conformation (Fig. 4). As indicated by the C–O distances (see ESI,† Table S2), the carboxyl moieties are fully protonated.
Compound 4 crystallises in the presence of DMF molecules which are found to be disordered. Crystals of 5 are exclusively composed of the tecton. Tecton 6 crystallises with CHCl3 solvent molecules, whereas compound 7 crystallizes with both H2O and CH2Cl2 molecules.
For compounds 4–7, the metrics for the macrocyclic backbone is close to the one observed for the parent compounds 1,152 (ref. 15a) and 3 (ref. 27) (see Table S2, ESI†). In the case of 5, one of the four tertiobutyl groups is found to be disordered (Fig. 3).
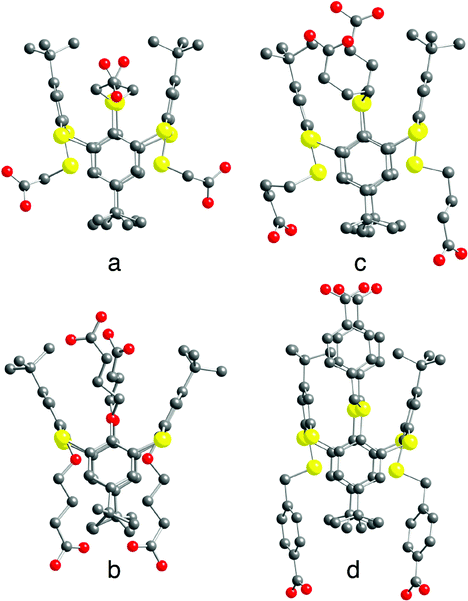 | ||
| Fig. 3 Crystal structures of tectons 4 (a), 5 (b), 6 (c) and 7 (d) in the 1,3-alternate conformation. H atoms, disordered tertiobutyl groups (5) and solvent molecules are not presented for clarity. For bond distances and angles, see the text and ESI,† Table S2. | ||
Formation of extended 1D tubular architectures by self-complementary tectons 4–8
Owing to the propensity of carboxylic acids to form a H-bonded dimeric complex (Fig. 2b) and due to the 1,3-A conformation adopted by the tetracarboxyl compounds 4–8, the latter might behave as self-complementary tectons and self-assemble into 1D H-bonded tubular networks (Fig. 2a).In the case of 4, the presence of disordered DMF molecules interacting with the carboxyl groups (O–O distances of 2.552(4)–2.806(7) Å) prevents the formation of dimeric H-bonded motives (Fig. 2b) and consequently, the generation of the extended 1D architecture. In this case, the isolated species is observed in the crystals.
For tectons 5–7, the tubular arrangement was observed. All three self-complementary tectons display the dimeric recognition pattern (Fig. 2b) and self-assemble into analogous 1D H-bonded networks (Fig. 2a) with the O–O bond distance in the 2.579(4)–2.686(5) Å range (Fig. 4) (see ESI,† Table S2). Whereas tubular architectures formed by tectons 5 (Fig. 4a and d) and 7 (Fig. 4c and f) are linear, for tecton 6 (Fig. 4b and e) a slightly zig-zag type arrangement is observed. Consequently, the diameter of the tubular networks formed by 5 and 7 of ca. 5 Å is substantially larger than the one observed for 6 (Fig. 4d–f).
