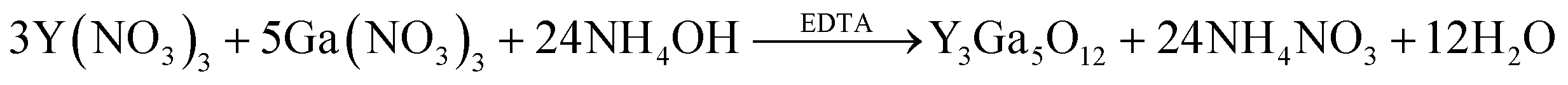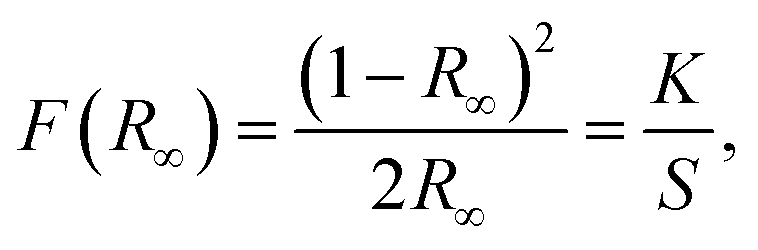DOI:
10.1039/C6CE01988A
(Paper)
CrystEngComm, 2016,
18, 8915-8925
Controlled synthesis of yttrium gallium garnet spherical nanostructures modified by silver oxide nanoparticles for enhanced photocatalytic properties†
Received
14th September 2016
, Accepted 17th October 2016
First published on 17th October 2016
Abstract
Spherical yttrium gallium garnet (Y3Ga5O12) samples were prepared by a single-step hydrothermal synthesis method, and these particles were coated with silver oxide (Ag2O) nanoparticles by following a facile wet chemical approach at ambient temperature. The structural, morphological, vibrational and optical properties of the as-prepared and Ag2O modified Y3Ga5O12 samples were analyzed. From Rietveld refinement of X-ray diffraction patterns, the crystal structure of the Y3Ga5O12 sample was refined. The morphological properties confirmed that the particles were spherical and their surfaces were covered with Ag2O nanoparticles. The Ag2O modified Y3Ga5O12 sample exhibited a relatively enhanced photocatalytic activity compared to the pure Y3Ga5O12 sample in Rhodamine B solution, suggesting that the introduction of Ag2O broadens the absorption of light and controls the photogenerated carrier recombination in Y3Ga5O12. These results provide a clear perspective on the synthesis of hybrid photocatalyst materials which have several ways of photogenerated carrier migration.
1. Introduction
Catalysis dates back to 19th century when a German researcher studied the effect of illumination of ZnO on the bleaching of Prussian blue.1 Meanwhile, many efforts have been devoted to understand and develop the photocatalysis process.2,3 After these successful research efforts, for the first time, Doodeve et al. demonstrated the potentiality of TiO2 as a photosensitizer for bleaching the dyes under ultraviolet (UV) light.4 Since then, great attention has been focused on understanding and improving the photocatalytic efficiency of TiO2 materials.5–9 The increasing environmental and energy issues in the present day world provoke researchers to synthesize new photocatalytic materials which can effectively degrade the pollutants under light irradiation. Heterogeneous photocatalysis is one type of catalysis which is of low cost and has great potential for applications in water and air purification.10,11 In spite of its advantages, the rapid recombination of electrons inside the lattice or on the surface of the photocatalyst limits its application and commercialization. Furthermore, to improve the efficiency, cocatalyst modified photocatalysis was studied as an effective strategy.12 The principle involved in the enhanced photocatalytic activity is the formation of a space-charge layer between the semiconductor and the cocatalyst, which enriches the charge separation rate.12,13 The cocatalyst plays an important role in the photocatalytic efficiency because it functions as an oxidation or reduction site to reduce the electron–hole pair recombination. Silver dioxide (Ag2O) has a wide range of applications in several industrial fields, as an electrode material, cleaning agent, preservative, colorant, and catalyst. It has a cubic structure with a = 4.72 Å and a reported band gap of approximately 1.2 eV. Ag2O has been studied by many researchers, and it acts as a good electron absorbing agent and as an efficient photosensitizer under UV and visible light irradiations, respectively.14,15 Thus, here, Ag2O is chosen as the cocatalyst for enhanced photocatalytic performance.
Recently, Jiang et al. have reported Y3Al5O12:Ce3+ (YAG:Ce3+) semiconductor particles mixed with TiO2 as an effective photocatalytic material.16 Similarly, Feng et al. also described the effect of YAG:Er3+ on the photocatalytic activity of TiO2 nanocrystals.17 The TiO2 particles showed enhanced photocatalysis with the addition of the semiconductor particles. More recently, Wang et al. have reported the influence of Ga-doped Er3+:YAG on the solar light photocatalytic activity of TiO2.18 It was demonstrated that Ga doping can widen the absorption range of visible or infrared light and give strong UV light emission which excites TiO2 as a photocatalyst. The solar light photocatalytic activity of Er3+:YAG in the degradation of organic dyes depends, to some extent, on Ga doping. In this context, we attempt to synthesize ternary compound Y3Ga5O12 garnet (YGG) samples. For the synthesis of ternary compounds, accurate reaction conditions should be maintained by precisely controlling the stoichiometry of the precursors. Although the stoichiometry of the precursors is precisely controlled, compound formation is not guaranteed since it is a multi-step process with intermediate stages. This process also requires relatively high temperatures which boost the formation of aggregates.19,20 The sol–gel process eliminates the problems of chemical inhomogeneity and the difficulties in the experimental process, and the products are obtained at unexpectedly low temperatures.21,22 On the contrary, the hydrothermal method paves the way for lowering the synthesis temperature, and a narrow grain size distribution with weak agglomeration is produced.23,24 Thus, the synthesis method in which the grinding and calcination processes are annihilated is required, and the products with less aggregation in a one step process are much anticipated.
To the best of our knowledge, there were no reports found on the hydrothermal synthesis of YGG nanoparticle samples. The photocatalytic properties of these materials were studied. In this work, to enhance the photocatalytic activity, the YGG nanoparticles were modified by Ag2O and the photocatalysis was performed using Rhodamine B (RhB) as an organic dye pollutant.
2. Experimental section
Materials
Yttrium nitrate hexahydrate (Y(NO3)3·6H2O), gallium nitrate hydrate (Ga(NO3)3·xH2O), ethylenediaminetetraacetic acid (EDTA), ammonia solution (NH4OH (28 wt%)), silver nitrate (AgNO3), and RhB were purchased from Sigma-Aldrich Co. All the chemicals were of high-purity grade and used as received without any further purification. The de-ionized (DI) water used in this experiment was obtained from a Milli-Q synthesis system (resistivity of 18.2 MΩ cm).
Synthesis of Y3Ga5O12 garnet nanoparticles.
For the synthesis of YGG nanoparticles, the precursor solution was prepared by adding stoichiometric amounts of yttrium nitrate hexahydrate, gallium nitrate hydrate and ammonia solution in 250 mL of DI water. After 10 min of continuous stirring, EDTA was added to the precursor solution, and then the solution was stirred for 1 h. The homogeneously mixed solution was transferred into an SUS-316 liner and placed in a stainless steel autoclave. Then, the temperature of the solution was raised to 200 °C from room temperature and maintained at this temperature for different reaction times. To study the growth mechanism of the nanoparticles, the experiment was repeated at different reaction time periods of 1, 3, 5, 10, 15, and 20 h. The proposed chemical reaction for the formation of YGG nanoparticles is shown below:| |  | (1) |
Synthesis of Ag2O modified Y3Ga5O12 garnet nanoparticles.
The Ag2O modified YGG nanoparticles were prepared by dispersing 0.5 g of as-prepared YGG nanoparticles into 10 ml of (0.1 M L−1) AgNO3 solution. The solution was stirred for 6 h under ambient atmospheric conditions, and the final product was collected and dried at 60 °C.
Characterization
The as-prepared samples were evaluated by using a field-emission scanning electron microscope (FE-SEM; LEO SUPRA 55, Carl Zeiss), a transmission electron microscope (TEM; JEM-2100F, JEOL), an X-ray diffractometer (XRD; Mac Science, M18XHF-SRA), and a Fourier transform infrared (FTIR) spectroscopy system. The luminescence properties were characterized by using a fluorospectrometer (FluoroMate FS-2, Scinco). The composition and chemical states of the Ag2O modified Y3Ga5O12 samples were characterized by using an X-ray photoelectron spectroscopy (XPS) system (Thermo Multi-Lab 2000 System).
Photocatalytic activity
The experiments intend to show the effect of the photocatalyst on organic pollutants under light illumination. Herein, RhB was used as an organic pollutant for conducting the photocatalytic experiments. The solution for photocatalysis was prepared by mixing 5 mg of dye in 1 L of DI water. Then, 0.05 g of photocatalyst material was added to 50 mL of 5 mg L−1 RhB dye solution. The solution was stirred under complete darkness for 30 min to reach an absorption balance. Subsequently, the solution was exposed to different light sources to observe the photocatalytic activity. UV light was obtained using a 300 W Xenon arc lamp (ORIEL Instruments) source with a short pass 400 nm filter (Edmund Optics, Barrington) and visible light with a cut-on 400 nm filter (FSQ-GG400, Newport Corporation). A solar simulator (SUN 3000, ABET) with a 300 W Xenon arc lamp was used for solar radiation. The solution was continuously stirred during the illumination process. The solution was then centrifuged to test the dye degradation percentage using a UV-vis spectrophotometer (CARY 300 Bio, Varian), and the experiment was conducted at different irradiation time periods.
