DOI:
10.1039/C6CE01078D
(Paper)
CrystEngComm, 2016,
18, 7965-7971
Single-crystal-to-single-crystal transformation of the desolvation of a cyclotriveratrylene–acetonitrile inclusion complex via a gating mechanism with subsequent polymorphism†
Received
8th May 2016
, Accepted 12th September 2016
First published on 12th September 2016
Abstract
A cyclotriveratrelyne–acetonitrile inclusion complex is shown to undergo a single-crystal-to-single-crystal transformation upon desolvation to an intermediate phase and it is furthermore demonstrated that the preparation conditions may introduce polymorphism of the apohost. We characterised the as-synthesized and desolvated polymorphs using single crystal (polymorph 1) and powder X-ray diffraction, thermal analysis and gas sorption. Successive data collections on the as-synthesized crystal at 50 °C show that the rotation of a single, unique methoxy group of the host molecule may allow evacuation of solvent molecules from the seemingly non-porous crystal.
Introduction
Cyclotriveratrylene (CTV), a cyclic trimer of veratrole, acts as a bowl-shaped, molecular host in its crown conformation, which is capable of binding ions or small organic/organometallic molecules.1–3 CTV (Scheme 1) has a triangular pyramid shape with the aromatic rings forming three faces of the pyramid and the methylene groups positioned at the apices.4–7 CTV is related to calix[n]arenes (where n is most commonly 4, 6 or 8) but is less common in terms of the variety of inclusion complexes formed and their diversity of applications.1,8 Molecules based on a CTV scaffold are common and have been used in a variety of applications in both soft materials and supramolecular chemistry.1,9 These include being used as constituents of discrete, cage-like, metallo-supramolecular assemblies, such as a self-entangled Solomon's cube10 and assemblies of triply interlocking cage-catenane species,11,12 and coordination polymers. CTV and its analogues have also been utilized as constituents in self-assembled and coordination supramolecular systems, and as precursors for cavitands and cryptophanes,1,13–19 where they bind smaller organometallic and organic guests.20–22 Their capability to host guests such as xenon, lanthanides and C60 and anionic C70 dimers has led to the growth of functionalized CTV derivatives for biomedical applications such as the distribution of fullerenes and their use in MRI-based diagnostic techniques.23–29 To date, very little work has been done on porous structures incorporating CTV or functionalized CTV. Porosity studies have been performed on conjugate microporous polymers and porous organic polymers that incorporate CTV.30,31 These have been shown to display modest hydrogen uptake of 0.5–0.8 wt% at 77 K and ∼1 bar with surface areas of 200–350 m2 g−1. Pd3L2 metallo-cryptophane cages with CTV-type L ligands, which are stabilized by a bis-N-heterocyclic carbene as an auxiliary cis-protecting ligand, have been shown to take up 1,2-dichlorobenzene or iodine via a single-crystal-to-single-crystal (SCSC) process.32 SCSC transformations allow the determination of crystal structures of materials after physical events such as desolvation/resolvation or polymorphic transformations and may thus yield important insights into mechanisms of transformation, which may help in the design of new materials.33 It is still a relatively rare phenomenon; however, increasing examples are being reported, especially for metal–organic frameworks (MOFs), including in our own laboratory.34,35 The higher prevalence for MOFs may be due to the greater degree of robustness of the crystal structure afforded by coordination bonds; nevertheless, examples of purely organic systems undergoing SCSC transformations in porosity studies have been reported.36,37 Herein we present, the synthesis of a CTV–acetonitrile inclusion compound, which is seemingly non-porous due to its isolated cavities. The inclusion compound can be desolvated into two different polymorphs, depending on the desolvation conditions. The solvated and desolvated forms have been characterized by X-ray diffraction, thermal analysis and gas sorption.
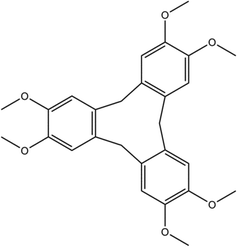 |
| | Scheme 1 Chemical structure of cyclotriveratrylene (1). | |
Experimental
Materials
A formaldehyde solution (>34.5 wt%) and veratrole (>99%) were purchased from Sigma Aldrich and used as received. CTV was synthesized via a literature method.38
Crystal growth
CTV (60 mg) was dissolved in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v acetonitrile:chloroform (5 mL) with stirring. Slow evaporation of the solvents leads to the appearance of crystals after 7–10 days.
:1 v/v acetonitrile:chloroform (5 mL) with stirring. Slow evaporation of the solvents leads to the appearance of crystals after 7–10 days.
Single crystal X-ray diffraction analysis and structure determination
Single crystals of suitable quality were selected and mounted on a cryoloop in oil. Data collections were carried out on a Bruker DUO APEX II CCD diffractometer using graphite monochromated MoKα (λ = 0.71073 Å) radiation with the crystal cooled to 173 K or 153 K using an Oxford Cryostream-700. Data reduction and cell refinement were performed using SAINT-Plus.39 The X-ray diffraction data were scaled and corrected for absorption effects using SADABS.40 The structure was solved by direct methods, implemented in SHELXS-97.41 Refinement, via the OLEX2 interface,42 proceeded by the full-matrix least-squares method, based on F2 values against all reflections as implemented in SHELXL-2014/7 with anisotropic displacement parameters for all non-H atoms.41 X-Seed and MERCURY were used for generating high-quality images using POV-RAY.43–45 PLATON was used to determine hydrogen bonding parameters as well as, together with MSROLL, void spaces using specified probe radii.46,47
Powder X-ray diffraction
Powder X-ray diffraction (PXRD) data were collected on a Bruker D8 Advance X-ray diffractometer with copper radiation (CuKα, λ = 1.5406 Å) at 30 kV and 40 mA. Each sample was scanned between 4 and 40° 2θ with a step size of 0.02°. MERCURY was used to calculate PXRD patterns from the single crystal data for comparison with experimentally obtained PXRD patterns.