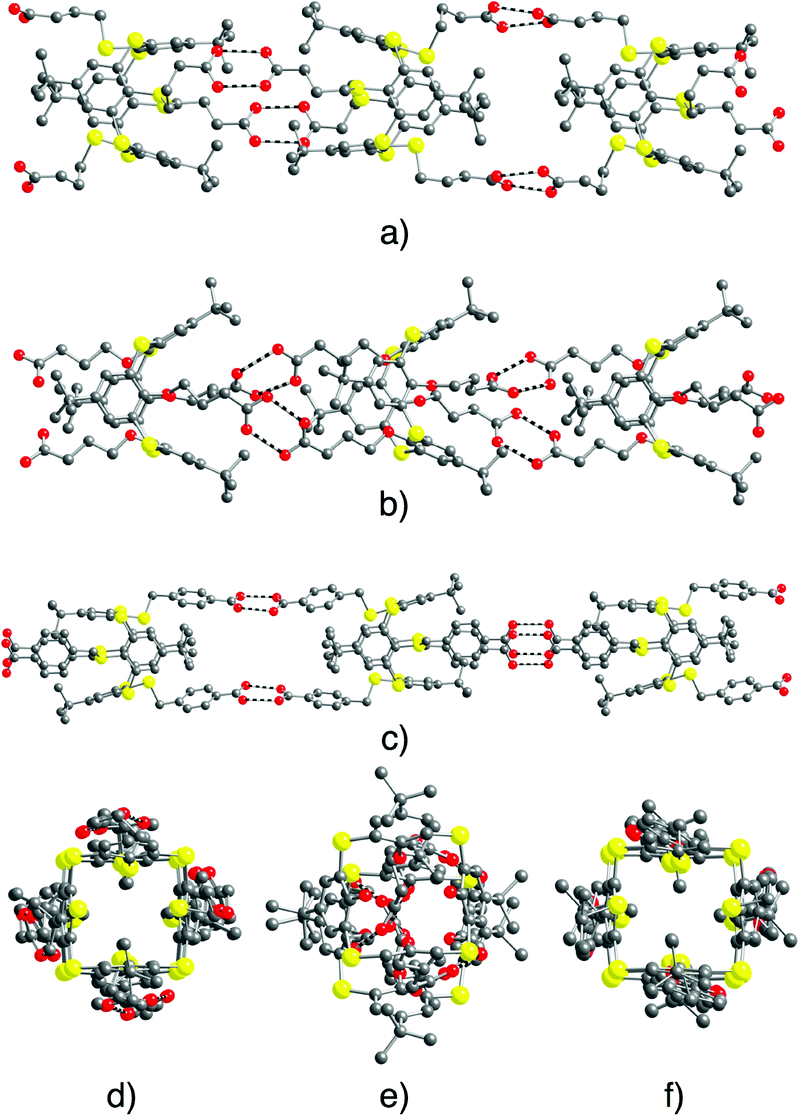 | ||
| Fig. 4 Solid state structures of 1D tubular H-bonded networks formed by self-complementary tectons 5–7. Views along the 1D network (a–c, respectively) and perpendicular views (d–f, respectively). H atoms, disordered tertiobutyl groups (in the case of tecton 5) and solvent molecules are not presented for clarity. For bond distances and angles, see the text and ESI,† Table S2. | ||
For all three cases, the 1D tubular architectures are packed in a parallel fashion.
For tecton 6, no specific interactions between CHCl3 solvent molecules and the network are found. For 7, however, whereas no interactions between CH2Cl2 solvent molecules and the network could be spotted, the water molecules do form H-bonds with the carboxylic acid moieties of the tecton (O–O distance of 3.197(6) Å).
Owing to the instability of the crystals of 4, 5 and 7 outside the crystallization mother liquor, no PXRD powder patterns could be recorded on the corresponding microcrystalline powder. In marked contrast, the microcrystalline powder of 6 was found to be stable and thus it could be investigated by PXRD. The study revealed a good match between the simulated and observed patterns (Fig. 5).
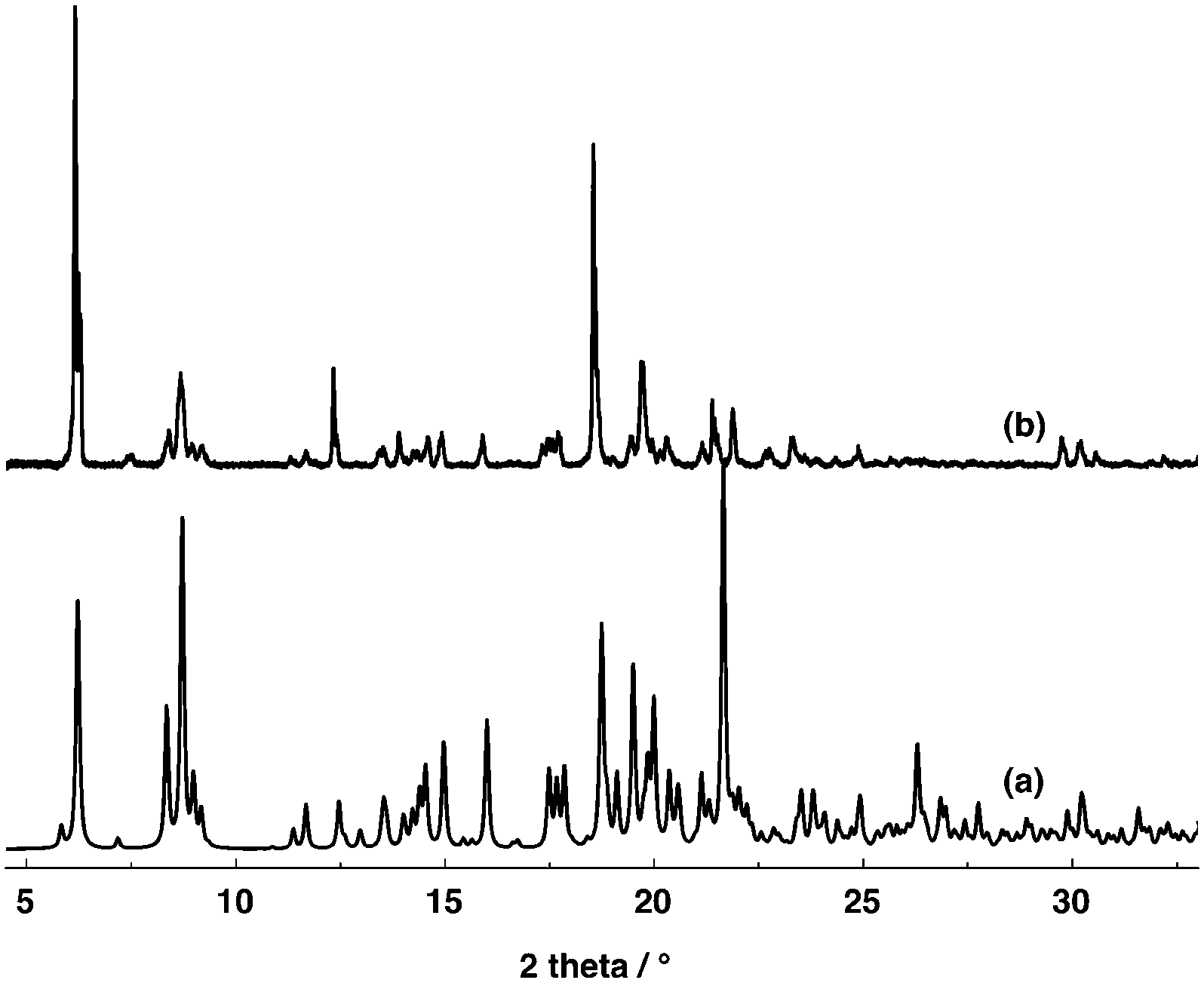 | ||
| Fig. 5 Simulated (a) and observed (b) PXRD powder patterns for compound 6. Discrepancies in peak intensity are due to the preferential orientation of the microcrystalline powder. | ||
Formation of extended networks by combinations of deprotonated compounds 4–8 and the dicationic H-bond donor tecton A2+
As stated in the introduction, the dicationic organic tecton A2+ (Fig. 1) is well suited to recognize two carboxylate moieties, through a dihapto mode of interaction (Fig. 2c). In other terms, tecton A2+ may be regarded as a molecular staple interconnecting two consecutive carboxylate groups by charge assisted H-bonds. This particular mode of interaction may be considered as a structural node of extended H-bonded networks (Fig. 2a) resulting from mutual bridging between deprotonated anionic tectons 4–8 bearing divergently oriented carboxylate moieties as H-bond acceptors and the H-bond donor cationic tecton A2+.The role played by the spacer connecting