3. Results and discussion
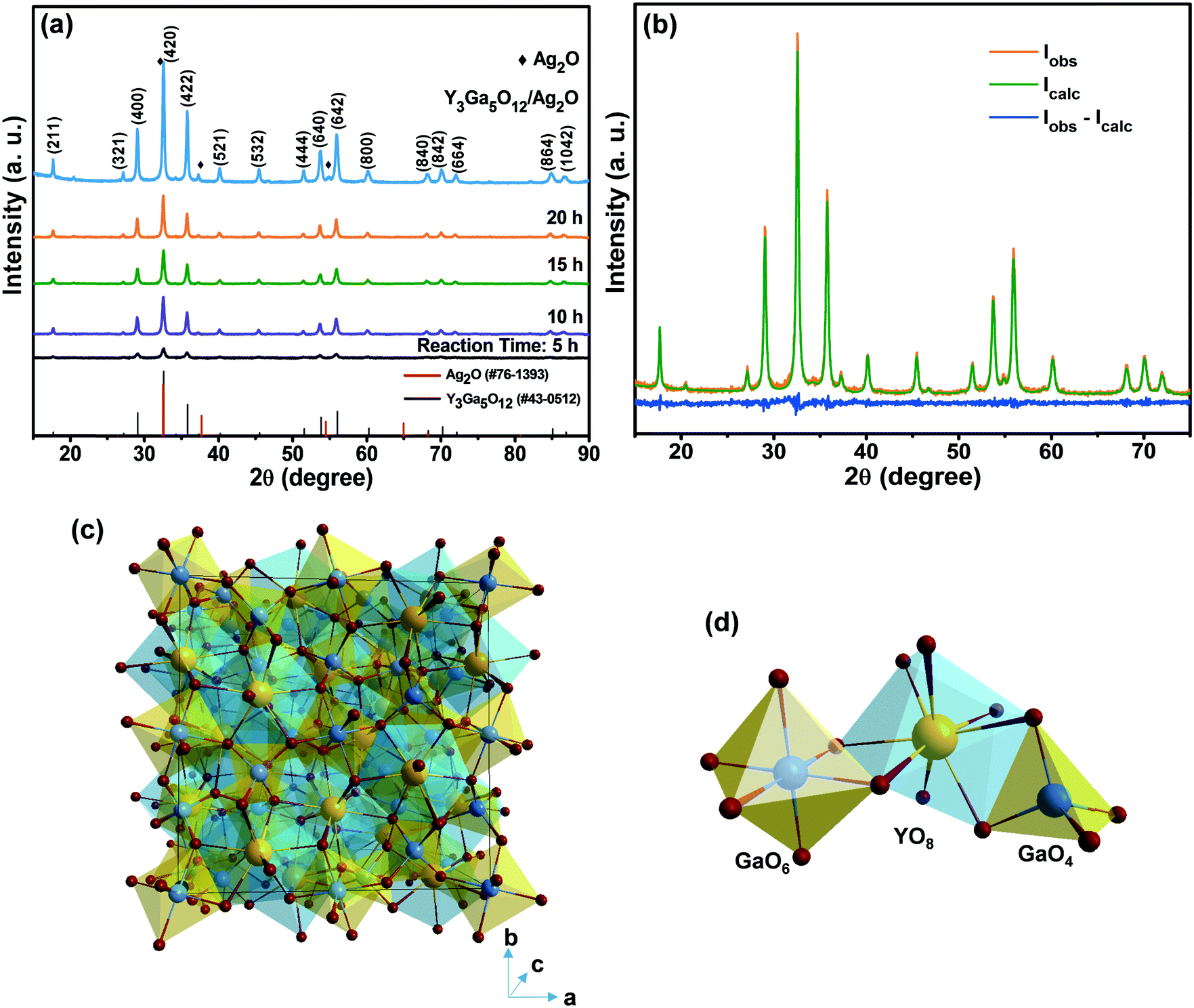
Fig. 1(a) shows the XRD patterns of the YGG samples synthesized at different reaction time periods of 5, 10, 15, and 20 h. The XRD patterns of all the YGG samples matched well with the standard JCPDS #43-0512, which confirms that the samples were crystallized in the cubic phase with the Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d (230) space group. There were no impurity peaks observed in the diffraction patterns which indicated that no mixed phases were formed, suggesting that the hydrothermal method is well suited for obtaining the pure phase YGG. The XRD pattern of the sample prepared at 5 h of reaction time exhibited the main diffraction peaks with weak intensity. This indicates the formation of YGG with low crystalline nature. As the reaction time increased, sharp and clear diffraction peaks appeared due to the improved crystalline nature of the YGG nanoparticles. The crystallite size was calculated by using the well-known Scherrer equation (Dhkl = 0.9λ/β
d (230) space group. There were no impurity peaks observed in the diffraction patterns which indicated that no mixed phases were formed, suggesting that the hydrothermal method is well suited for obtaining the pure phase YGG. The XRD pattern of the sample prepared at 5 h of reaction time exhibited the main diffraction peaks with weak intensity. This indicates the formation of YGG with low crystalline nature. As the reaction time increased, sharp and clear diffraction peaks appeared due to the improved crystalline nature of the YGG nanoparticles. The crystallite size was calculated by using the well-known Scherrer equation (Dhkl = 0.9λ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ) by taking the dominant diffraction peaks from the XRD pattern. The average crystallite sizes of the samples prepared at different growth time periods were found to be 18.4 nm (5 h), 25.2 nm (10 h), 29.1 nm (15 h), and 34.7 nm (20 h), respectively. The XRD pattern of Ag2O modified YGG nanoparticles is shown in Fig. 1(a) which is highly consistent with that of the YGG sample, implying that the Ag2O nanoparticles on the surface of spherical YGG nanoparticles have no effect on the crystal structure of YGG. Small peaks of Ag2O were observed in the XRD pattern of the Ag2O modified YGG, and the JCPDS card values of Ag2O (#76-1393) are also presented in Fig. 1(a). The XRD pattern was refined by the Rietveld method using FullProf software, as can be seen in Fig. 1(b). The observed peaks were in accordance with the calculated data with a small difference found in the intensity of a few peaks. The crystal structure parameters and the fitting parameters (χ2, Rp, Rwp) are presented in Table 1. These results reveal that the crystal structures of YGG nanoparticles are analogous to the bulk counterparts. For instance, the cell parameter (a = 12.27 Å) for the bulk sample matches well that for the nanoparticle sample (a = 12.299 Å). The crystal structure of YGG is shown in Fig. 1(c). The crystal structure contains GaO4 tetrahedra and GaO6 octahedra which are connected by corner sharing oxygen ions, leading to the formation of dodecahedral cavities which are filled with Y3+ ions. As a result, three distinct crystallographic cation sites are found in the YGG sample. In other words, the Ga3+ ions occupy the tetrahedral S4 site (24(d)) and the octahedral S6 site (16(a)), and the Y3+ ions occupy the dodecahedral D2 site (coordination number = 8). Each tetrahedral Ga3+ is connected to four octahedral Ga3+ (four fold coordination), and each octahedral Ga3+ is connected to six tetrahedral Ga3+ (six fold coordination). These Y3+ dodecahedron, Ga3+ tetrahedron, and Ga3+ octahedron are shown in Fig. 1(d). The individual anion (O2−) is connected to a pair of dodecahedra, a single tetrahedron, and a single octahedron occupying the 96(h) site.
cosθ) by taking the dominant diffraction peaks from the XRD pattern. The average crystallite sizes of the samples prepared at different growth time periods were found to be 18.4 nm (5 h), 25.2 nm (10 h), 29.1 nm (15 h), and 34.7 nm (20 h), respectively. The XRD pattern of Ag2O modified YGG nanoparticles is shown in Fig. 1(a) which is highly consistent with that of the YGG sample, implying that the Ag2O nanoparticles on the surface of spherical YGG nanoparticles have no effect on the crystal structure of YGG. Small peaks of Ag2O were observed in the XRD pattern of the Ag2O modified YGG, and the JCPDS card values of Ag2O (#76-1393) are also presented in Fig. 1(a). The XRD pattern was refined by the Rietveld method using FullProf software, as can be seen in Fig. 1(b). The observed peaks were in accordance with the calculated data with a small difference found in the intensity of a few peaks. The crystal structure parameters and the fitting parameters (χ2, Rp, Rwp) are presented in Table 1. These results reveal that the crystal structures of YGG nanoparticles are analogous to the bulk counterparts. For instance, the cell parameter (a = 12.27 Å) for the bulk sample matches well that for the nanoparticle sample (a = 12.299 Å). The crystal structure of YGG is shown in Fig. 1(c). The crystal structure contains GaO4 tetrahedra and GaO6 octahedra which are connected by corner sharing oxygen ions, leading to the formation of dodecahedral cavities which are filled with Y3+ ions. As a result, three distinct crystallographic cation sites are found in the YGG sample. In other words, the Ga3+ ions occupy the tetrahedral S4 site (24(d)) and the octahedral S6 site (16(a)), and the Y3+ ions occupy the dodecahedral D2 site (coordination number = 8). Each tetrahedral Ga3+ is connected to four octahedral Ga3+ (four fold coordination), and each octahedral Ga3+ is connected to six tetrahedral Ga3+ (six fold coordination). These Y3+ dodecahedron, Ga3+ tetrahedron, and Ga3+ octahedron are shown in Fig. 1(d). The individual anion (O2−) is connected to a pair of dodecahedra, a single tetrahedron, and a single octahedron occupying the 96(h) site.