Thermal analysis
Hot-stage microscopy (HSM) was performed using a Nikon SMZ-10 stereoscopic microscope fitted with a Linkam THMS600 hot stage and a Linkam TP92 control unit. Samples were placed under silicone oil on a cover slip and heated at 10 °C min−1. Thermal events were monitored with a Sony Digital Hyper HAD colour video camera and captured using the Soft Imaging System program analySIS. Differential scanning calorimetry (DSC) was performed using a DSC Q200 instrument. The sample (∼4 mg) was weighed directly into a pierced aluminium pan on an analytical balance. Thermogravimetric analysis (TGA) was performed using a TGA Q500 instrument. The sample (∼3.7 mg) was weighed directly into open aluminium crucibles. During the DSC and TGA experiments, the sample holder was continuously purged with a dry nitrogen flow of 60 mL min−1, whilst a heating rate of 10 °C min−1 was employed.
Results & discussion
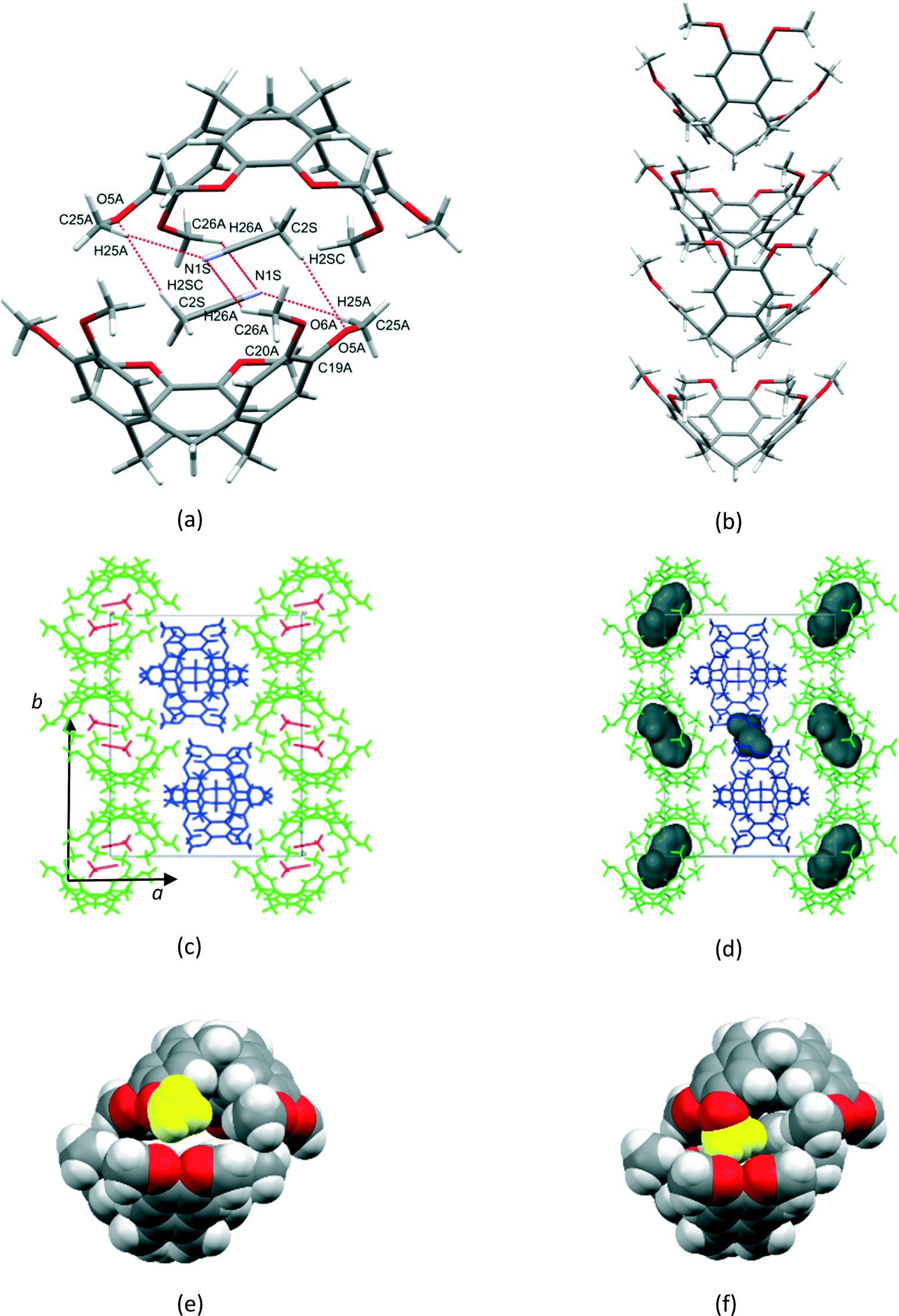
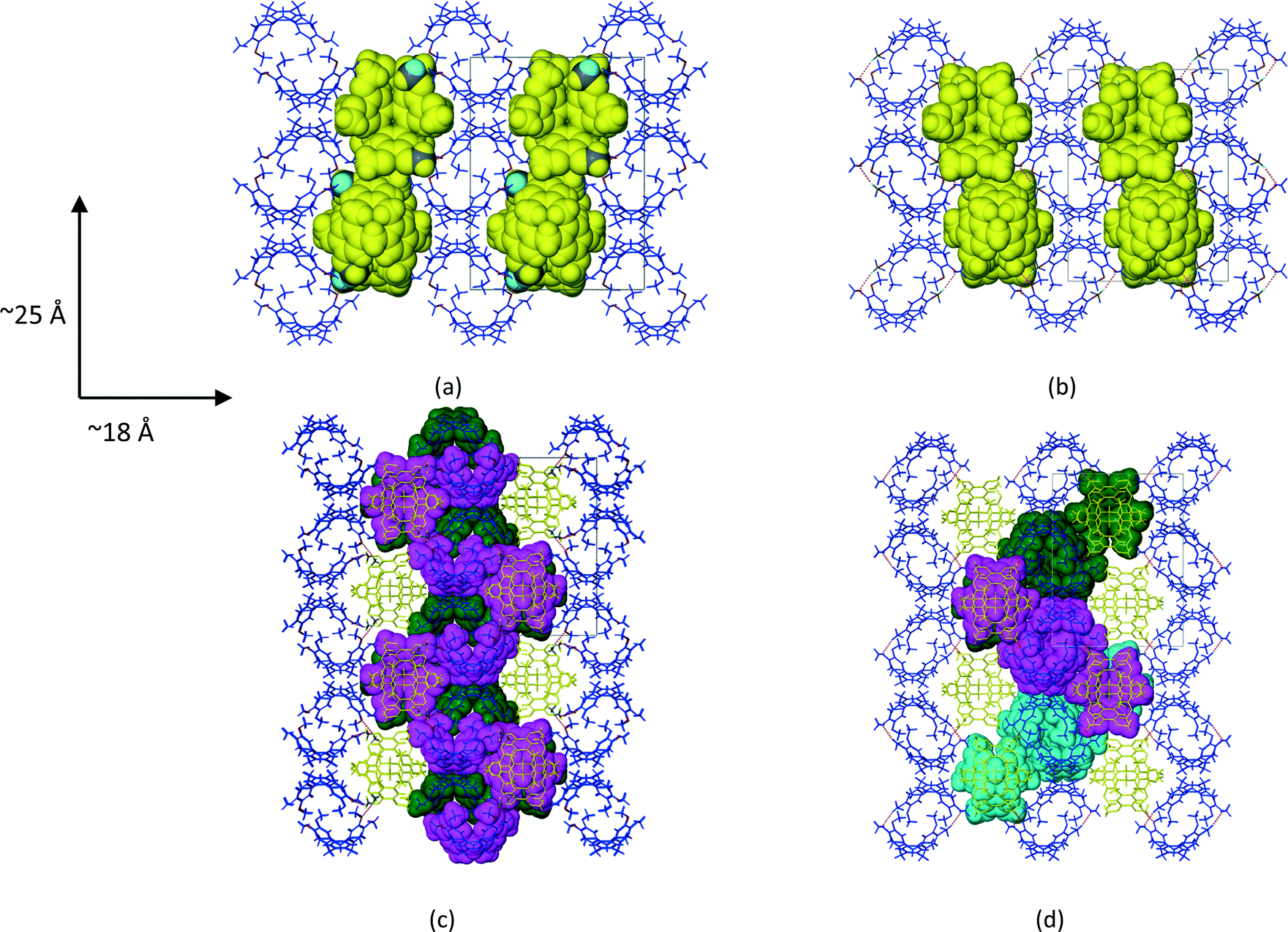
1a crystallized from a 1:1 v/v mixture of acetonitrile and chloroform. Single crystal X-ray structure analyses show that the asymmetric unit (ASU) of 1a consists of two CTV molecules and one acetonitrile molecule (chloroform was excluded from the crystal structure but was added for solubilisation of 1). The two unique CTV molecules play two different structural roles in the crystal structure with one forming centrosymmetric, dimeric capsules which encapsulate two acetonitrile molecules per dimer (Fig. 1a), whilst the second self-includes to form columns of stacked CTV molecules (Fig. 1b). The dimers and stacked columns are arranged in 2D layers in the bc plane which stack along the a-axis (Fig. 1c). No classical hydrogen bonds are present in the structure of 1a; however, five unique C–H⋯O hydrogen bonds seem to play an important role in host–solvent and host–host interactions (Table S1†). The centrosymmetric dimer is indirectly held together by six host–solvent C–H⋯O/C–H⋯N hydrogen bonds. Four of these hydrogen bonds (C25A–H25A⋯N1S and C26A–H26A⋯N1S) involve the acetonitrile nitrogen atom acting as a