the four carboxylic acid moieties to the calixarene backbone in compounds 4–8 was systematically investigated. Furthermore, combinations of their deprotonated analogues, generated using 4 eq. of NEt3, as base, with tecton A2+ (2 eq.) were also studied (see the Experimental section). Among several attempts, the following crystalline materials [(43−)2-(A2+)3], [52−-A2+], [(74−)-(A2+)2] and [84−-(A2+)2] were obtained and analysed by X-ray diffraction on single crystals (see Table S5, ESI†).
For the combination of 4 with A2+ in the presence of 4 eq. of NEt3, probably because of the short nature of the spacer (–CH2–) and the presence of tertiobutyl groups, the expected tubular architecture (Fig. 2a) was not observed in the obtained crystals under the crystallization conditions used (see the Experimental section). Indeed, instead of the projected 1/2 stoichiometry [(44−)-(A2+)2], the calixarene derivative was partially deprotonated and found to be in its trianionic form. This was substantiated by C–O distances ranging from 1.187(9) to 1.291(8) Å (see Table S3 in the ESI†). In the crystal, a 2/3 stoichiometry between the trianionic compound 43− and the dicationic tecton A2+, leading to [(43−)2-(A2+)3], is observed. The metrics observed for 43− is close to the one observed for 4 and the structural parameters observed for A2+ are close to those already reported.29
[(43−)2-(A2+)3] crystallises in the presence of DMF and water solvent molecules. Interestingly, the interaction between all three carboxylate moieties of 43 and the dicationic tecton A2+, as expected from the design of A2+, takes place through a dihapto mode of H-bonding with N⋯O distances in the range of 2.669(10) to 2.818(10) Å (see ESI,† Table S3). Owing to the 3/2 anion/cation ratio, the overall architecture is a deformed honeycomb 2D H-bonded network in the xOy plane (Fig. 6). The four DMF and the H2O molecules interact with each other and with the H-bonded network. The DMF molecule is H-bonded with the water molecule with a O⋯O distance of 2.786(15) Å. Furthermore, the water molecule interacts with a carboxylate group (O⋯O distance of 2.784(22) Å). Consecutive 2D H-bonded planes are packed along the c axis with solvent molecules lying between the planes.