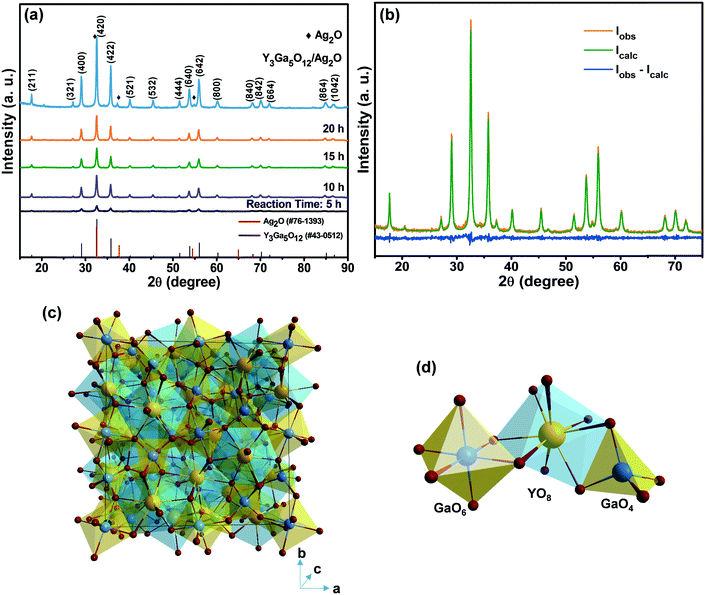 |
| | Fig. 1 (a) XRD patterns of the YGG samples prepared at different growth time periods and the Ag2O modified sample. (b) Rietveld refinement of YGG. (c) Unit cell structure of YGG. (d) Bonding arrangements of Y–O, Ga–O, GaO6, YO8, and GaO4. | |
Table 1 Crystallographic and Rietveld refinement data for the YGG structure
| Crystallographic data |
| Formula |
Y3Ga5O12 |
| Crystal system |
Cubic |
| Space group |
Iad (230) |
|
a (Å) |
12.299 |
| Volume (Å3) |
1860.72 |
| Rietveld data |
| Program |
FullProf suite |
| Range |
15–75° |
| Step |
0.02 |
|
R
p
|
12.7 |
|
R
wp
|
17.1 |
|
χ
2
|
6.51 |
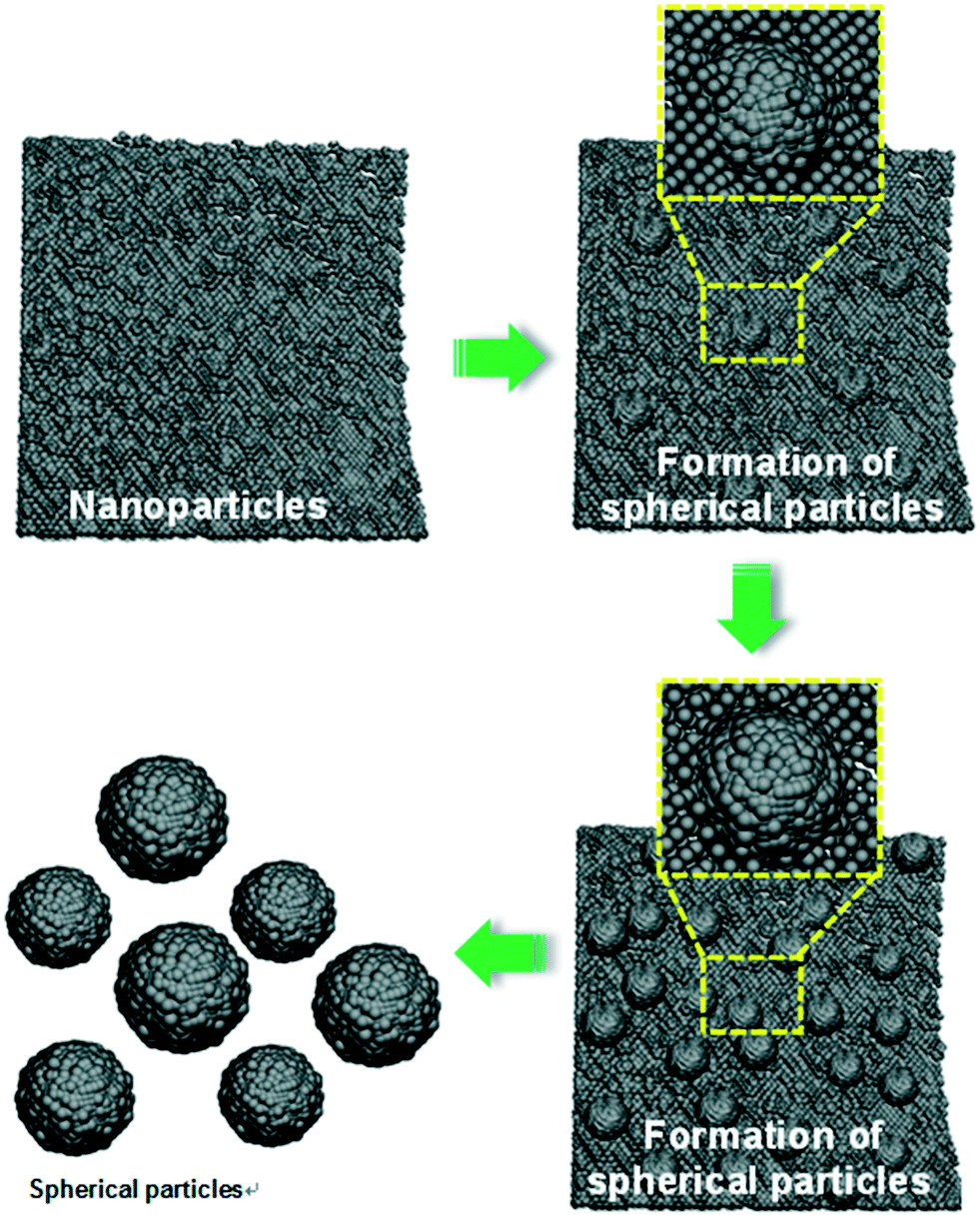
Fig. 2 shows the FE-SEM images of the YGG samples prepared at different growth time periods of 1, 3, 5, and 20 h, respectively. From the SEM images, it is clear that spherical nanoparticles were formed through the variation of growth time. In order to understand the role of EDTA, the YGG samples were prepared with and without EDTA. Without EDTA, the YGG sample exhibited the formation of nanosheets as shown in Fig. S1† and its morphology is similar to the shape found in the previous literature.25 When EDTA was introduced, the formation of smaller quasi-spherical shaped nanoparticles was observed.26,27 The formation of the nanoparticles is attributed to the adsorption of EDTA on the surface of YGG nuclei. The absorbed EDTA slows down the growth of YGG nuclei and produces quasi-spherical nanoparticles. Therefore, the particles further try to assemble/aggregate, resulting in the formation of spherical nanoparticles of sizes in the range of 100–300 nm. From these results, it is evident that EDTA plays an important role in the formation of spherical nanoparticles. The SEM images taken for the samples prepared without and with EDTA at 1 h of reaction time revealed a clear change in the morphology as can be seen in Fig. S1.† At 1 h of reaction time, many small quasi-spherical shaped nanoparticles were formed as shown in Fig. 2(a). When the reaction time was increased to 3 h, the aggregation/assemblage of nanoparticles occurred to minimize the surface energy (Fig. 2(b)). When the reaction time was increased to 5 h, the aggregation/assemblage process was accelerated and the amount of spherical shaped nanoparticles increased at the expense of smaller primary nanoparticles (Fig. 2(c)). When the reaction time was further increased, definite spherical shaped particles were seen due to the coalescence of primary nanoparticles and the enhanced aggregation/assemblage process. However, at longer reaction time (20 h), the spherical nanoparticles became bigger due to the dominant Ostwald ripening process, as shown in Fig. 2(d). The SEM images at longer reaction times of 10, 15 and 20 h showing the Ostwald ripening process are displayed in Fig. S2.† From the obtained results, we are able to describe the growth mechanism of YGG spherical nanoparticles by involving the sequence of simultaneous nucleation, aggregation/assembly, and Ostwald ripening process.
 |
| | Fig. 2 FE-SEM images of the YGG samples synthesized with EDTA at different growth time periods of (a) 1 h, (b) 3 h, (c) 5 h, and (d) 20 h. | |
Fig. 3 shows the schematic diagram for the growth mechanism of YGG spherical nanoparticles. Initially, a huge number of small nanoparticles are formed due to nucleation. As the reaction time increases, the nanoparticles try to assemble/aggregate. At higher reaction time, the assemblage/aggregation process is accelerated and almost spherical shaped particles are observed. Finally, when the reaction time reaches 20 h, definite spherical shaped YGG nanoparticles are formed due to the aggregation/assemblage and Ostwald ripening process.