bifurcated hydrogen bond acceptor for host methyl hydrogen bond donors, whilst the remaining two hydrogen bonds (C2S–H2SC⋯O5A) involve the host methoxy oxygen atom acting as a hydrogen bond acceptor for a methyl hydrogen bond donor of the solvent molecule. In addition, the acetonitrile molecule also forms two unique C–H⋯π interactions with the dimer CTV molecule (Table S1†). Subjecting crystals of 1a under a dynamic vacuum at 90 °C for 24 h resulted in the desolvated form 2a, a process which occurred via a SCSC transformation. The asymmetric unit of 2a consists of two crystallographically independent CTV molecules, as was the case for 1a; however, the absence of appreciable electron density in the difference Fourier map indicated the absence of acetonitrile. This was confirmed by thermogravimetric analysis, whilst the SQUEEZE routine in PLATON indicated a residual electron density of only 6 electrons per unit cell. The void space for 2a (Fig. 2d) calculated by PLATON (using a probe radius of 1.2 Å) is 4.6% per unit cell and is largely due to the cavity of the dimer, with a much smaller portion of this space (0.8% per unit cell) due to the space between the columns of stacked CTV molecules. This 4.6% void space is smaller than the 7.2% potential void space calculated for 1a if the acetonitrile molecules are artificially removed, which indicates that structural changes occurred upon solvent removal. The voids are isolated in 2a (as well as in 1a with the solvent molecules artificially removed) and it is reasonable to suggest that the 1a to 2a transition involves evacuation of the acetonitrile molecules along the ∼10 Å axis with at least one unique rotatable methoxy groups acting as ‘gates’. We show that this is plausible in a simple fashion by taking one methoxy group of a dimer CTV molecule in 1a (O6A–C26A) and artificially rotating it by 75° about the C20A–O6A bond so that C26A points away from the dimer cavity. This resulted in the void space of two neighbouring cavities connecting (Fig. S1†). The hypothesis is also supported by the fact that the two corresponding methoxy groups in