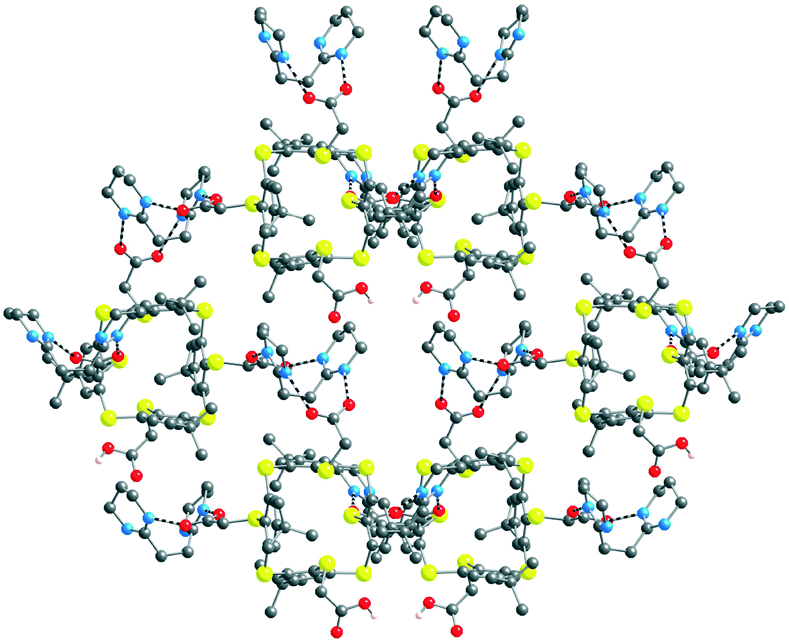 | ||
| Fig. 6 Solid state structures of the 2D honeycomb type H-bonded [(43−)2-(A2+)3] network formed by complementary tectons 43− and A2+. H atoms, DMF and water solvent molecules are not presented for clarity. For bond distances and angles, see the text and ESI,† Table S3. | ||
Owing to the decomposition of crystals of [(43−)2-(A2+)3] in air, no PXRD measurements could be performed.
For the combination of 5 with A2+ in the presence of 4 eq. of NEt3, again the formation of the tubular architecture (Fig. 2a) was not observed under the crystallization conditions used (see the Experimental section). The calixarene-based tecton was in its dianionic state (52−) resulting from the partial deprotonation of two out of the four carboxylic acid moieties. This was reflected by C–O distances ranging from 1.204(9) to 1.316(8) Å (see ESI,† Table S3).
Again, the metrics observed for 52− is close to the one observed for 5 and structural parameters for A2+ are similar to those reported.29 In the crystal, a 1/1 stoichiometry between the dianionic compound 52− and the dicationic tecton A2+ leading to [52−-A2+] is observed. The latter crystallises in the presence of 1 MeOH and 1 H2O solvent molecule. The two deprotonated carboxyl groups are in trans disposition, thus divergently oriented, and located above and under the main plane of the calix backbone.
The overall structure is a 1D H-bonded network (Fig. 7). The connectivity pattern between the components (tectons 52−, A2+ and H2O and MeOH solvent molecules) is rather complex.
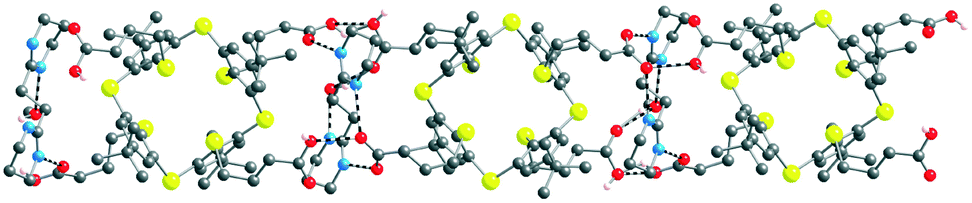 | ||
| Fig. 7 Solid state structures of the 1D H-bonded [52−-A2+] network formed by complementary tectons 52− and A2+. H atoms, MeOH and H2O solvent molecules are not presented for clarity. For bond distances and angles, see the text and ESI,† Table S3. | ||
In marked contrast with the abovementioned combination of 43− with A2+, for [52−-A2+], interactions between carboxylate groups and the dicationic tecton takes place through a monohapto mode of H-bonding (dON of 2.725(8) and 2.815(10) Å) (see ESI,† Table S3). Furthermore, tecton A2+ also forms H-bonds of NHO type with the carboxylic acid groups of 52− (dON of 2.791(7) Å). The cationic tecton A2+ is also connected with MeOH by a H bond (dON of 2.780(11) Å). Owing to the presence of both carboxylic acid and carboxylate moieties, several H-bonded patterns are observed between them: carboxylic–carboxylate (dOO of 2.508(7) Å), carboxylic–carboxylic (dOO of 2.481(9) Å). Furthermore, the MeOH molecule, in addition to A2+, is also H-bonded to a carboxylate group (dOO of 2.685(10) Å).
The 1D arrangements are packed in a parallel fashion along the a and b axes. Water molecules are located between 1D networks without specific interactions with them.
Again, due to the decomposition of crystals of [52−-A2+] in air, no PXRD measurements could be performed.
The combination of A2+ in the presence of 4 eq. of NEt3, with either 7 or 8, afforded crystals which were investigated by X-ray diffraction on single crystals. The structural study revealed that both 7 and 8 are fully deprotonated and behave as tetra anionic tectons (74− and 84−) (see ESI,† Table S3). The anion/cation stoichiometry is ½, leading to [X4−-(A2+)2] (X = 7 or 8). The metrics observed for 74− and for 84− is similar to those for 7 and reported for 8.27 Structural parameters for the dicationic tecton A2+ within the networks are close to the one reported.29
For [74−-(A2+)2], the crystal, in addition to the cationic and anionic tectons, contains two water molecules. For the anionic partner 74−, two out of the four phenyl groups bearing the carboxylate moieties are found to be disordered.