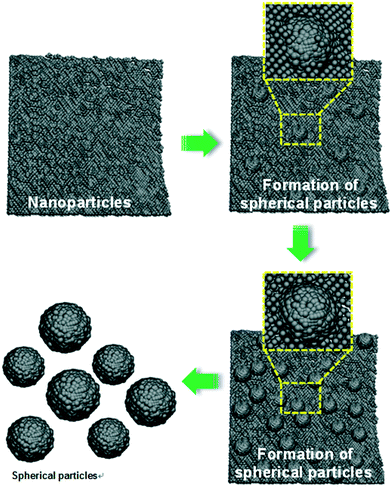 |
| | Fig. 3 Schematic diagram showing the formation of spherical YGG nanoparticles. | |
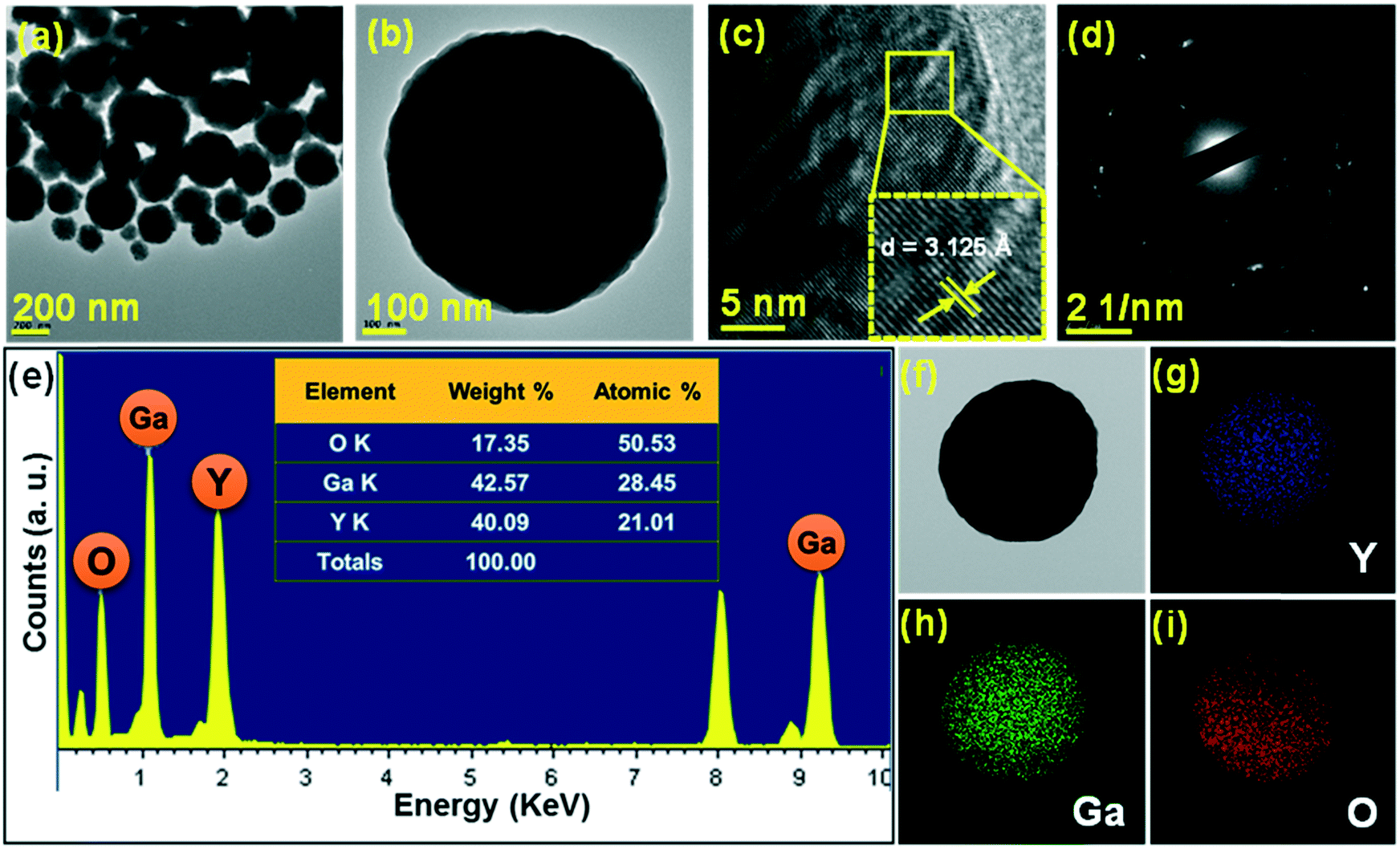
Furthermore, the analysis of crystal and morphological properties was carried out by performing TEM measurements as shown in Fig. 4. The distinct spherical nanoparticle formation was observed as shown in the low-magnification TEM image (Fig. 4(a)). The magnified image of a single spherical nanoparticle is shown in Fig. 4(b). The high-resolution TEM (HR-TEM) image taken for the sample revealed that the nanoparticles which aggregate to form spherical structures are highly crystalline and these crystalline nanoparticles assemble to form spherical structures. The lattice fringes appeared clearly due to the crystalline nature of the nanoparticles. The d-spacing calculated from the HR-TEM image was 3.125 Å (Fig. 4(c)), and it matched well with the XRD data corresponding to the (400) plane. The selective area electron diffraction (SAED) pattern (Fig. 4(d)) displayed a ring pattern which is formed by many dots, suggesting that the spherical particles formed are nanocrystalline in nature.28 The spatial distribution of elements in the YGG nanoparticles was studied by elemental mapping and energy dispersive X-ray (EDX) analysis. Fig. 4(e) shows the EDX spectrum obtained for a single spherical nanoparticle. The table inside the EDX spectrum shows the weight% and atomic% distributions of the elements present in the spherical nanoparticle sample. To establish the chemical distribution in the spherical nanoparticle, elemental mapping was carried out for the single spherical nanoparticle, as shown in Fig. 4(f–i). This complete two-dimensional image of chemical zonation depicts the uniform distribution of Y, Ga and O elements in the spherical nanoparticles.
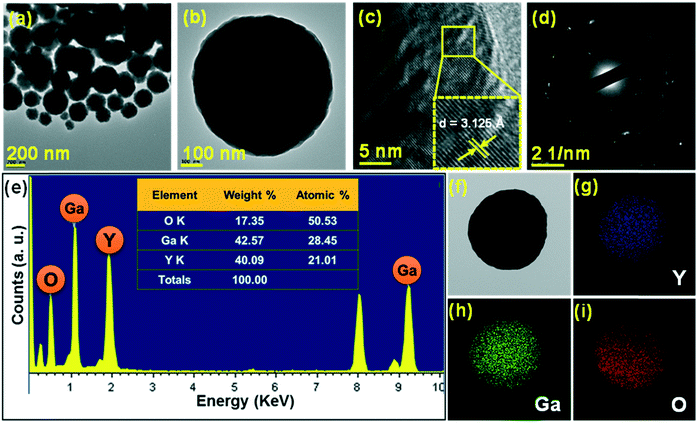 |
| | Fig. 4 (a) Low-magnification TEM image, (b) high-magnification TEM image, (c) HR-TEM image, (d) SAED pattern, and (e) EDX spectrum of the YGG sample prepared at 20 h of reaction time. (f) TEM image and elemental mappings of (g) yttrium (Y), (h) gallium (Ga), and (i) oxygen (O). | |
FTIR spectra within the wavenumber range of 4000–450 cm−1, for samples prepared at different hydrothermal growth time periods of 5, 10, 15, and 20 h, respectively, are shown in Fig. S3.† From the FTIR spectra, the broad band in the longer wavenumber region (3200 cm−1) is due to the H–O–H stretching vibrations of the water molecule.29 The sharp and intense absorption peak around 1384 cm−1 is related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration because of the use of EDTA in the growth process.23 The band around 1597 cm−1 is due to the O–H stretching vibration, and N–H deformation vibrations are also observed due to the use of ammonia in the experiment.30 Below 1000 cm−1, we can observe two bands related to the Ga–O stretching and Ga–O–Ga bending modes. The intense band around 625 cm−1 is due to the Ga–O stretching vibration, and the weak band below 500 cm−1 corresponds to the Ga–O–Ga bending vibration.31–33
O stretching vibration because of the use of EDTA in the growth process.23 The band around 1597 cm−1 is due to the O–H stretching vibration, and N–H deformation vibrations are also observed due to the use of ammonia in the experiment.30 Below 1000 cm−1, we can observe two bands related to the Ga–O stretching and Ga–O–Ga bending modes. The intense band around 625 cm−1 is due to the Ga–O stretching vibration, and the weak band below 500 cm−1 corresponds to the Ga–O–Ga bending vibration.31–33