the actual structures are found in significantly different orientations in 1a and 2a as indicated by the torsion angles C26A–O6A–C20A–C19A and C7A–O1A–C1AS–C2A of −170.0(1)° and −80.4(1)° for 1a and 2a, respectively, indicating that this methoxy group is rotated in the crystal structures. In 2a the corresponding methoxy group O1A–C7A points towards the interior of the dimer cavity which is allowed by the absence of the acetonitrile solvent molecule (Fig. 1e and f). In 1a two unique C–H⋯O hydrogen bonds (C16B–H16D⋯O1A and C25B–H25E⋯O5A) serve to link the two unique CTV molecules so that 1D, zigzag strands of connected CTV molecules of both types are formed along the ∼25 Å axis (Fig. 2c). In 2a, CTV molecules of a dimer are now directly connected to each other by one unique C–H⋯O hydrogen bond (C8A–H8AB⋯O5A), indicative of the lateral shift of these molecules. A second unique C–H⋯O hydrogen bond (C7B–H7BB⋯O6A) connects the dimer to a ‘stack’ CTV molecule on both sides of the dimer. Unlike in 1a, these ‘stack’ CTV molecules are not connected to a second dimer resulting in hydrogen-bonded tetramers instead (Fig. 2d and S2†). These tetramers which are related to each other by the crystallographic 21 axis along the b-direction (Fig. 2d and Table 1).
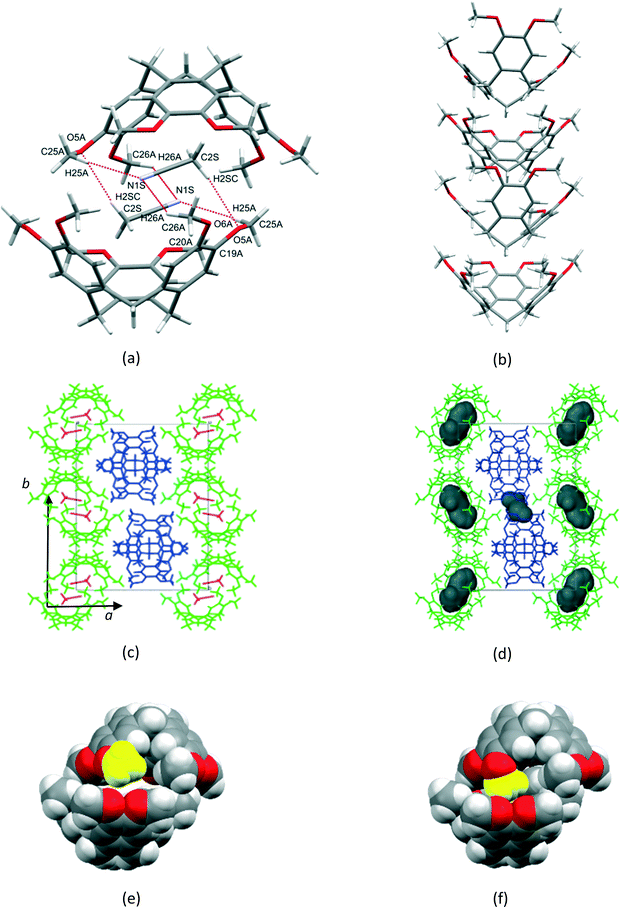 |
| | Fig. 1 (a) Centrosymmetric dimer formed by ASU CTV molecule 1 (b) self-inclusion of ASU CTV molecule 2 (c) crystal packing in 1a (d) crystal packing in 2a showing isolated voids (e) dimer in 1a with the methyl group coloured in yellow corresponding to methyl group in (f) 2a that is rotated towards the interior of the cavity. | |
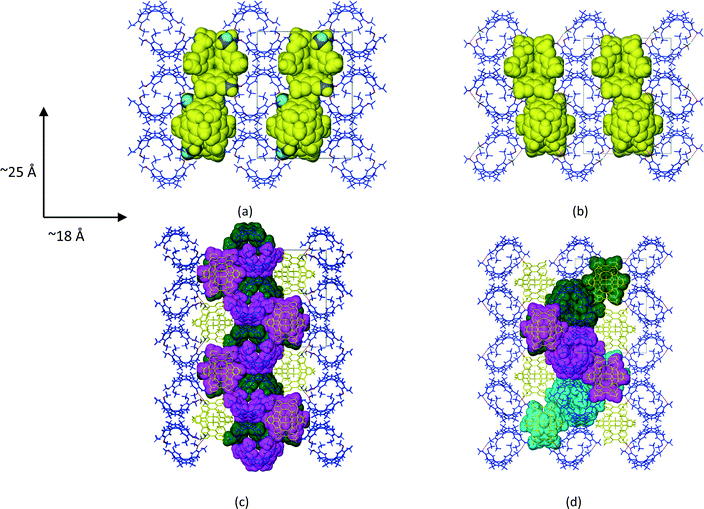 |
| | Fig. 2 Packing diagrams of CTV molecules in (a) 1a and (b) 2a with ‘stacked’ CTV molecules shown in space-filling mode. (c) CTV molecules in 1a and (d) 2a connected by C–H⋯O hydrogen bonding shown in space-filling mode and the same colour. All views are down the ∼10 Å axis. | |
Table 1 Crystal data and refinement parameters for 1a, 2a and 3a
|
|
1a
|
2a
|
| Empirical formula |
C28H31.5N0.5O6 |
C27H30O6 |
| Formula weight |