In both cases, as a result of the design of the complementary anionic and cationic tectons behaving as H-bond acceptor and donor sites, respectively, a dihapto mode of H-bonding with the N⋯O distance in the range of 2.625(15)–2.816(13) Å is observed (Fig. 8) (see ESI,† Table S3). Consequently, the mutual interconnection of the anionic tectons 74− and 84− and the cationic tecton A2+ leads to the formation of 1D tubular H-bonded networks (Fig. 2a). For [74−-(A2+)2], the 1D network runs along the b axis (Fig. 8a and c). For [84−-(A2+)2], the 1D network running along the a axis is not linear but corrugated (Fig. 8b and d).
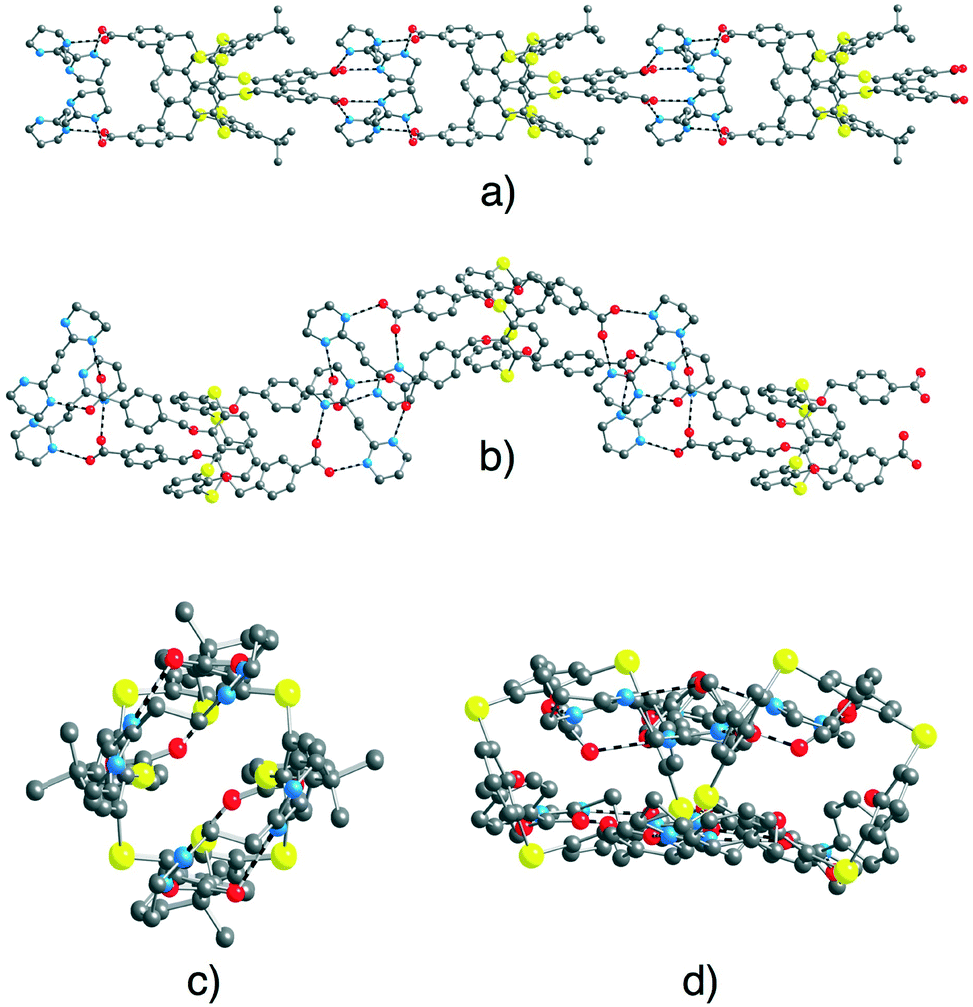 | ||
| Fig. 8 Solid state structures of 1D H-bonded networks formed by [74−-(A2+)2] (perpendicular (a) and parallel (c) views of the extended arrangement) and by [84−-(A2+)2] (perpendicular (b) and parallel (d) views of the extended arrangement). For [74−(A2+)2] disordered phenyl groups, H2O solvent molecules and H atoms are not presented for clarity. For bond distances and angles, see the text and ESI,† Table S3. | ||
The 1D networks are packed in a parallel fashion along the other two space directions (a and c for [74−-(A2+)2] and b and c for [84−-(A2+)2]).
For [74−-(A2+)2], the water molecules are located between the 1D chains and interact with the carboxylate groups of tecton 74− through H bonds with O–O distances of 2.138(15), 2.844(14) and 2.914(11) Å (see ESI,† Table S3).