The morphologies of the Ag2O modified YGG powders were characterized by SEM and TEM as shown in Fig. 5. Fig. 5(a) shows the schematic diagram for the formation of Ag2O nanoparticles around the surface of spherical YGG nanoparticles. The SEM image of Ag2O modified YGG nanoparticles is shown in Fig. 5(b). This indicates that the Ag2O nanoparticles were coated on the surface of all the spherical YGG nanoparticles. Fig. 5(c)–(f) show the TEM images of the Ag2O modified YGG sample. In the TEM image (Fig. 5(c)), it can be seen that the Ag2O nanoparticles were all over the surface of spherical YGG nanoparticles. The HR-TEM image is shown in Fig. 5(d), which displays two different lattice fringes for YGG and Ag2O. The lattice fringes were magnified as shown in Fig. 5(e) and (f), and the planes matched well to the JCPDS card values of YGG (#43-0512) and Ag2O (#76-1393), respectively. Elemental mappings of the Ag2O modified YGG sample are shown in Fig. 5(g–k), indicating the uniform distribution of Y, Ga, O, and Ag elements in the sample. The spatial distribution of elements was studied by the EDX spectrum which is shown in Fig. S4.† The linear compositional variation of the Ag2O modified YGG was further supported by the line scan mapping as shown in Fig. S5.†
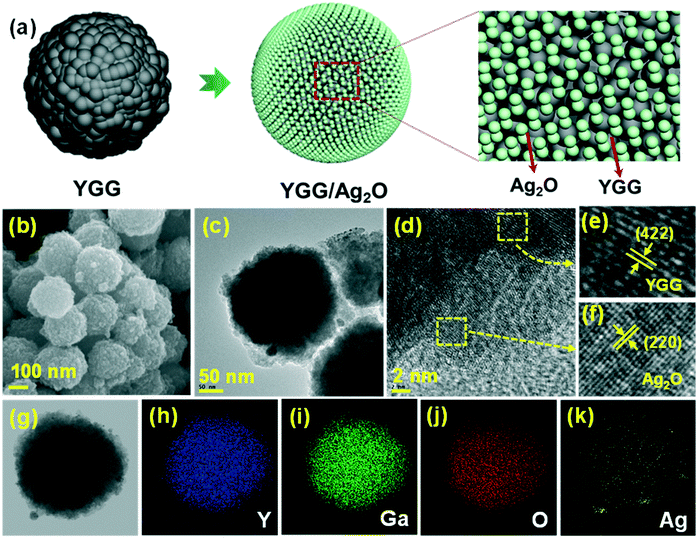 |
| | Fig. 5 (a) Schematic diagram showing the arrangement of Ag2O nanoparticles on YGG nanospheres. (b) SEM image, (c–f) TEM images, and (g–k) elemental mappings. | |
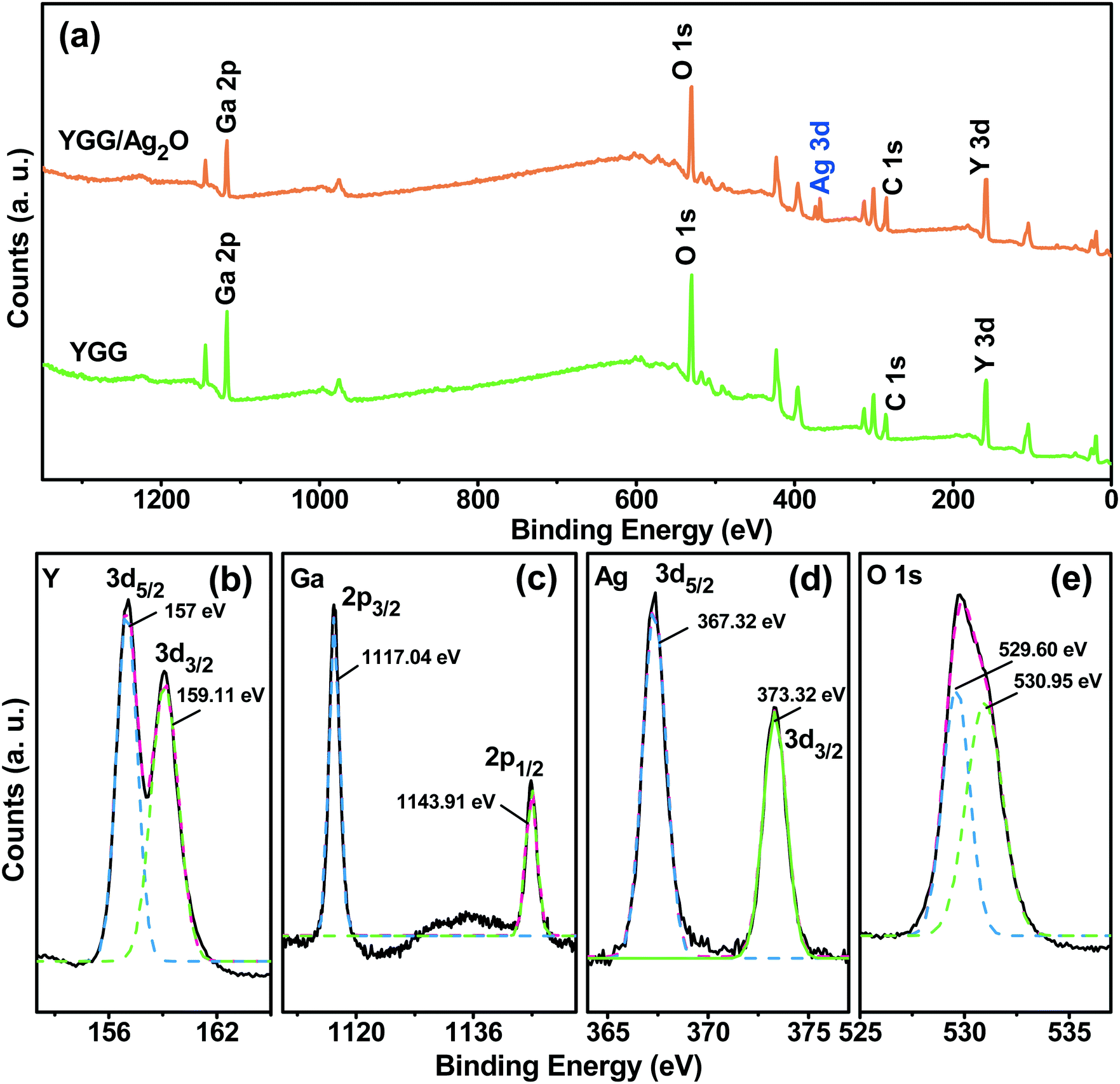
Fig. 6 shows the XPS spectra of the YGG and Ag2O modified YGG samples to study the electronic states of the elements and elemental compositions. The survey scan spectra shown in Fig. 6(a) exhibited distinct elements of Y, Ga, and O for the YGG, while the Ag2O modified YGG sample showed an extra element, Ag, along with Y, Ga, and O, signifying the presence of Ag2O. The trace amounts of C in the XPS spectra are due to the extrinsic hydrocarbon during the XPS measurement. Fig. 6(b)–(e) show the high-resolution XPS curves of the Y 3d, Ga 2p, Ag 3d, and O 1s for the corresponding samples. Herein, all the spectral curves were fitted with a Gaussian method. As shown in Fig. 6(b), the peaks at binding energy values of 157 and 159.11 eV for Y 3d5/2 and 3d3/2 revealed the +3 oxidation state of Y. The spin orbit splitting of 2.11 eV is observed, and it is in agreement with previous reports.34 In addition, the Ga 2p spectrum exhibited the spin–orbit splitting into 2p3/2 and 2p1/2 at binding energy values of 1117.8 and 1143.5 eV, and the O 1s peak at 530.39 eV was observed, indicating that the Ga exists in the +3 oxidation state. The high-resolution spectrum in Fig. 6(d) shows two distinct peaks at 368.2 and 374.6 eV, which are attributed to the Ag 3d5/2 and 3d3/2 spin–orbit doublets of the Ag 3d. In general, the oxidation states of Ag(I) and Ag(0) are mainly observed in the binding energy values of 367.5–368.4 eV, which confirms that the Ag exists in the +1 oxidation state and it is similar to the values reported for Ag+ in Ag2O.35 Clearly, the asymmetric O 1s spectrum of the Ag2O modified YGG showed two peaks (Fig. 6(e)), i.e. one main peak at 529.6 eV along with a shoulder peak at 530.95 eV unlike the O 1s of YGG which indicates the formation of Ag2O.36,37 The O 1s spectra of the YGG and the Ag2O modified YGG are shown in Fig. S6.† The quantitative analysis of atomic percentage (at%) of elements present in the YGG and Ag2O modified YGG samples is shown in Table S1.†
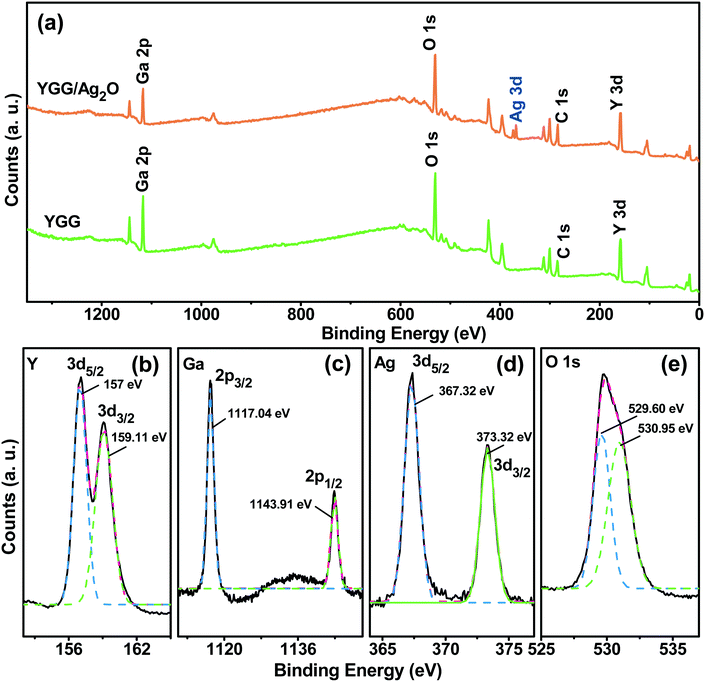 |
| | Fig. 6 (a) Survey scan XPS spectra of the YGG and Ag2O modified YGG samples, and high-magnification XPS spectra of (b) Y 3d, (c) Ga 2p, (d) Ag 3d, and (e) O 1s. | |