471.04 |
450.51 |
| Temperature/K |
173.15 |
153.15 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/c |
P21/c |
|
a/Å |
18.7531(8) |
18.606(2) |
|
b/Å |
25.0061(11) |
24.551(3) |
|
c/Å |
10.5276(5) |
10.4027(13) |
|
α/° |
90 |
90 |
|
β/° |
91.2590(10) |
90.819(3) |
|
γ/° |
90 |
90 |
| Volume/Å3 |
4935.6(4) |
4751.5(10) |
|
Z
|
8 |
8 |
|
ρ
calc g cm−3 |
1.268 |
1.26 |
|
μ/mm−1 |
0.089 |
0.088 |
|
F(000) |
2008 |
1920 |
| Crystal size/mm3 |
0.30 × 0.20 × 0.10 |
0.6 × 0.33 × 0.24 |
| Radiation |
MoKα (λ = 0.71073 Å) |
MoKα (λ = 0.71073 Å) |
| 2Θ range for data collection/° |
2.714 to 59.034 |
2.746 to 52.944 |
| Index ranges |
−25 ≤ h ≤ 25, −34 ≤ k ≤ 32, −14 ≤ l ≤ 14 |
−23 ≤ h ≤ 22, −30 ≤ k ≤ 30, −12 ≤ l ≤ 13 |
| Reflections collected |
112892 |
48505 |
| Independent reflections |
13782 [Rint = 0.0792, Rsigma = 0.0468] |
9790 [Rint = 0.0362, Rsigma = 0.0310] |
| Data/restraints/parameters |
13782/0/635 |
9790/0/607 |
| Goodness-of-fit on F2 |
1.023 |
1.027 |
| Final R indexes [I ≧ 2σ (I)] |
R
1 = 0.0476, wR2 = 0.1117 |
R
1 = 0.0444, wR2 = 0.1005 |
| Final R indexes [all data] |
R
1 = 0.0925, wR2 = 0.1322 |
R
1 = 0.0696, wR2 = 0.1122 |
| Largest diff. peak/hole/e Å−3 |
0.36/−0.25 |
0.68/−0.38 |
Variable temperature X-ray diffraction studies
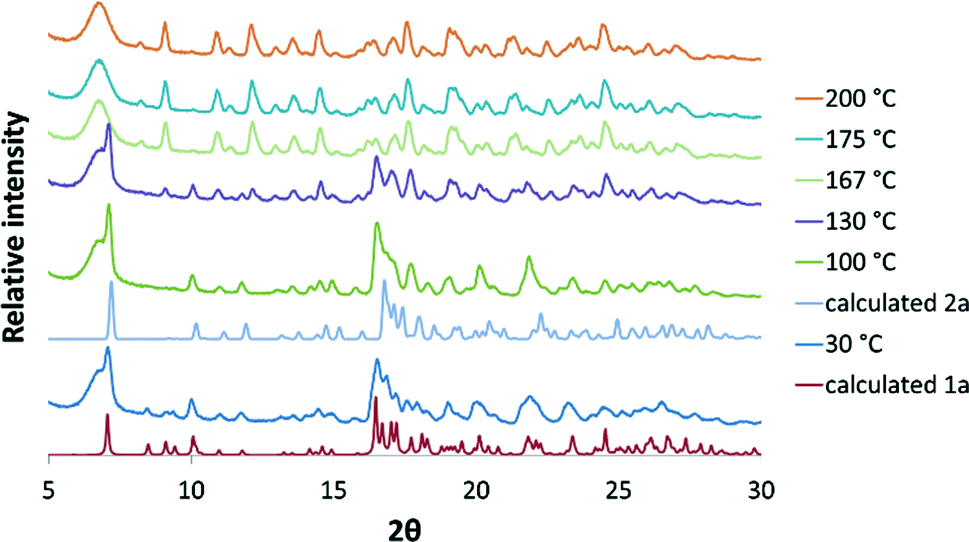
The solvated form 1a, could also be prepared by gentle grinding of CTV in the presence of acetonitrile (without chloroform) for approximately 20 minutes. Variable temperature PXRD experiments (VT-PXRD) performed on 1a, prepared in this manner, over the temperature range 30–210 °C indicated that the sample is crystalline over the entire temperature range of analysis (Fig. 3). The 30 °C PXRD pattern agrees closely with the calculated PXRD pattern from the single crystal structure of 1a, indicating that this method of producing 1a was successful. At 100 °C the three peaks at 8.4, 8.9 and 9.2 degrees 2θ are reduced and the pattern matches that of the calculated 2a pattern more closely. From 130–200 °C the gradual appearance of peaks at 8.2 and 9.1 degrees 2θ indicates that 2a transforms to another polymorph 3a.