The PXRD analysis of the microcrystalline powder of [74−-(A2+)2] revealed a good match between the simulated and experimental diffractograms (Fig. 9). For [84−-(A2+)2], unfortunately due to the instability of crystals in air, no PXRD investigation could be carried out.
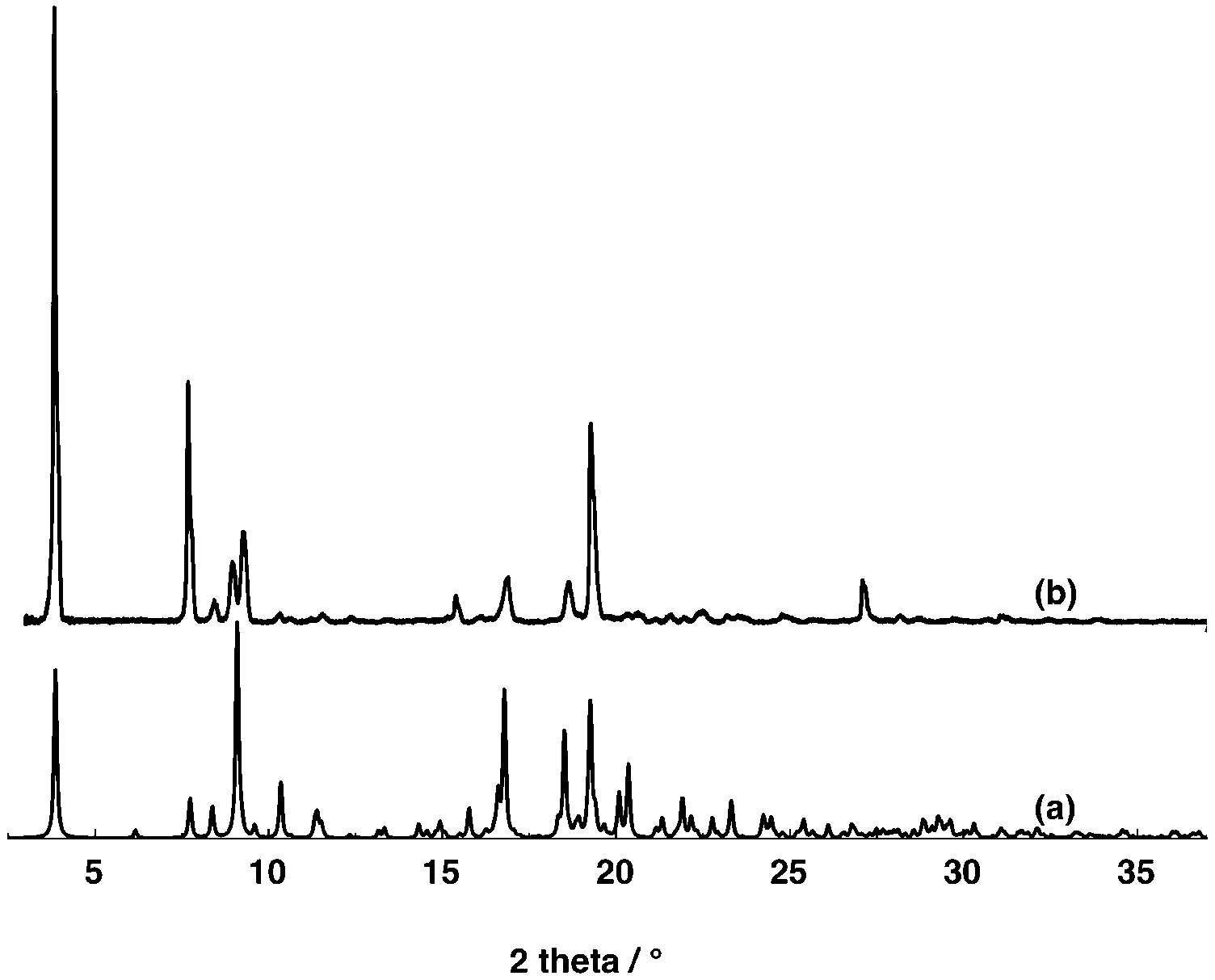 | ||
| Fig. 9 Simulated (a) and observed (b) PXRD powder patterns for compound [74−-(A2+)2]. Discrepancies in peak intensity are due to the preferential orientation of the microcrystalline powder. | ||
Conclusions
The aim of this systematic study was to investigate the formation of tubular 1D H-bonded networks in the crystalline phase. For that purpose, a series of thiacalix[4]arene derivatives blocked in the 1,3-alternate conformation and bearing four carboxylic acids have been designed and studied in the solid state. Owing to the presence of divergently oriented carboxylic acid groups, compounds 4–8 may, in principle, behave as self-complementary H-bond donor (OH moiety) and acceptor (CO group) tectons. Under used crystallization conditions, among the five compounds investigated, only tectons 5–7 afforded crystals which were investigated by X-ray diffraction on single crystals. For all three cases, as expected from their self-complementary nature, 1D H-bonded tubular architectures were obtained (Fig. 2a). These arrangements result from mutual bridging of consecutive tectons through COOH–COOH H-bonded nodes (Fig. 2b).