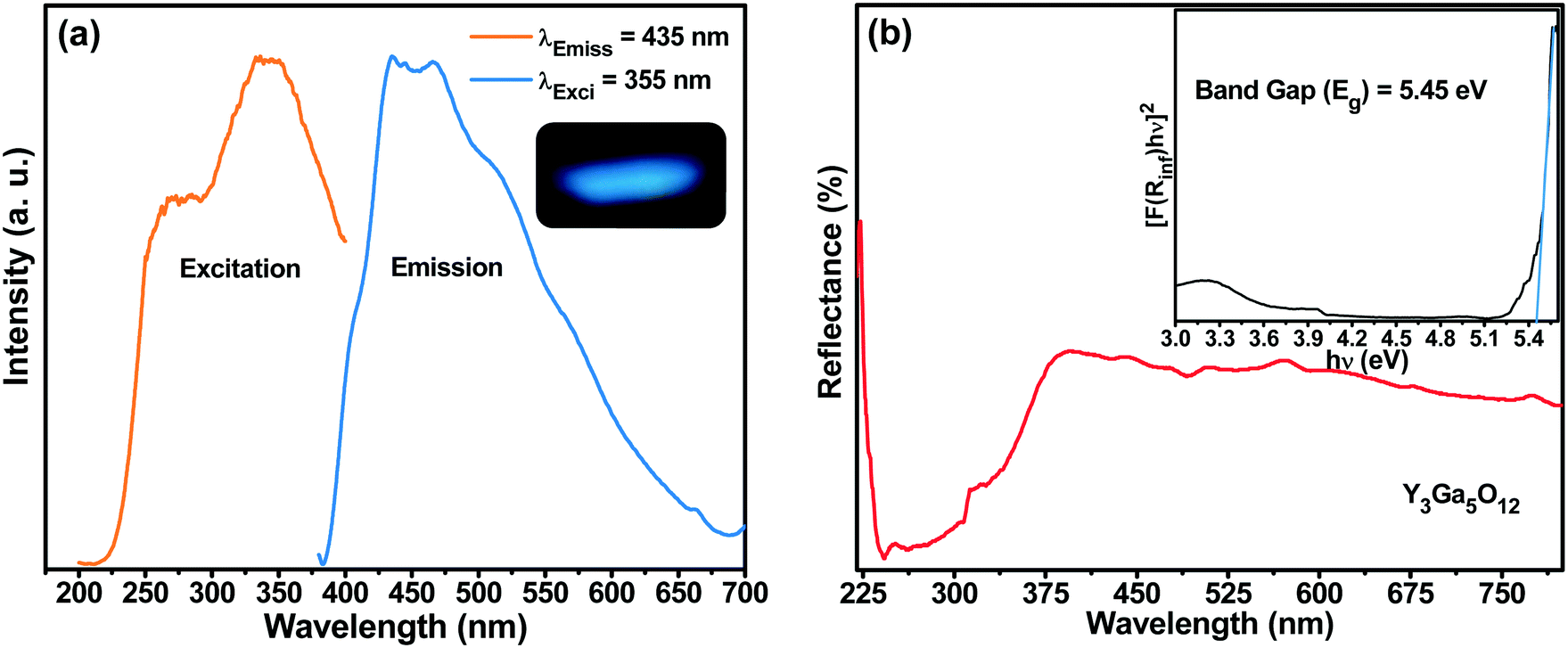
The photoluminescence (PL) excitation (PLE) and emission spectra of the as-prepared spherical YGG nanoparticle sample are shown in Fig. 7(a). The PLE spectrum was measured within the wavelength range of 200 to 415 nm by monitoring the emission wavelength of 435 nm. The excitation spectrum shows a broad band centered at a wavelength of 355 nm. Likewise, the PL emission spectrum was observed at an excitation wavelength of 355 nm. A broad band from 390 to 650 nm centered at 435 nm was observed in the blue region of the emission spectrum. A similar blue emission by the host lattice has also been reported in YGG:Dy3+ phosphors by Chengli et al. This is attributed to the presence of self-activated Ga–O luminescent centers in the host lattice.38 In addition, there were other previous reports where they have shown the same broad band in the blue region of the PL emission spectrum. Krishna et al. showed that the blue emission from ZnGa2O4 crystals is due to the distorted octahedral Ga–O groups which act as self-activated luminescent centers.39 The (Gd2O3)x(Ga2O3)(1−x) thin films exhibited a broad band which stretches from 350 to 500 nm, and the emission peak changed with the x concentration. As the x concentration was increased to ∼37.5%, the emission peak was shifted from 430 to 397 nm and it remained constant with further increase.40 Lü et al. also reported the blue emission from the Gd3Ga5O12 host lattice when observed at an excitation wavelength of 335 nm.32 Furthermore, in reports on Ga2O3 under UV light illumination, the blue emission was observed due to the presence of oxygen vacancies.41–43 The facts mentioned above imply that the broadband blue emission observed for the as-prepared YGG nanoparticle sample is attributed to the Ga–O groups existing in the host lattice.
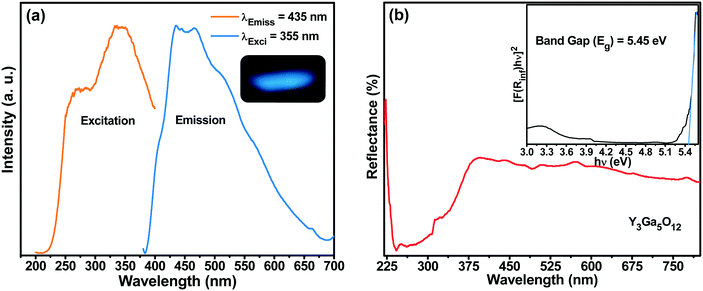 |
| | Fig. 7 (a) PLE and PL emission spectra of the YGG sample (the inset shows the emission color of the sample). (b) DR spectra of the YGG sample (the inset shows the band gap of the samples). | |
Fig. 7(b) shows the diffused reflectance (DR) spectrum within the range of 220–800 nm for the YGG nanoparticle sample. The band observed below 400 nm is due to the host material. Based on the DR spectrum, the band gap of the sample was calculated from the well-known Kubelka–Munk theory. The Kubelka–Munk function F(R∞) is given as follows:44
| | 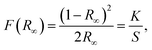 | (2) |
where
R∞ is the diffuse reflectance of the sample. It is the ratio of scattered light from a layer of sample and an ideal non-absorbing reference sample as a function of wavelength (
λ) (
R∞ =
Rsample/
Rreference).
K is the absorption coefficient and
S is the scattering coefficient. The well-known Tauc relation which relates the energy band gap (
Eg) and the linear absorption coefficient (
α) of a material is given below:
where
ϑ is the photon energy and
A1 is constant.
K becomes 2
α when the material scatters in a perfectly diffuse manner.
S is considered as a constant with respect to
λ, and utilizing
eqn (2) and
(3), the following equation can be obtained:
| | | (F(R∞)hϑ)2 = A2(hϑ − Eg)n. | (4) |
Here, the values of
n = 1, 3, 4, 6 for direct allowed, direct forbidden, indirect allowed and indirect forbidden transitions, respectively, and
n = 2 for non-metallic materials. The plot is drawn between (
F(
R∞)
hϑ)
n and
hϑ for different values of
n and it fits best for
n = 1, suggesting the nature of band transitions as direct.
44 The band gap was found to be 5.45 eV as shown in the inset of
Fig. 7(b).