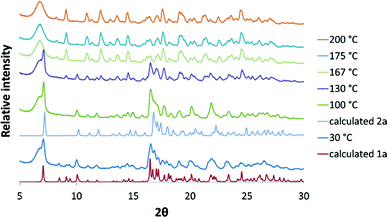 |
| | Fig. 3 VT-PXRD patterns in the range 30–200 °C compared with the calculated patterns of 1a and 2a. | |
We also performed in situ variable-temperature single crystal X-ray diffraction (SCXRD) experiments on 1a in order to investigate the possibility of following the SCSC transformation to 2a and subsequently 3ain situ. Heating was achieved by the flow of hot dry air over the crystal using the diffractometer's cryostream. At first we collected unit cell parameters at temperatures from 25–110 °C. It was clear that after 100 °C that the crystal was no longer a single crystal as indicated by the drastic loss of intensity of the diffraction spots. Keeping in mind that the experimental conditions are very different from how 2a was produced initially (90 °C under vacuum vs. the flow of continuous dry air over the crystal), we adjusted our approach by doing successive data collections at 50 °C on the same crystal (2 hours required for a complete data set). We then completely solved and refined the host structure and subjected each structure to a SQUEEZE analysis to ascertain the electron count per unit cell for each data collection. We present these results in Fig. 4 and Table S2.† Successive data collections show a continuous reduction in the electron count per unit cell indicative of the loss of the acetonitrile from the crystal. The count after the 24 hours (12 data collections) was 18 electrons per unit cell which is slightly more than the 6 electrons per unit cell calculated for the structure of the crystal subjected to a temperature of 90° under vacuum for 24 hours. Furthermore, after 6 hours the position of C26A had to be modelled over two positions (second position labelled C27A) indicating dynamic rotation of this methyl group. The electron density of C27A points towards the interior of the dimer cavity (as in the case of 2a in Fig. 1f) as this space becomes available to allow a degree of self-inclusion. The fact that the rotation away from the dimer cavity is not observed, as suggested in Fig. S1† for the cavities to ‘connect’, indicates that this rotation is short-lived compared to the rotation towards the interior of the cavity. Nevertheless, the observed disorder supports a ‘gating’ hypothesis for movement of molecules in and out of the structure. The in situ SCXRD experiments prove that the transformation from 1a to 2a can occur via a SCSC transformation under the appropriate conditions. We could not show that this could be the case for the transition from 2a to 3a and could therefore not determine the structure of the latter. However, we suggest that 3a is probably a higher-density form than 2a by virtue of a greater degree of self-inclusion.
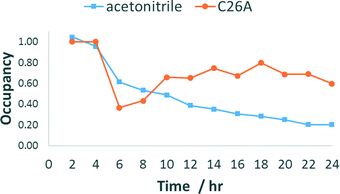 |
| | Fig. 4 Occupancy of acetonitrile and C26A position after successive in situ 2 hour SCXRD data collections at 50 °C. Note that acetonitrile occupancy was calculated from a SQUEEZE electron count per unit cell analysis and the C26 occupancy was refined in SHELXL-2014. | |
Thermal analysis
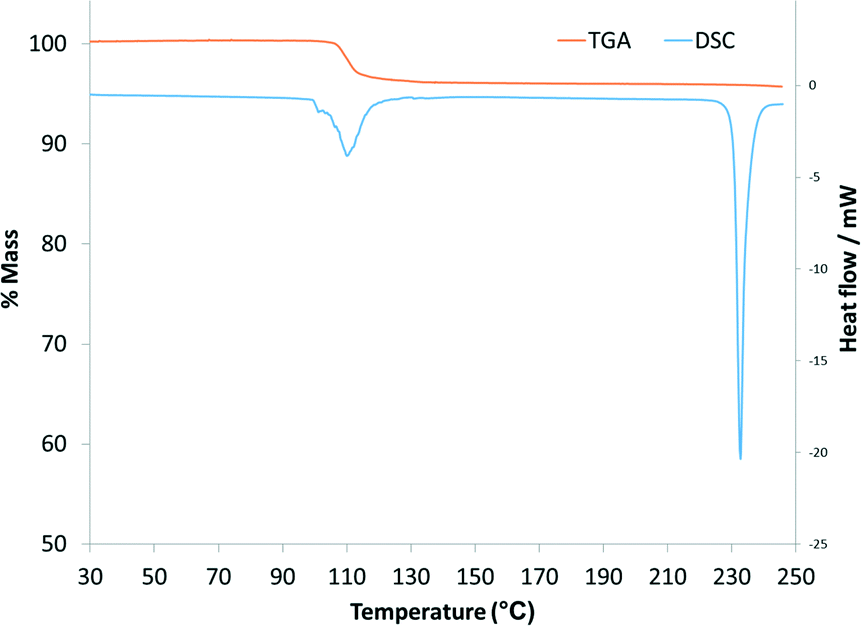
HSM photographs (Fig. 5) of crystals of 1a allow visual indication of their desolvation as bubbles are formed in the silicone oil upon heating. The first bubble appears at 60 °C, with more vigorous bubbling occurring at 110 °C accompanied by the crystals becoming opaque which is an indication of the loss of monocrystallinity. DSC analysis (Fig. 6) show a broad endotherm, with a shoulder, in the temperature range 100–120 °C, representing desolvation and concomitant rearrangement of the host molecules to form 3a. In a separate experiment, this rearrangement was confirmed by heating crystals on the DSC equipment to 130 °C and subjecting them to PXRD analysis (Fig. S4†). The experimental conditions for the VT-PXRD were less ‘harsh’ by slower heating of 1a by recording patterns at 20–25 °C intervals from room temperature which allowed 2a to observed. The TGA thermogram of 1a (Fig. 6) shows a single-step mass loss of 4.12% (calculated 4.39%) in the temperature range 30–150 °C corresponding to one acetonitrile molecule per asymmetric unit.