Upon deprotonation of the self-complementary neutral tectons 4–8 with NEt3, i.e. transformation of the carboxylic acid moieties into carboxylates, anionic tectons were generated in situ. Their propensity to form H-bonded networks in the presence of the dicationic H-bond donor A2+, designed to behave as a molecular staple interconnecting two carboxylates moieties (Fig. 2c), was studied. Under used conditions, i.e. combinations of compounds 4–8 as tetra-acids with 4 eq. of NEt3 and (A2+, 2TsO−), depending on the nature of the spacer connecting the carboxylate unit to the calix backbone, different deprotonated species are obtained. Indeed, whereas in the case compound 4 the latter was in its 43− form, for compound 5, the dianionic form 52− was obtained. The other two compounds 7 and 8 were fully deprotonated in X4− (X = 7 or 8).
The combination of 43− and A2+, the network [(43−)2-(A2+)3], is a deformed honeycomb 2D H-bonded network (Fig. 6). On the other hand, the combination of 52− and A2+, leads to a non-tubular 1D H-bonded network [52−-A2+] (Fig. 7). Finally, for 74− and 84−, their combinations with A2+ lead to a 1/2 stoichiometry and the formation of the expected (Fig. 2a) 1D tubular H-bonded networks, [74−-(A2+)2] and [84−-(A2+)2] (Fig. 8).
Acknowledgements
The Russian Science Foundation (Grant No 15-13-30006), the University of Strasbourg, the International Centre for Frontier Research in Chemistry (icFRC), the Labex CSC (ANR-10-LABX- 0026 CSC) within the Investissement d'Avenir program ANR-10-IDEX-0002-02, the Institut Universitaire de France and the CNRS are acknowledged for financial support.References
- (a) P. De Santis, S. Morosetti and R. Rizzo, Macromolecules, 1974, 7, 52 CrossRef CAS; (b) J. D. Lear, Z. R. Wasserman and W. F. DeGrado, Science, 1988, 240, 117 Search PubMed; (c) M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324 CrossRef CAS PubMed; (d) D. H. Lee and M. R. Ghadiri, in Comprehensive Supramolecular Chemistry, ed. J. L. Atwood, J. E. D. Davies, D. D. Macnicol, J.-P. Sauvage, M. W. Hosseini and F. Vögtle, Pergamon, 1996, vol. 9, p. 451 Search PubMed.
- (a) V. Semetey, D. Rognan, C. Hemmerlin, R. Graff, J.-P. Briand, M. Marraud and G. Guichard, Angew. Chem., Int. Ed., 2002, 41, 1893 CrossRef CAS; (b) N. Sakai and S. Matile, Chem. Commun., 2003, 2514 RSC.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401 CrossRef CAS PubMed.
- C. Kaes, M. W. Hosseini, C. E. F. Rickard, B. W. Skelton and A. White, Angew. Chem., Int. Ed., 1998, 37, 920 CrossRef CAS.
- (a) O. J. Gelling, F. van Bolhuis and B. L. Feringa, J. Chem. Soc., Chem. Commun., 1991, 917 RSC; (b) Y. Dai, T. J. Katz and D. A. Nichols, Angew. Chem., Int. Ed. Engl., 1996, 35, 2109 CrossRef CAS; (c) B. Wu, W.-J. Zhang, S.-Y. Yu and X.-T. Wu, Chem. Commun., 1997, 1795 CAS.
- M. J. Hannon, C. L. Painting and W. Errington, Chem. Commun., 1997, 1805 RSC.
- M. Loï, M. W. Hosseini, A. Jouaiti, A. De Cian and J. Fischer, Eur. J. Inorg. Chem., 1999, 1981 CrossRef.
- W. L. Leong and J. J. Vittal, Chem. Rev., 2011, 111, 688 CrossRef CAS PubMed.
- (a) J.-M. Lehn, J. Malthête and A.-M. Levelut, J. Chem. Soc., Chem. Commun., 1985, 1794 RSC; (b) C. Mertesdorf and H. Ringsdorf, Mol. Cryst. Liq. Cryst., 1989, 5, 1757 CrossRef CAS; (c) V. Percec, G. Johansson, J. A. Heck, G. Ungar and S. V. Betty, J. Chem. Soc., Perkin Trans. 1, 1993, 1411 RSC; (d) T. Komori and S. Shinkai, Chem. Lett., 1993, 1455 CrossRef CAS.
- U. F. Kragten, M. F. M. Roks and R. J. M. Nolte, J. Chem. Soc., Chem. Commun., 1985, 1275 RSC.
- (a) S. Mann, Nature, 1993, 365, 499 CrossRef CAS; (b) M. W. Hosseini, Acc. Chem. Res., 2005, 38, 313 CrossRef CAS PubMed.