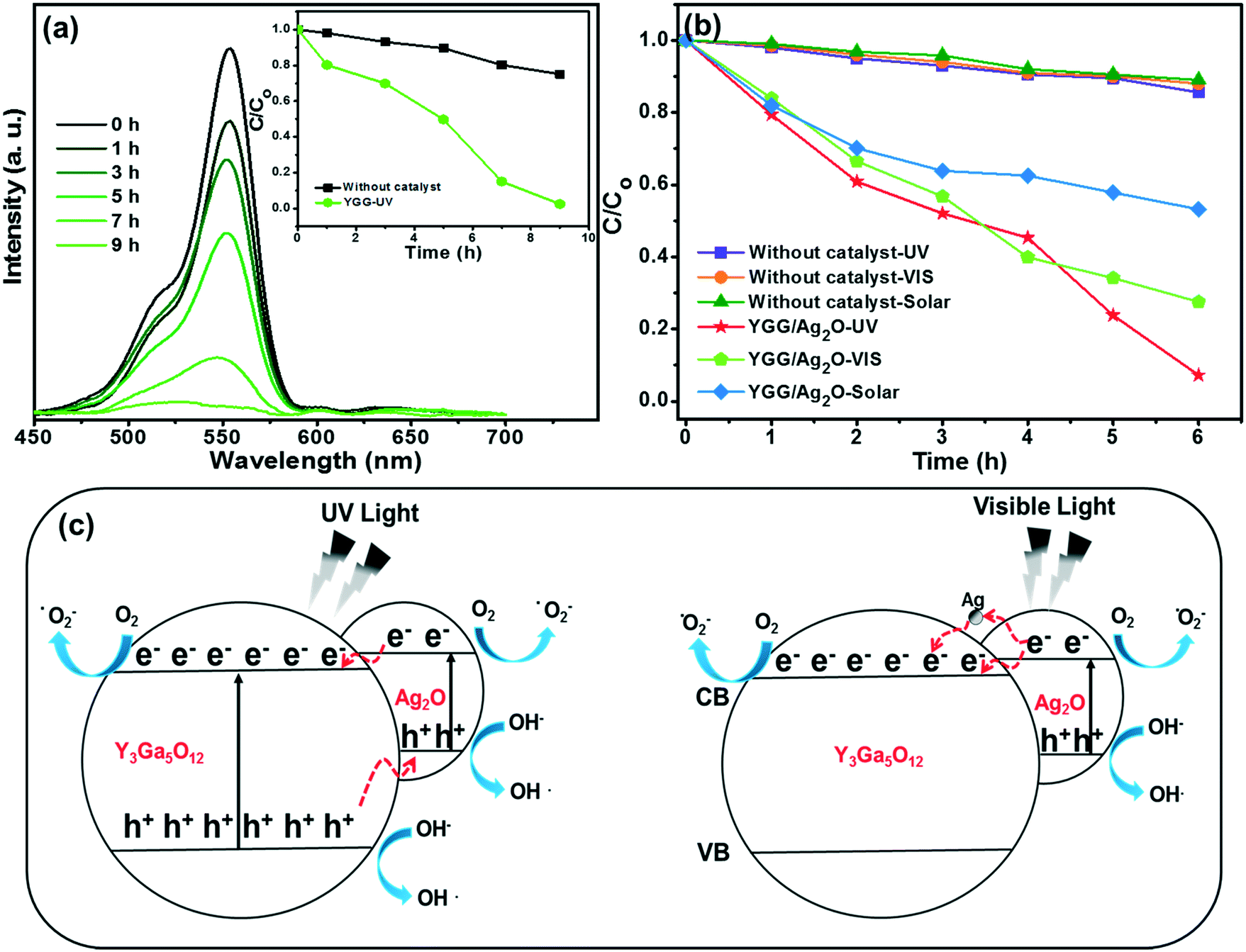
In order to establish the photocatalytic activity of YGG nanoparticles, the YGG sample prepared at the reaction time of 20 h was chosen. Fig. 8(a) shows the absorption spectra of the RhB dye solution with the YGG photocatalyst under UV light irradiation. The absorption peak of the dye was observed in the wavelength range of 450 to 600 nm centered at 553 nm. The peak intensity decreased as the irradiation time increased, and the absorption peak almost disappeared at 9 h of irradiation time, indicating the complete degradation of the RhB dye. The photodegradation rate (C/Co) in the presence and absence of the photocatalyst is shown in the inset of Fig. 8(a). Here, C and Co are the intensities of the RhB absorption peak in the presence and absence of UV light. The photodegradation rates of 97.5 and 25% were obtained for the RhB dye solutions with and without the YGG photocatalyst at the irradiation time of 9 h. The photodegradation of the dye in the presence of YGG takes a long time of 9 h for its complete degradation. To enhance the degradation efficiency, the YGG particles were modified with Ag2O. Fig. 8(b) shows the photocatalytic activity of the Ag2O modified YGG sample under UV light irradiation. As shown in Fig. 8(b), it is clear that the photodegradation efficiency was improved and the dye was degraded within the time of 6 h. The reaction kinetics of photocatalytic degradation can be obtained by the pseudo-first order rate equation:
where
k is the rate constant and
t is the irradiation time. The plot was drawn between ln(
C/
Co) and irradiation time and fitted linearly as shown in Fig. S8.
† The estimated rate constant and linear correlation coefficient (
R2) are shown in Table S2.
† The
R2 values were found to be greater than 0.8988, supporting the pseudo-first order rate kinetics. When UV light is irradiated, the YGG spherical nanoparticles are excited and generate electrons (e
−) and holes (h
+). These e
−–h
+ pairs recombine quickly; hence, the photocatalytic activity is very low for YGG spherical nanoparticles. When the Ag
2O modified YGG sample was irradiated with UV light, both the YGG and Ag
2O get excited. The photogenerated e
− move from the conduction band of Ag
2O to YGG and h
+ from the valance band of YGG to Ag
2O, thus efficiently separating e
− and h
+. The e
− and h
+ separation was further confirmed by taking the PL spectra of the YGG and the Ag
2O modified YGG. The PL spectrum actually corresponds to the e
− and h
+ transfer behavior, consequently symbolizing the recombination and separation of e
− and h
+. The PL intensity of the Ag
2O modified YGG is less than that of the original YGG sample, as shown in Fig. S7.
† This suggests that the recombination of exciton pairs was limited. Therefore, the excited e
− and h
+ lifetime can be extended, giving rise to higher photocatalytic activity than the as-prepared YGG spherical nanoparticles. Later on, the e
− react with adsorbed H
2O and O
2 and produce superoxide radical anions like O
![[2 with combining dot above]](https://www.rsc.org/images/entities/char_0032_0307.gif) −
−. The h
+ were held by OH
− and H
2O, resulting in the production of OH species (strong oxidizing agent). Moreover, the loaded Ag
2O generates O
2 species which elevate the creation of OH species, and enhances the photocatalytic activity under UV irradiation.
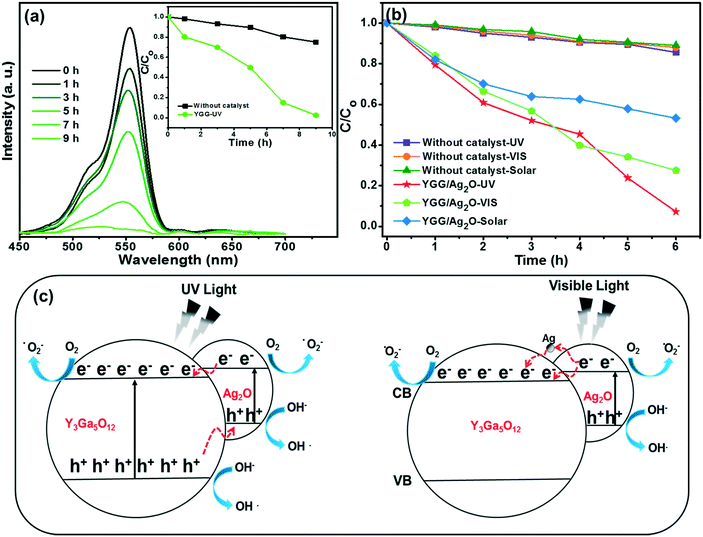 |
| | Fig. 8 (a) UV/vis absorption spectra of the RhB solution in the presence of YGG spherical nanoparticles under UV radiation. The inset of (a) shows the photodegradation rate curves. (b) Photodegradation rates of the RhB dye solution in the presence and absence of Ag2O modified YGG nanoparticles under different light radiations. (c) Photocatalysis mechanism under UV and visible light. | |
Subsequently, when the Ag2O modified YGG particles are illuminated with visible light, Ag2O alone gets excited due to its small band gap (1.2 eV). The excited e− move from the conduction band of Ag2O to YGG and the h+ remain in the valence band, thus separating them efficiently. Furthermore, the photogenerated e− are transferred to lattice Ag+ due to the higher positive potential of Ag+/Ag. The Ag+ acts as a sacrificial reagent for the efficient evolution of oxygen. In the present work, the trace amount of Ag+ in Ag2O was reduced to metallic Ag in situ by photogenerated e− during the experiment. The e− are transferred from Ag2O to metallic Ag and then to YGG, leading to the separation of e− and h+. Therefore, photocatalytic activity is observed under visible light. The possible mechanism supposed for enhanced photocatalytic activity is schematically shown in Fig. 8(c). At the same time, the relevant formula reactions are as follows:45–47
| Y3Ga5O12 + hv → Y3Ga5O12 + e− + h+ |
| Ag2O + hv → Ag2O + e− + h+ |
The photocatalytic activity of the Ag2O modified YGG sample was also tested under simulated solar radiation as shown in Fig. 8(c). Among the different light sources used in this experiment, the visible light has the shortest penetration depth, and thus the masking effect was major.48 Besides, the simulated solar radiation contains both UV light and visible light. It may affect significantly the photocatalytic activity of the Ag2O modified YGG sample. On the other hand, since the simulated solar radiation contains a larger portion of visible light than UV light, the simulated solar radiation cannot penetrate efficiently into the sample, leading to lower photocatalytic activity. Therefore, efficient photocatalytic activity is observed under UV light irradiation for the Ag2O modified YGG sample. This indicates that the improved photocatalytic activity is primarily due to efficient absorption of light and photoinduced charge separation.
4. Conclusions
The YGG spherical nanoparticles were successfully prepared by a single-step hydrothermal process. Under 355 nm excitation, the YGG sample exhibited a broadband blue emission with a band maximum at 435 nm. This is attributed to the self-activated Ga–O luminescent centers present in the host lattice. The photocatalytic activity of this sample was examined with the RhB dye solution under UV light irradiation. To enhance the photocatalytic activity, Ag2O was loaded onto the surface of YGG spherical nanoparticles. The Ag2O modified YGG spherical nanoparticles showed enhanced photocatalytic activity under UV light, and they also revealed photocatalytic activity under visible and solar light. The improved photocatalytic activity was due to the effective charge separation. The present work is expected to furnish the development of new complex oxide semiconductor materials and the use of a cocatalyst for its effective degradation of organic pollutants.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) Grant funded by the Korean government (MISP) (No. 2015R1A5A1037656 and No. 2013R1A1A2010037).