 |
| | Fig. 5 Hot stage microscope photographs of compound 1a under silicone oil at various temperatures (bubbles indicate the release of solvent). | |
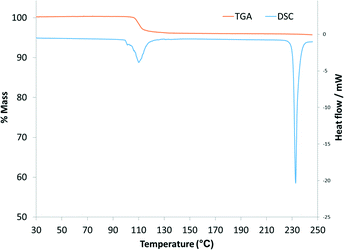 |
| | Fig. 6 DSC and TGA thermograms of 1a. | |
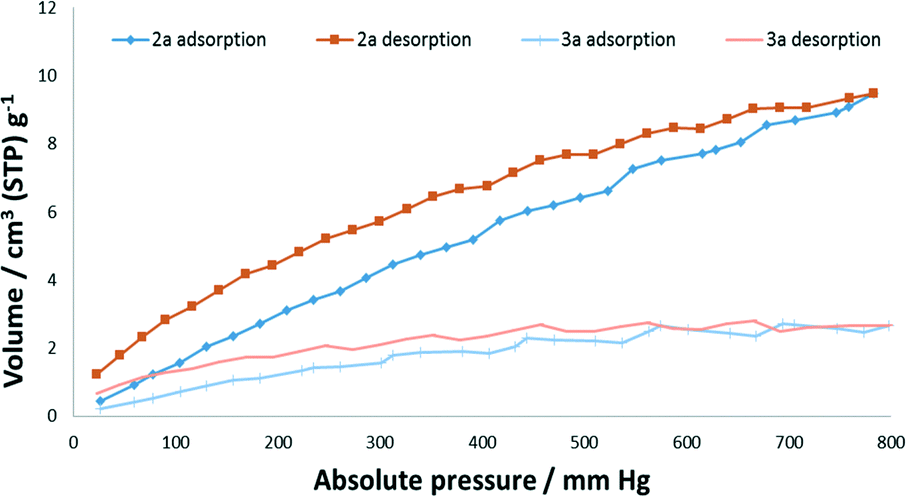
Gas sorption studies
Forms 2a and 3a (preparation and confirmation in Fig. S4†) were subjected to gas sorption analyses from vacuum up to 700–800 mm Hg absolute pressure. Nitrogen sorption at 77 K was negligible for both forms, which we suspect is due to nitrogen not being able to penetrate the structures at these modest pressures. Carbon dioxide sorption was significant for the pressure and temperature (273 K) of analysis (Fig. 7). The sorption of 9.47 cm3 (STP) g−1 at 783 mm Hg for 2a corresponds to ∼0.4 CO2 molecules per asymmetric unit. This sorption occurs despite an obstructed path for CO2 sorption indicating that CO2 is able to induce rotation of the methyl groups for sorption to occur. The ability of CO2 to induce structural changes in many seemingly non-porous materials, often selectively, has been attributed to its quadrupole moment. However, in a recent study combining in situ PXRD and SCXRD experiments with computational studies, weak van der Waals interactions were also shown to be important for a metal–organic framework.48 The transformation of 2a to 3a is from a single crystal to a polycrystalline material (as confirmed by in situ SCXRD and VT-PXRD experiments) and therefore would involve an increase in surface area and therefore a relative increase in sorption compared to if it was via an SCSC transformation. However, we believe that 3a is probably a higher-density form of 2a and therefore this factor negates the effect of the increase in surface area. Another interesting phenomenon was that for both 2a and 3a hysteresis was present indicating that this material is able to hold on to carbon dioxide at low pressures.
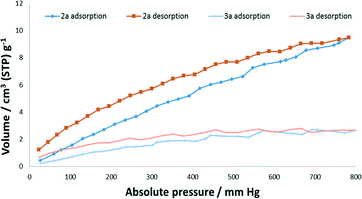 |
| | Fig. 7 CO2 sorption isotherms of 2a and 3a at 273 K. | |
Conclusions
We have isolated a new CTV inclusion compound with acetonitrile, 1a, obtained from a mixture of acetonitrile and chloroform. The CTV molecules are found to be present as dimers, which include acetonitrile, and self-included molecules which stack into columns. The solvate 1a could be desolvated into polymorphs 2a and 3a, with the transition to 2a occurring via a single-crystal-to-single-crystal transformation. The desolvation of 1a probably involves cooperative rotation of the methoxy methyl groups of the dimer CTV molecules with the columns of stacked CTV molecules imparting stability on the structure so that monocrystallinity is retained. Carbon dioxide gas sorption analysis show appreciable sorption for 2a despite the modest pressures employed and the modest void space calculated for 2a, whilst 3a also shows carbon dioxide sorption indicating that the proposed ‘closed’ structure is able to ‘open up’ on exposure to this gas.
Acknowledgements
RMP and CLO thank the National Research Foundation of South Africa and the University of Cape Town for financial support.
Notes and references
- M. J. Hardie, Chem. Soc. Rev., 2010, 39, 516–527 RSC.
-
A. Collet, J.-P. Dutasta, B. Lozach and J. Canciell, in Supramolecular Chemistry I – Directed Synthesis and Molecular Recognition, ed. E. Weber, Springer, Berlin, 1993, ch. 3, vol. 165, pp. 103–129 Search PubMed.
- T. Brotin and J.-P. Dutasta, Chem. Rev., 2009, 109, 88–130 CrossRef CAS PubMed.
- A. S. Lindsey, Chem. Ind., 1963, 823 CAS.
- H. Erdtman, R. Ryhage and F. Haglid, Acta Chem. Scand., 1964, 18, 1249–1254 CrossRef CAS.
- A. S. Lindsey, J. Chem. Soc., 1965, 1685–1692 RSC.