- (a) M. Simard, D. Su and J. D. Wuest, J. Am. Chem. Soc., 1991, 113, 4696 CrossRef CAS; (b) M. W. Hosseini, CrystEngComm, 2004, 6, 318 RSC; (c) M. W. Hosseini, Chem. Commun., 2005, 582 Search PubMed.
- (a) C. D. Gutsche, Monographs in Supramolecular Chemistry, Calixarenes revisited, ed. J. F. Stoddart, R.S.C., London, 1998 Search PubMed; (b) Calixarenes 2001, ed. J. Vicens, Z. Asfari, J. M. Harrowfield and V. Böhmer, Kluwer, 2001 Search PubMed.
- (a) H. Kumagai, M. Hasegawa, S. Miyanari, Y. Sugawa, Y. Sato, T. Hori, S. Ueda, H. Kamiyama and S. Miyano, Tetrahedron Lett., 1997, 38, 3971 CrossRef CAS; (b) T. Sone, Y. Ohba, K. Moriya, H. Kumada and K. Ito, Tetrahedron, 1997, 53, 10689 CrossRef CAS.
- (a) H. Akdas, L. Bringel, E. Graf, M. W. Hosseini, G. Mislin, J. Pansanel, A. De Cian and J. Fischer, Tetrahedron Lett., 1998, 39, 2311 CrossRef CAS; (b) M. W. Hosseini, ACS Series, Calixarene Molecules for Separation, ed. G. J. Lumetta, R. D. Rogers and A. S. Gopalan, 2000, vol. 557, p. 296 Search PubMed.
- (a) G. Mislin, E. Graf, M. W. Hosseini, A. De Cian, N. Kyritsakas and J. Fischer, Chem. Commun., 1998, 2545 RSC; (b) G. Laugel, E. Graf, M. W. Hosseini, J.-M. Planeix and N. Kyritsakas, New J. Chem., 2006, 30, 1340 RSC.
- C. Klein, E. Graf, M. W. Hosseini, A. De Cian and J. Fischer, Chem. Commun., 2000, 239 RSC.
- (a) M. W. Hosseini, R. Ruppert, P. Schaeffer, A. De Cian, N. Kyritsakas and J. Fischer, J. Chem. Soc., Chem. Commun., 1994, 2135 RSC; (b) O. Félix, M. W. Hosseini, A. De Cian and J. Fischer, New J. Chem., 1998, 22, 1389 RSC; (c) O. Félix, M. W. Hosseini, A. De Cian and J. Fischer, New J. Chem., 1998, 22, 1389 RSC.
- M. W. Hosseini, Coord. Chem. Rev., 2003, 240, 157 CrossRef CAS.
- (a) K. T. Holman, S. M. Martin, D. P. Parker and M. D. Ward, J. Am. Chem. Soc., 2001, 123, 4421 CrossRef CAS PubMed; (b) K. T. Holman, A. M. Pivovar and M. D. Ward, Science, 2001, 294, 1907 CrossRef CAS PubMed; (c) S. A. Dalrymple and G. K. H. Shimizu, J. Am. Chem. Soc., 2007, 129, 12114 CrossRef CAS PubMed.
- K.-M. Park, E. Lee, C. S. Park and S. S. Lee, Inorg. Chem., 2011, 50, 12085 CrossRef CAS PubMed.
- H. Akdas, W. Jaunky, E. Graf, M. W. Hosseini, A. De Cian and J. Fischer, Tetrahedron Lett., 2000, 41, 3601 CrossRef CAS.
- (a) X. Li, S. L. Gong, W. P. Yang, Y. Y. Chen and X. G. Meng, Tetrahedron, 2008, 64, 6230 CrossRef CAS; (b) Y. Li, W. Yang, Y. Chen and S. Gong, CrystEngComm, 2011, 13, 259 RSC; (c) Y. Li, W. Yang, R. Guo, Y. Chen and S. Gong, CrystEngComm, 2012, 14, 1455 RSC.
- E. Lee, H. Ju, S. Sung Lee and K.-M. Park, Cryst. Growth Des., 2013, 13, 992 CAS.
- G. M. Sheldrick, Program for Crystal Structure Solution, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- H. Akdas, E. Graf, M. W. Hosseini, A. De Cian and N. Kyritsakas-Gruber, C. R. Chim., 2003, 6, 565 CrossRef CAS.
- P. Rao, M. W. Hosseini, A. De Cian and J. Fischer, Chem. Commun., 1999, 2169 RSC.
- D. Appelhans, V. Stastny, H. Komber, D. Voigt, B. Voit, P. Lhotak and I. Stibor, Tetrahedron Lett., 2004, 45, 7145 CrossRef CAS.
- G. Brand, M. W. Hosseini, R. Ruppert, A. De Cian, J. Fischer and N. Kyritsakas, New J. Chem., 1995, 19, 9 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray diffraction data, tables of selected bonds and angles. CCDC 1504484–1504490 and 1505426. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce02026g |
| This journal is © The Royal Society of Chemistry 2016 |