References
-
J. Coronado, in Design of Advanced Photocatalytic Materials for Energy and Environmental Applications, ed. J. M. Coronado, F. Fresno, M. D. Hernández-Alonso and R. Portela, Springer, London, 2013, ch. 1, pp. 1–4, DOI:10.1007/978-1-4471-5061-9_1.
- L. Bruner and J. Kozak, Z. Elektrochem. Angew. Phys. Chem., 1911, 17, 354–360 CAS.
- E. Keidel, Farben-Ztg., 1929, 34, 1242–1243 CAS.
- C. Doodeve and J. Kitchener, Trans. Faraday Soc., 1938, 34, 902–912 RSC.
- I. Izumi, W. W. Dunn, K. O. Wilbourn, F.-R. F. Fan and A. J. Bard, J. Phys. Chem., 1980, 84, 3207–3210 CrossRef CAS.
- M. Anpo, Pure Appl. Chem., 2000, 72, 1265 CAS.
- Z. Lin, A. Orlov, R. M. Lambert and M. C. Payne, J. Phys. Chem. B, 2005, 109, 20948–20952 CrossRef CAS PubMed.
- X.-Y. Zhang, H.-P. Li, X.-L. Cui and Y. Lin, J. Mater. Chem., 2010, 20, 2801–2806 RSC.
- J. Fei and J. Li, Adv. Mater., 2015, 27, 314–319 CrossRef CAS PubMed.
- A. Ibhadon and P. Fitzpatrick, Catalysts, 2013, 3, 189 CrossRef CAS.
- C. S. Lima, K. A. Batista, A. García Rodríguez, J. R. Souza and K. F. Fernandes, Sol. Energy, 2015, 114, 105–113 CrossRef CAS.
- H. Yu, R. Liu, X. Wang, P. Wang and J. Yu, Appl. Catal., B, 2012, 111–112, 326–333 CrossRef CAS.
- H. Chu, X. Liu, J. Liu, J. Li, T. Wu, H. Li, W. Lei, Y. Xu and L. Pan, Mater. Sci. Eng., B, 2016, 211, 128–134 CrossRef CAS.
- P. Kamal Kumar, G. Ramesh and P. K. Giri, Nanotechnology, 2016, 27, 315703 CrossRef PubMed.
- R. Kumar, R. El-Shishtawy and M. Barakat, Catalysts, 2016, 6, 76 CrossRef.
- X. Jiang, Y. Wang and C. Pan, J. Alloys Compd., 2011, 509, L137–L141 CrossRef CAS.
- G. Feng, S. Liu, Z. Xiu, Y. Zhang, J. Yu, Y. Chen, P. Wang and X. Yu, J. Phys. Chem. C, 2008, 112, 13692–13699 CAS.
- J. Wang, Y. Li, L. Zhang, J. Q. Gao, B. X. Wang, Q. Yang and P. Fan, Russ. J. Phys. Chem. A, 2014, 88, 149–157 CrossRef CAS.
- H. Shi, C. Zhu, J. Huang, J. Chen, D. Chen, W. Wang, F. Wang, Y. Cao and X. Yuan, Opt. Mater. Express, 2014, 4, 649–655 CrossRef.
- H. K. Yang and J. H. Jeong, J. Phys. Chem. C, 2010, 114, 226–230 CAS.
- L. K. Bharat, L. S. Reddy and J. S. Yu, Mater. Lett., 2015, 144, 85–89 CrossRef CAS.
- H. Wu, Y. Hu, W. Zhang, F. Kang, N. Li and G. Ju, J. Sol-Gel Sci. Technol., 2012, 62, 227–233 CrossRef CAS.
- L. Krishna Bharat, B. Vengala Rao and J. S. Yu, Chem. Eng. J., 2014, 255, 205–213 CrossRef CAS.
- X. Jiang, Y. Pan, S. Huang, X. A. Chen, J. Wang and G. Liu, J. Mater. Chem. C, 2014, 2, 2301–2306 RSC.
- Q. Chen and X. Shen, Cryst. Growth Des., 2010, 10, 3838–3842 CAS.
- X. Yang, Z. Fu, G. Liu, C. Zhang, Y. Wei, Z. Wu and T. Sheng, RSC Adv., 2015, 5, 70220–70228 RSC.
-
R. Viswanatha and D. D. Sarma, in Nanomaterials Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2007, ch. 4, pp. 139–170, DOI:10.1002/9783527611362.
- L. Krishna Bharat, J. Y. Park and J. S. Yu, Chem. Eng. J., 2014, 240, 179–186 CrossRef CAS.
- A. C. Taş, P. J. Majewski and F. Aldinger, J. Am. Ceram. Soc., 2002, 85, 1421–1429 CrossRef.
- L. Krishna Bharat, S. H. Lee and J. S. Yu, Mater. Res. Bull., 2014, 53, 49–53 CrossRef CAS.
- V. Venkatramu, S. F. Leon-Luis, U. R. Rodriguez-Mendoza, V. Monteseguro, F. J. Manjon, A. D. Lozano-Gorrin, R. Valiente, D. Navarro-Urrios, C. K. Jayasankar, A. Munoz and V. Lavin, J. Mater. Chem., 2012, 22, 13788–13799 RSC.
- W. Lü, H. Zhou, G. Chen, J. Li, Z. Zhu, Z. You and C. Tu, J. Phys. Chem. C, 2009, 113, 3844–3849 Search PubMed.
- V. Monteseguro, M. Rathaiah, K. Linganna, A. D. Lozano-Gorrín, M. A. Hernández-Rodríguez, I. R. Martín, P. Babu, U. R. Rodríguez-Mendoza, F. J. Manjón, A. Muñoz, C. K. Jayasankar, V. Venkatramu and V. Lavín, Opt. Mater. Express, 2015, 5, 1661–1673 CrossRef.
- D. Majumdar and D. Chatterjee, J. Appl. Phys., 1991, 70, 988–992 CrossRef CAS.
- O. Ahmed Zelekew and D.-H. Kuo, Phys. Chem. Chem. Phys., 2016, 18, 4405–4414 RSC.
- C.-M. Liu, J.-W. Liu, G.-Y. Zhang, J.-B. Zhang, Q.-S. Wu, Y.-Y. Xu and Y.-Q. Sun, RSC Adv., 2015, 5, 32333–32342 RSC.
- N. Liang, M. Wang, L. Jin, S. Huang, W. Chen, M. Xu, Q. He, J. Zai, N. Fang and X. Qian, ACS Appl. Mater. Interfaces, 2014, 6, 11698–11705 CAS.
- C. Sun and C. Tu, Mater. Express, 2012, 2, 145–150 CrossRef CAS.
- K. M. Krishna, G. Anoop and M. K. Jayaraj, J. Electrochem. Soc., 2007, 154, J310–J313 CrossRef CAS.
- X. Xu, Z. Xu, Y. Hou, Y. Su, X. Xu, X. Wang and W. Tong, Appl. Phys. Lett., 2000, 77, 672–674 CrossRef CAS.
- G. Li, C. Peng, C. Li, P. Yang, Z. Hou, Y. Fan, Z. Cheng and J. Lin, Inorg. Chem., 2010, 49, 1449–1457 CrossRef CAS PubMed.
- V. Chirmanov, P. C. Stanish, A. Layek and P. V. Radovanovic, J. Phys. Chem. C, 2015, 119, 5687–5696 CAS.
- B. K. Kang, H. D. Lim, S. R. Mang, K. M. Song, M. K. Jung, S.-W. Kim and D. H. Yoon, Langmuir, 2015, 31, 833–838 CrossRef CAS PubMed.
- S. K. Sharma, S. Som, R. Jain and A. K. Kunti, J. Lumin., 2015, 159, 317–324 CrossRef CAS.
- S. Gu, Y. Chen, X. Yuan, H. Wang, X. Chen, Y. Liu, Q. Jiang, Z. Wu and G. Zeng, RSC Adv., 2015, 5, 79556–79564 RSC.
- S. Ma, J. Xue, Y. Zhou and Z. Zhang, J. Mater. Chem. A, 2014, 2, 7272–7280 CAS.
- W. Zhou, H. Liu, J. Wang, D. Liu, G. Du and J. Cui, ACS Appl. Mater. Interfaces, 2010, 2, 2385–2392 CAS.
- W. R. Siah, H. O. Lintang, M. Shamsuddin, H. Yoshida and L. Yuliati, Catal. Sci. Technol., 2016, 6, 5079–5087 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FE-SEM images of YGG samples synthesized in the absence and presence of EDTA, high-magnification FE-SEM images at different reaction times, FTIR spectra of YGG nanoparticles prepared at different reaction times, EDX spectrum, line scan mapping, and XPS spectrum of Ag2O modified YGG nanoparticles; PL spectra of YGG and Ag2O modified YGG; a table consisting of XPS survey data (at%) for the elements of YGG and Ag2O modified YGG. See DOI: 10.1039/c6ce01988a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a