- A. B. Morrison and G. W. Smith, J. Chem. Soc., 1965, 3864–3965 Search PubMed.
- F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef.
- J. Gabard and A. Collet, J. Chem. Soc., Chem. Commun., 1981, 1137–1139 RSC.
- T. K. Ronson, J. Fisher, L. P. Harding, P. J. Rizkallah, J. E. Warren and M. J. Hardie, Nat. Chem., 2009, 1, 212–216 CrossRef CAS PubMed.
- A. Westcott, J. Fisher, L. P. Harding, P. Rizkallah and M. J. Hardie, J. Am. Chem. Soc., 2008, 130, 2950–2951 CrossRef CAS PubMed.
- J. J. Henkelis, T. K. Ronson, L. P. Harding and M. J. Hardie, Chem. Commun., 2011, 47, 6560–6562 RSC.
- A. Collet, Tetrahedron, 1987, 43, 5725–5759 CrossRef CAS.
- M. J. Hardie, R. Ahmad and C. J. Sumby, New J. Chem., 2005, 29, 1231–1240 RSC.
- M. J. Hardie and C. L. Raston, Angew. Chem., Int. Ed., 2000, 39, 3835–3839 CrossRef CAS.
- S. T. Mough, J. C. Goeltz and K. T. Holman, Angew. Chem., Int. Ed., 2004, 43, 5631–5635 CrossRef CAS PubMed.
- A. Arduini, F. Calzavacca, D. Demuru, A. Pochini and A. Secchi, J. Org. Chem., 2004, 69, 1386–1388 CrossRef CAS PubMed.
- C. J. Sumby, J. Fisher, T. J. Prior and M. J. Hardie, Chem. – Eur. J., 2006, 12, 2945–2959 CrossRef CAS PubMed.
- J. A. Wytko, C. Boudon, J. Weiss and M. Gross, Inorg. Chem., 1996, 35, 4469–4477 CrossRef CAS PubMed.
- K. T. Holman, G. W. Orr, J. L. Atwood and J. W. Steed, Chem. Commun., 1998, 2109–2110 RSC.
- M. Staffilani, G. Bonvicini, J. W. Steed, K. T. Holman, J. L. Atwood and M. R. J. Elsegood, Organometallics, 1998, 17, 1732–1740 CrossRef CAS.
- R. Ahmad and M. J. Hardie, Supramol. Chem., 2006, 18, 29–38 CrossRef CAS.
- J. W. Steed, P. C. Junk, J. L. Atwood, M. J. Barnes, C. L. Raston and R. S. Burkhalter, J. Am. Chem. Soc., 1994, 116, 10346–10347 CrossRef CAS.
- D. V. Konarev, S. S. Khasanov, I. I. Vorontsov, G. Saito, M. Y. Antipin, A. Otsuka and R. N. Lyubovskaya, Chem. Commun., 2002, 2548–2549 RSC.
- Y. Rio and J. F. Nierengarten, Tetrahedron Lett., 2002, 43, 4321–4324 CrossRef CAS.
- R. O. Zachary, N. Dorjderem, C. H. Richard and P. B. Daniel, Nanotechnology, 2011, 22, 275611 CrossRef PubMed.
- T. Brotin and J.-P. Dutasta, Eur. J. Org. Chem., 2003, 973–984 CrossRef CAS.
- M. J. Hardie and C. J. Sumby, Inorg. Chem., 2004, 43, 6872–6874 CrossRef CAS PubMed.
- O. Taratula and I. J. Dmochowski, Curr. Opin. Chem. Biol., 2010, 14, 97–104 CrossRef CAS PubMed.
- X. Han, L. Li, Z. Huang, J. Liu and Q. Zheng, Chin. J. Chem., 2013, 31, 617–623 CrossRef CAS.
- J. Song, Z. Huang and Q. Zheng, Chin. J. Chem., 2013, 31, 577–581 CrossRef CAS.
- J. J. Henkelis, C. J. Carruthers, S. E. Chambers, R. Clowes, A. I. Cooper, J. Fisher and M. J. Hardie, J. Am. Chem. Soc., 2014, 136, 14393–14396 CrossRef CAS PubMed.
- L. J. Barbour, Aust. J. Chem., 2006, 59, 595–596 CrossRef CAS.
- A. Husain, M. Ellwart, S. A. Bourne, L. Ohrstrom and C. L. Oliver, Cryst. Growth Des., 2013, 13, 1526–1534 CAS.
- T. Tahier and C. L. Oliver, CrystEngComm, 2015, 17, 8946–8956 RSC.
- J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000–1002 CrossRef CAS PubMed.
- C. M. Kane, O. Ugono, L. J. Barbour and K. T. Holman, Chem. Mater., 2015, 27, 7337–7354 CrossRef CAS.
- H. Zhang and J. L. Atwood, J. Crystallogr. Spectrosc. Res., 1990, 20, 465–470 CrossRef CAS.
-
Bruker (2007), Bruker AXS Inc., Madison, Wisconsin USA, 2007 Search PubMed.
-
G. M. Sheldrick, SADABS, Bruker AXS Inc., Madison, Wisconsin USA, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
-
Persistence of Vision Raytracer Pty. Ltd., 2004, 3.6 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- M. L. Connolly, J. Mol. Graphics, 1993, 11, 139–141 CrossRef CAS PubMed.
- S.-M. Hyun, J. H. Lee, G. Y. Jung, Y. K. Kim, T. K. Kim, S. Jeoung, S. K. Kwak, D. Moon and H. R. Moon, Inorg. Chem., 2016, 55, 1920–1925 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1478610 and 1478611. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce01078d |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence