
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the initial stages of lipid-mediated haemozoin nucleation†
David
Kuter
a,
Roxanne
Mohunlal
b,
Sharné-Maré
Fitzroy
a,
Constance
Asher
b,
Peter J.
Smith
c,
Timothy J.
Egan
*b and
Katherine A.
de Villiers
 *a
*a
aDepartment of Chemistry and Polymer Science, Stellenbosch University, Private Bag X1, Matieland 7602, South Africa. E-mail: kdev@sun.ac.za
bDepartment of Chemistry, University of Cape Town, Rondebosch 7701, South Africa. E-mail: timothy.egan@uct.ac.za
cDivision of Pharmacology, Department of Medicine, University of Cape Town Medical School, Observatory 7925, South Africa
First published on 20th May 2016
Abstract
Lipid-mediated haemozoin (Hz) formation was investigated using experimental and computational techniques. Glycerolipids (predominantly diacylglycerols), not phospholipids, were found entrapped by Hz crystals isolated from Plasmodium falciparum and consequently are proposed to nucleate Hz crystals in vivo. The initial stages of Hz formation were investigated using molecular dynamics simulations which involved modelling ferrihaem species pertinent to Hz formation (π–π and μ-propionato dimers) at lipid–water interfaces comprising a representative glycerolipid 1,2-dioleoyl-sn-glycerol (DOG) or phospholipid 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC). Simulations showed that π–π dimers partition into both lipids and remain near the hydrated lipid–water interface. Dimer aggregation, which was only observed in DOG, is proposed to be crucial for nucleating Hz. Indeed, β-haematin (synthetic Hz) production was found experimentally to be greater in DOG than DOPC (65 vs. 24%). Density functional theory calculations indicate that iron coordination by the carboxylate group of the propionate side chain, not the propionic acid group, is feasible in the aggregate. This can only occur, however, if ferrihaem is in a zwitterionic form with an axial water ligand, one propionic acid and one propionate side chain. UV-visible spectroscopy indicates that the presence of solvent water molecules is crucial for maintaining this species over a tautomeric form consisting of two propionic acid groups and an axial hydroxide ligand. Anhydrous conditions promote the latter tautomer which is unlikely to form Hz. A mechanism describing the initial stages of Hz formation is proposed based on these findings, which supports proposals that Hz crystal growth occurs in the lipid sub-phase.
Introduction
Haemozoin (Hz) biocrystallisation is an essential haem detoxification pathway utilised by certain pathogenic blood-feeding organisms including Plasmodium falciparum (causative agent of malaria),1Schistosoma mansoni (causative agent of bilharzia),2Rhodnius prolixus (vector of Trypanosoma cruzi, the causative agent of Chagas disease)3 and others.4,5 Since this process is unique to these pathogenic organisms and not the human host, it has been considered an attractive drug target and diagnostic marker. Consequently, Hz has been the focus of much investigation. The seminal paper of Pagola et al. confirmed that synthetic Hz (β-haematin) crystals are composed of μ-propionato ferrihaem dimers that are hydrogen-bonded via propionic acid side chains.1 While the structure of Hz is well established, the mechanism of its formation in vivo is still a matter of much contention. In this regard, there is debate as to whether proteins6,7 or lipids8–10 mediate Hz biocrystallisation. Strong evidence has been provided in support of the latter. Indeed, lipids in the absence of proteins are able to produce β-haematin in vitro under aqueous biomimetic conditions.9–14 Moreover, Hz crystals observed via microscopy have been found in close contact with lipids in P. falciparum12,15 and R. prolixus3 as well as in S. mansoni16 and other trematodes.5Considerable research has been devoted to uncovering the role that lipids play in facilitating Hz biocrystallisation. Early studies implicated phospholipids as the mediators of Hz formation in vivo in P. falciparum,8,11 with later work proposing that the digestive vacuole (DV) membrane provides a scaffold for its crystallisation.17 Indeed, crystals aligned with the inner DV membrane in P. falciparum have been reported15 and Hz associated with perimicrovillar membranes in the gut content of R. prolixus has also been observed.3 On the other hand, Hz has been found in close proximity to glycerolipid droplets in both P. falciparum12,18 and S. mansoni,3 which has prompted the alternate hypothesis that neutral glycerolipids instead are responsible for mediating Hz formation in vivo. Presently, there is no consensus whether phospholipids or glycerolipids are the promoters of Hz biocrystallisation. Indeed, unsaturated lipids of both classes have been shown to facilitate β-haematin formation in vitro.13,14,19,20 Furthermore, thin layer chromatography (TLC) has identified both glycerolipids and phospholipids associated with the surface of Hz crystals isolated from P. falciparum, with the former being present in larger quantities than the latter.12,18
In addition to the uncertainty surrounding the type of lipid that promotes Hz formation, the mechanism by which lipids mediate its crystallisation is also unclear. A templated growth mechanism at the lipid–water interface has been proposed which suggests that growth is directed towards the aqueous medium.15 This hypothesis is based on close complementarity between the packing of lipid headgroups at the air–water interface and propionic acid side chains exposed at the (100) face in Hz crystals.21 An alternative proposal is that the Hz formation takes place within the non-aqueous lipid sub-phase.10,19,22 While recent evidence from atomic force microscopy that has described crystal growth in this manner in n-octanol is convincing,23 there is still a lack of structural knowledge regarding the behaviour of ferrihaem species pertinent to Hz formation at lipid–water interfaces.
In this work, we attempt to address the above uncertainties surrounding lipid-mediated Hz formation using a combination of experimental and computational techniques. To determine whether phospholipids, glycerolipids or a combination thereof are associated with Hz in vivo, crystals were isolated from P. falciparum and subjected to TLC analysis. Glycerolipids (specifically diacylglycerols), not phospholipids, were found to be entrapped, suggesting that they nucleate Hz crystals. Insight into this process was obtained by modelling ferrihaem species at a glycerolipid (1,2-dioleoyl-sn-glycerol, DOG)–water as well as at a phospholipid (1,2-dioleoyl-sn-glycero-3-phosphatidylcholine, DOPC)–water interface. A ferrihaem π–π tetramer aggregate that readily formed in DOG closely resembles the packing observed in Hz crystals and is proposed to represent the initial stage of Hz nucleation. Iron–carboxylate bond formation in the aggregate was subsequently investigated using density functional theory (DFT) calculations. These indicate that iron coordination is possible in the tetramer aggregate in a low dielectric medium by a propionate, not propionic acid, side chain. The presence of some water molecules, however, was found via UV-visible spectroscopy to be essential in stabilizing the tautomer of ferrihaem that is a prerequisite for Hz formation. Based on the findings from computation and experiment, a general mechanism describing the initial stages of Hz nucleation is presented.
Experimental
Parasite culturing
Chloroquine-sensitive (NF54) P. falciparum was maintained in a continuous culture following the methods of Trager and Jensen with minor modifications.24 Parasites were maintained at a 2% haematocrit concentration in a medium comprising 10.4 g L−1 RPMI 1640 with glutamine (excluding NaHCO3), 4 g L−1 glucose, 6 g L−1 HEPES, 0.088 g L−1 hypoxanthine, 1.2 mL L−1 gentamycin and 5 g L−1 Albumax II. Prior to use, 16.8 mL of 5% (w/v) NaHCO3 was added to 400 mL of medium. Cultures were gassed with an air mixture comprising 3% O2, 4% CO2 and 93% N2 for 2 min and were then incubated at 37 °C. The culture medium was changed daily and parasite growth carefully monitored using 10% (v/v) Giemsa staining.25 Parasite cultures were synchronised by treatment with 5% (w/v) D-sorbitol during the ring stage of their life cycle and sub-cultured using O+ red blood cells during their trophozoite stage. Mature trophozoites were isolated by treating pelleted parasitised red blood cells with 1% (w/v) saponin at room temperature for 3 min and aspirating the supernatant obtained after 10 min of centrifugation at a relative centrifugal force of 1500g. The resulting trophozoite pellet was washed four times with 150 mM phosphate-buffered saline or until the supernatant was clear. DVs were isolated from the trophozoites by trituration and Hz crystals extracted by multiple freeze–thaw cycles as previously described.26TLC analysis
Hz crystals isolated from trophozoites were extensively washed using a 4% (w/v) solution of sodium dodecyl sulphate followed by 150 mM phosphate-buffered saline and were then dissolved in 0.1 M NaOH. Lipids were extracted from washings or dissolved crystals using chloroform/methanol (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and were separated using TLC as described by Jackson et al.18 and Pisciotta et al.12 Briefly, this involved separating phospholipids using a chloroform/methanol/water (50:20:3 v/v) mobile phase, followed by neutral lipids in a hexane/diethyl ether/acetic acid mobile phase (16:10:1 v/v). Lipids used as standards were obtained from Sigma-Aldrich. They were prepared at a concentration of 3.31 mM in either acetone/methanol (1:9 v/v) or chloroform/methanol (2:1 v/v) and were subjected to the same TLC system as used above (see Fig. S1†). TLC plates were stained with 0.05% (w/v) primuline in acetone/water (4:1 v/v) and visualized using a UV lamp at 365 nm. β-Haematin (25 mg) prepared from an acetate solution (see below) was incubated in 1 mL of parasite lysate or in 1 mL of the glycerolipid mixture reported by Pisciotta et al.12 at 37 °C for 15 minutes. The mixtures were allowed to equilibrate at room temperature for a further 15 minutes before crystals were collected via centrifugation. These were extensively washed in the same manner as that described for Hz isolated from parasites. To determine the effect of NaOH on the lipid profile, parasite lysate or 3.31 mM DOPC was treated with 0.1 M NaOH in a ratio of 2:1 (v/v) and separated using TLC in the same manner as described above. Washed Hz crystals were imaged as previously reported.26
:1 v/v) and were separated using TLC as described by Jackson et al.18 and Pisciotta et al.12 Briefly, this involved separating phospholipids using a chloroform/methanol/water (50:20:3 v/v) mobile phase, followed by neutral lipids in a hexane/diethyl ether/acetic acid mobile phase (16:10:1 v/v). Lipids used as standards were obtained from Sigma-Aldrich. They were prepared at a concentration of 3.31 mM in either acetone/methanol (1:9 v/v) or chloroform/methanol (2:1 v/v) and were subjected to the same TLC system as used above (see Fig. S1†). TLC plates were stained with 0.05% (w/v) primuline in acetone/water (4:1 v/v) and visualized using a UV lamp at 365 nm. β-Haematin (25 mg) prepared from an acetate solution (see below) was incubated in 1 mL of parasite lysate or in 1 mL of the glycerolipid mixture reported by Pisciotta et al.12 at 37 °C for 15 minutes. The mixtures were allowed to equilibrate at room temperature for a further 15 minutes before crystals were collected via centrifugation. These were extensively washed in the same manner as that described for Hz isolated from parasites. To determine the effect of NaOH on the lipid profile, parasite lysate or 3.31 mM DOPC was treated with 0.1 M NaOH in a ratio of 2:1 (v/v) and separated using TLC in the same manner as described above. Washed Hz crystals were imaged as previously reported.26
Acetate-mediated β-haematin production
β-Haematin was prepared using a procedure previously reported with slight modifications.27 Haemin (90 mg, Fluka) was dissolved in 10 mL of NaOH (0.1 M) and neutralized with 1 mL of HCl (1 M). To this, 9.25 mL of acetate buffer (9.7 M, pH 4.8) was added and the mixture was incubated for 1 hour at 60 °C. After incubation, the reaction was quenched with water and the mixture was cooled over ice. The resulting precipitate was collected via filtration and extensively washed with water. To remove any unreacted haemin, the air-dried precipitate was placed in a 15 mL Falcon tube with 1 mL of an aqueous pyridine solution consisting of 5% (v/v) pyridine, 40% (v/v) acetone and 0.02 M HEPES (pH 7.4). This well-shaken mixture was diluted to 10 mL with water, centrifuged for 10 minutes and the supernatant discarded. The resulting precipitate was washed with water until the supernatant was clear. Finally, the precipitate was collected via filtration and left to dry over P2O5. The identity of the product as β-haematin was confirmed using infrared spectroscopy. The powder X-ray diffraction pattern of β-haematin was measured using a Bruker D8 Advance powder X-ray diffractometer with CuKα radiation (λ = 1.5418 Å). Surface image analysis of the crystals was performed using a Zeiss MERLIN FEG® scanning electron microscope equipped with a GEMINI II® column. An operating voltage of 5 kV, a working distance of 3.6 mm and an I-Probe of approximately 250 pA were used. The sample was coated with a thin layer of gold, prior to analysis, in order to establish conductivity.Lipid-mediated β-haematin production
β-Haematin was produced by modifying a previously reported procedure for use in 24-well plates.28 A haematin stock solution (3.2 mM) was prepared by dissolving 5 mg of haematin (Sigma) in 1 mL of NaOH (0.1 M) and diluting with 1.5 mL of an acetone/methanol (1:9 v/v) solution. A 2 μL aliquot was added to 200 μL of a DOG or DOPC stock solution (1.6 mM in 1:9 acetone/methanol) and mixed. This solution was then added to 1 mL of citric acid buffer (50 mM, pH 4.8) in a 24-well plate which had been pre-incubated at 37 °C for at least 45 minutes. After 4 hours of incubation at 37 °C, the reaction was quenched by addition of 240 μL of a pyridine solution consisting of 30 mL of pyridine, 40 mL of acetone, 10 mL of HEPES buffer (2 M, pH 7.5) and 20 mL of water. A 200 μL aliquot was removed from the reaction mixture after 10 minutes and placed in a 96-well plate which was read at 405 nm using a Thermo Scientific Multiskan GO spectrophotometric plate reader. The yield of β-haematin was determined from the concentration of unreacted ferrihaem measured as the pyridine complex in accordance with a previously reported procedure by measuring the absorbance relative to that obtained after a 0 h incubation period.28
UV-visible spectrophotometry
UV-visible spectra were recorded on a Varian Cary 100 spectrophotometer at 25 °C under an anhydrous argon atmosphere so as to minimise solvent hygroscopy. A 30 μM haematin (Sigma) solution was prepared in rigorously dried methanol to which aliquots of buffered solutions were dispensed using a Hamilton syringe. The spectra were recorded following each addition and have been corrected for dilution. Aqueous solutions were buffered to pH 10 and 6.5 using 0.2 M CHES and HEPES, respectively. Rigorously dried methanol was prepared according to a literature procedure29 and was stored over 4 Å molecular sieves.Molecular dynamics simulations
π–π dimeric and μ-propionato dimeric ferrihaem species were separately simulated at a solvated DOPC or DOG bilayer–water interface consisting of 184 lipid and approx. 13000 TIP3P water molecules. The phospholipid bilayer was constructed using the CHARMM-GUI interface30–34 around two identical ferrihaem dimers oriented such that their porphyrin rings were parallel to the bilayer surface. One dimer was positioned in the centre of the bilayer and the other was placed in the water component near the interface. While glycerolipids do not inherently form bilayers, for ease of simulation, a DOG bilayer was constructed by editing the coordinates of the phospholipid system (see the ESI†). This system represents a reasonable approximation of the surface of a lipid since ferrihaem molecules only interacted at one surface and did not span the bilayer. An alternative approach of using a micelle was considered, however, the curvature at the surface of a micelle is an inappropriate model for the lipid surface which is almost flat on the molecular scale. Force field parameters for the neutral ferrihaem π–π dimer were taken from a previous study and adjusted slightly (see the ESI† for details), while those for the neutral μ-propionato ferrihaem dimer were optimized in a similar manner to that previously described (see the ESI† for details).35 Solvated bilayers were equilibrated in a step-wise manner for a total of 375 ps using the CHARMM software package (version 36)30 according to the restrained equilibration procedure described by Jo et al.34 Further energy minimization (1000 steps) and equilibration (10 ns) were conducted using the NAMD software package (version 2.9)36 with a 1 fs time-step. Production dynamics were recorded over 100 ns using a 2 fs time-step. All simulations were conducted at 310 K under an isothermal–isobaric ensemble. Long-range electrostatic interactions were calculated using the particle-mesh Ewald method with a cut-off of 16 Å and van der Waals interactions were decreased smoothly to zero between 10 and 12 Å. Details regarding analysis of trajectories can be found in the ESI.†
DFT calculations
DFT calculations were performed using iron(III) porphine as a model for ferrihaem. Starting structures of the iron(III) porphine complexes were created by editing the geometry optimized structure of H2O–Fe(III) protoporphyrin IX previously reported.37 Calculations were conducted using the Gaussian software package38 and followed the approach of Durrant for the investigation of Fe(III) porphine coordinating ligands.39 Briefly, this involved performing in vacuo geometry optimizations on all possible iron spin states (5/2, 3/2 and 1/2) using the B3LYP functional and LANL2DZ basis set (B3LYP/LANL2DZ). In all cases, a quadratic convergence procedure was required to assist self-consistent field convergence. Frequency calculations were then performed on resulting geometries to ensure true minima were obtained and the lowest energy (zero point corrected) spin state structures were identified. Single point energy calculations on these lowest energy structures in the presence of the SMD implicit solvent model40 with n-octanol dielectric followed using the B3LYP functional and 6-311+G(d,p) basis set, B3LYP/6-311+G(d,p), and an ultrafine integration grid. The final energies of molecules solvated by implicit n-octanol were calculated by adding B3LYP/LANL2DZ zero point energy corrections to B3LYP/6-311+G(d,p) single point energies. To determine whether coordination to Fe(III) porphine would occur in a lipid environment if acetic acid and acetate molecules were initially positioned at a distance of 3.4 Å from the iron centre, geometry optimisations (B3LYP/LANL2DZ) of these molecules were conducted in the presence of implicit n-octanol (SMD).Results and discussion
Lipids associated with Hz crystals
Previously, Pisciotta et al. identified lipids associated with Hz crystals isolated from P. falciparum.12 In that study, Hz was extracted using sucrose cushion centrifugation in an effort to minimize lipid loss during the extraction procedure. Because this method of isolation is gentle, the possibility exists that the membrane material from the parasite DV could have contaminated the isolated crystals. Indeed, a minor phospholipid component was observed by TLC analysis of isolated Hz crystals. To identify lipids that are associated with Hz in the DV and to minimize contamination by cellular debris, we have obtained crystals from isolated P. falciparum DVs using trituration and freeze–thawing methods. TLC analysis was performed using the non-destructive lipid-specific fluorescent dye, primuline, for visualization.41 Isolated crystals were extensively washed with sodium dodecyl sulphate detergent and phosphate-buffered saline solutions in order to remove all lipids associated with the surface of the crystals. Extraction of the washing solutions into the organic solvent, followed by TLC separation, indicated that these surface-associated lipids are mainly diacylglycerols, triacylglycerols and phospholipids (Fig. 1a). A minor monoacylglycerol component was also detected. A similar lipid profile was reported by Pisciotta et al. although the relative abundance of the monoacylglycerols and diacylglycerols we obtained appears to be somewhat lower and higher, respectively.12 This observed difference is likely a result of the different methods used to isolate the crystals. Extensively washed Hz crystals having no detectable lipids associated with the surface (Fig. S2†) were then dissolved in sodium hydroxide after which extraction with the organic solvent was performed on the solution. TLC analysis of this organic phase clearly indicates that glycerolipids, not phospholipids, were present (Fig. 1b). Diacylglycerols appeared to be the most abundant, while monoacylglycerols were the least abundant. The absence of phospholipids is not an artefact of headgroup dephosphorylation during the dissolution process since there was no disappearance of phospholipid TLC spots in parasite lysate treated with 0.1 M sodium hydroxide over the same timescale (Fig. S3†). Nor did the addition of 0.1 M sodium hydroxide to a solution containing the phospholipid DOPC affect its TLC profile (Fig. S3†).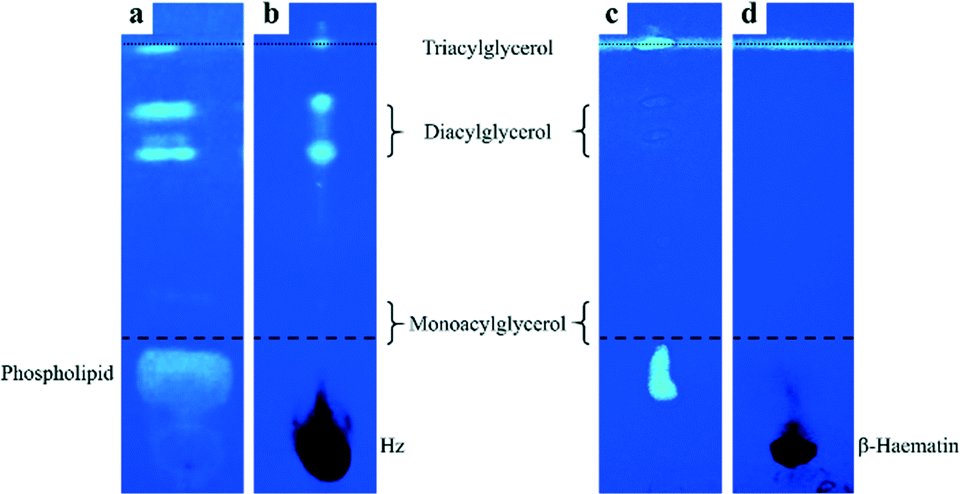 | ||
| Fig. 1 TLC silica gel plates stained with primuline to identify lipids associated with Hz crystals isolated from P. falciparum (a and b) and β-haematin incubated in parasite lysate (c and d). TLC analysis of contents extracted from the first wash (a and c) and from extensively washed and then dissolved (b and d) crystals. Dashed and dotted lines indicate the first and second solvent fronts, respectively. Note: diacylglycerol spots in (c) are very weak. | ||
The observation that glycerolipids were present within dissolved crystals is not likely to be a consequence of their incorporation into the crystal lattice of Hz itself. This would cause disruption of the crystal which is not observed. Transmission electron micrographs of the isolated and washed crystals show that Hz is highly crystalline and that it exhibits remarkably regular lattice fringes (Fig. S5†). Indeed, this observation has been previously reported.26 Incorporation of lipids in such a manner, in fact, would not be expected to occur since Hz crystals themselves are non-porous. The possibility was then considered that these glycerolipids arose as an artefact of the isolation procedure in which strongly adhering cellular debris simply could not be removed from the surface of crystals by extensive washing. To investigate this possibility, β-haematin was chemically prepared in the absence of lipids. This crystalline material (Fig. S6†) was incubated in parasite lysate from which parasite-derived Hz had been precipitated. Analysis of the sample of β-haematin incubated in parasite lysate was then conducted in the same manner as that used for natural Hz crystals. While surface washings from β-haematin crystals showed a similar TLC profile to that observed for natural Hz crystals albeit with much weaker diacylglycerols lipid spots (Fig. 1c), no lipids were detected when the extensively washed β-haematin crystals were then dissolved (Fig. 1d). Even when β-haematin crystals where incubated in a mixture of glycerolipids reportedly present in P. falciparum,12 no lipids were detected within the dissolved crystals (Fig. S4†). These findings definitively show that the glycerolipids detected within the dissolved Hz crystals were not merely strongly adhered to the surface. Instead, the glycerolipids were probably entrapped in empty spaces between Hz crystals during their growth in the DV. Indeed, slit-shaped macropores have been very recently reported to be present in β-haematin.42 The presence of these lipids in isolated washed haemozoin strongly suggests that it is these lipids that have a role in nucleating Hz crystals.
Modelling Hz nucleation
The initial stages of Hz nucleation at a lipid–water interface are not well understood at the molecular level. In order to probe this, the behaviour of ferrihaem species pertinent to Hz formation was investigated at aqueous solvated lipid interfaces using molecular dynamics (MD) simulation. Two specific ferrihaem species were separately studied, namely the μ-propionato (Fig. 2a) and π–π dimer (Fig. 2b). The former species represents the smallest molecular subunit of Hz, while the latter is considered the dominant form of ferrihaem in an aqueous solution under biologically relevant conditions.43 These molecules were simulated separately at both a neutral glycerolipid–water and a phospholipid–water interface. In the case of the former, DOG (Fig. 2c) was selected for simulation based on the findings reported above which indicate that diacylglycerols are the major glycerolipid component entrapped by Hz crystals. This specific glycerolipid has been identified in association with sucrose-purified Hz12 and its structural isomer (1,3-dioleoyl-sn-glycerol) is known to efficiently mediate β-haematin formation in vitro.20 The phospholipid DOPC (Fig. 2c) was chosen for simulation due to its structural similarity to DOG and based on reports that phosphocholine-containing phospholipids are a major component of parasite membranes.44,45 The validity of using DOPC and DOG as model systems in MD simulations was confirmed by determining their ability to form β-haematin. Both lipids successfully mediated β-haematin formation although in our experiment, DOPC was found to produce a much lower yield than DOG (24 vs. 65%, Fig. S7†).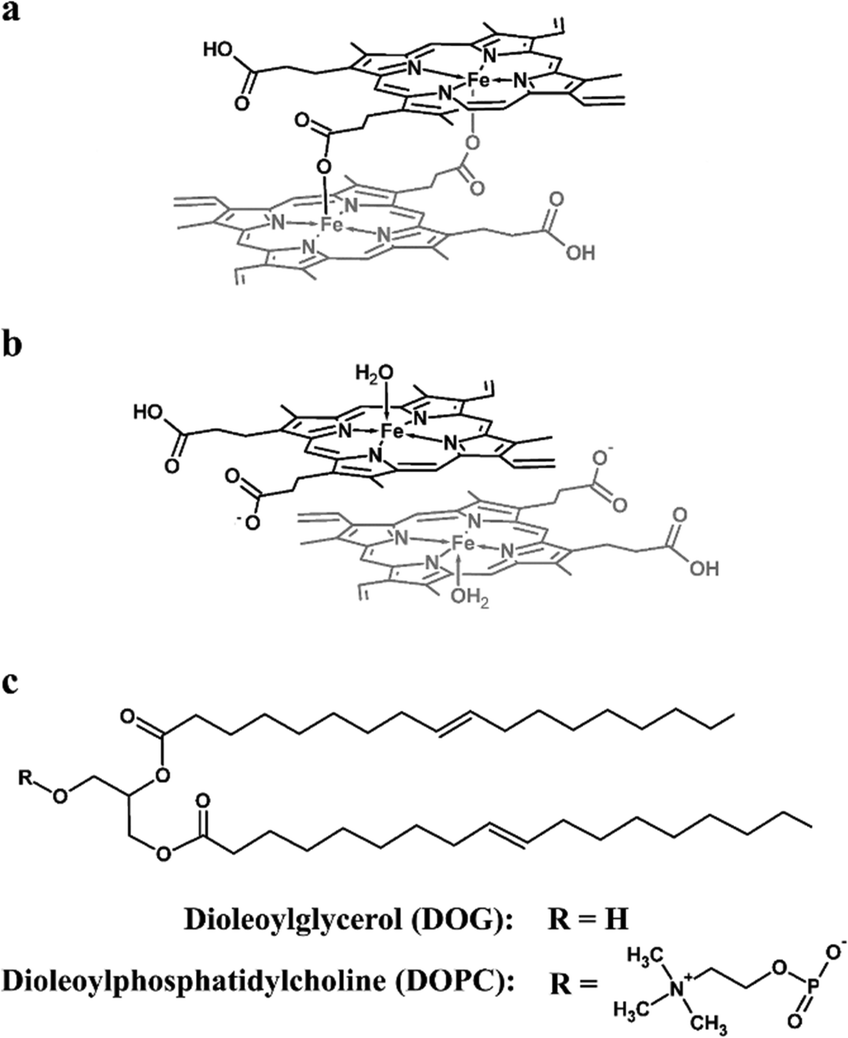 | ||
| Fig. 2 Molecular structures of (a) μ-propionato dimeric ferrihaem, (b) π–π dimeric ferrihaem and (c) the lipids 1,2-dioleoyl-sn-glycerol (DOG) and 1,2-dioleoyl-sn-glycero-3-phosphatidylcholine (DOPC) used in MD simulations. | ||
MD simulations of the π–π and μ-propionato dimers at DOG–water and DOPC–water interfaces indicate that the preferred location of both ferrihaem species over the majority of the simulation is within the lipid sub-phase. This preference occurs regardless of whether the species is initially placed in the aqueous phase or within the lipid component. In the case of the former, the dimers partition within 25 ns into the lipid (Fig. S11†) and do not re-enter the bulk aqueous phase thereafter. On the other hand, ferrihaem species initially positioned within the lipid remain in this phase over the entire course of the simulation. These observations are consistent with the lipophilic nature of ferrihaem (logP = 2.6)19 as well as its reported enhanced solubility in citric acid buffer saturated n-octanol compared to an acidic aqueous solution.46 Interestingly, the non-covalently associated ferrihaem π–π dimer was found to remain associated over the entire course of the simulation, even when partitioned into the lipid. While both ferrihaem dimers exhibit an affinity for the lipid sub-phase, differences were observed in the extent of their immersion. The π–π dimer was found to be located just below the hydrophilic lipid headgroup region near the ester groups (Fig. 3a–c) in both DOPC–water and DOG–water interfaces. Here it is able to maximize favourable hydrogen bonding in the case of the hydrophilic moieties of the ferrihaem molecule while simultaneously accommodating the more non-polar portions (vinyl and methyl groups) in the lipid tail region. Indeed, frequent contacts were observed between solvent water molecules and the axial water ligands as well as the propionate and propionic acid side chains of the π–π dimer (Table 1). Notable hydrogen bonding contacts were also found to exist between the carbonyl ester groups and axial water ligands, however little interaction was observed with the phospholipid or glycerolipid headgroups themselves. In general, hydrogen bonding contacts were greater in number at the DOG interface than in DOPC, but this does not appear to be a result of the differences in ferrihaem penetration depth in the two lipids. Rather, the absence of bulky phosphatidylcholine groups in DOG permits greater hydration of its glycerol headgroups. In contrast to the π–π dimer, the μ-propionato dimer penetrated deeper towards the lipid interior (Fig. 3d–f), suggesting that this particular ferrihaem species is more hydrophobic. This can be rationalized on the basis of hydrogen bonding contacts identified above which, for the π–π dimer, showed significant solvation around the axial water ligand and propionate side chains (Table 1). The formation of the μ-propionato dimer, however, necessarily requires the dissociation of the axial water ligand and coordination of the propionate groups. This causes no change in the formal molecular charge but effectively removes two of the most hydrated groups in the molecule, leading to a marked decrease in solvation and thus a more hydrophobic character. Indeed, substantially fewer hydrogen bonding contacts are observed for the μ-propionato dimer (Table 1).
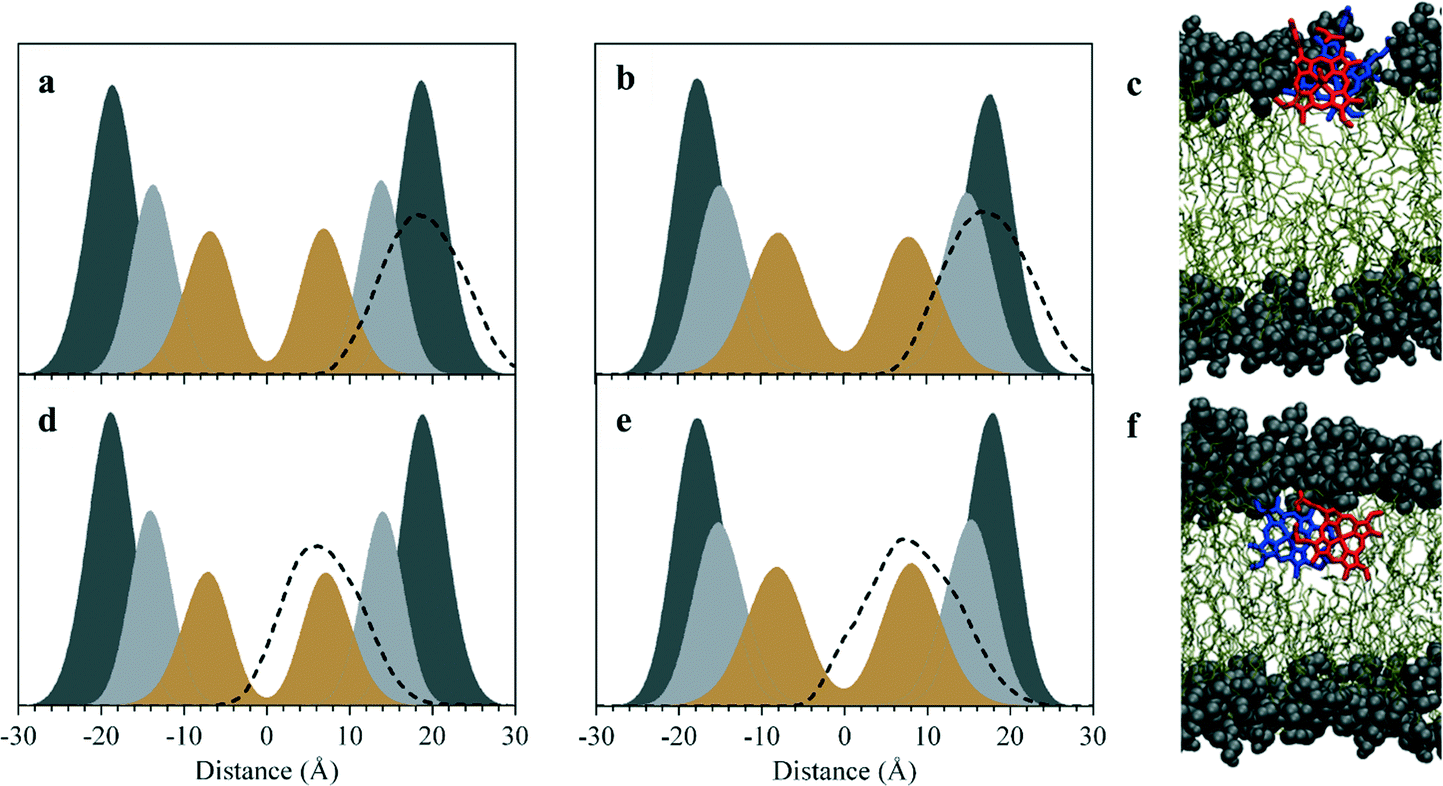 | ||
| Fig. 3 Lipid penetration by ferrihaem species. Density profiles along the z-axis indicating the depth of penetration (dashed lines) by π–π dimeric (a and b) and μ-propionato dimeric species (d and e) into DOPC (left plots) and DOG (right plots). Distances are referenced relative to modelled bilayer centres. Phosphatidylcholine and glycerol headgroups are shaded dark grey, ester groups light grey and double-bonded carbon atoms in the lipid tail in brown. Data have been scaled to aid visualization. (c and f) Representative snapshots taken from the MD simulations in the glycerolipid system showing π–π and μ-propionato dimers, respectively. To aid visualization, water molecules have been omitted, glycerol headgroups and ester groups have been depicted as spheres, and ferrihaem constituents of dimers are coloured in red and blue. | ||
| Interaction type | DOPC | DOG | ||
|---|---|---|---|---|
| π–πb | μ-Propionatoc | π–πd | μ-Propionatod | |
a Average contacts over the duration of the simulation while in the lipid phase.
b 26–100 ns.
c 4–100 ns.
d 0–100 ns.
e Interaction with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of free and coordinated propionate groups of π–π and μ-propionato species, respectively. O of free and coordinated propionate groups of π–π and μ-propionato species, respectively.
|
||||
| Solvent water: | ||||
| COOH⋯OH2 | 0.83 | 1.12 | 1.42 | 0.63 |
| COCOOH⋯H2O |
0.90 | 0.13 | 1.61 | 0.10 |
| COCOO⋯H2Oe |
3.71 | 0.37 | 4.93 | 0.09 |
| Fe–OaxH2⋯OH2 | 1.50 | — | 1.64 | — |
| Fe–Oax⋯H2O | 0.03 | — | 0.07 | — |
| Lipid headgroups: | ||||
| COOH⋯OPO3 |
0.41 | 0.03 | — | — |
| COCOO⋯N+(CH3)3 |
0.03 | — | — | — |
| COCOO⋯HOglycerole |
— | — | 0.17 | 0.02 |
| COCOOH⋯HOglycerol |
— | — | 0.06 | 0.00 |
| Fe–OaxH2⋯OPO3 |
0.26 | — | — | — |
| Lipid ester groups: | ||||
| COOH⋯OCester |
0.28 | 0.43 | 0.13 | 0.28 |
| Fe–OaxH2⋯OCester |
1.71 | — | 1.62 | — |
These findings do not support the proposed mechanism of templated growth of the Hz crystal which suggests that molecular recognition between the propionic acid groups of the μ-propionato dimer and lipid headgroups facilitates crystal growth in the aqueous phase.21 There is no evidence of the π–π dimer in the aqueous phase having a conformation that could facilitate the formation of the μ-propionato species and, even if the latter were formed in small amounts, simulations show no propensity of the propionic acid groups of this species to interact with lipid headgroups to any great degree (Table 1). On the other hand, the observation that the μ-propionato dimer is located towards the interior of both lipids supports proposals that Hz crystallisation occurs within the lipid sub-phase.23,46 Consequently, the ability of ferrihaem to penetrate the lipid must be crucial to Hz formation. Indeed, the ability of unsaturated phospholipids to mediate β-haematin formation has been related to their ability to exist in the fluid state rather than in a gel phase.13,14 Such findings can only be accounted for if lipid immersion is a prerequisite for Hz formation.
While the different headgroups of DOG and DOPC did not dramatically influence the positioning of a particular ferrihaem species, they do appear to affect aggregation of the π–π dimer. In the phospholipid, little interaction is observed between pairs of π–π dimers over the course of the simulation. In contrast, π–π dimers at the interface between water and the glycerolipid were found to rapidly aggregate and form a tetramer aggregate (Fig. 4a). Since DOG was found to be more efficient at producing β-haematin than DOPC (Fig. S7†), the role of this aggregate in Hz formation was explored further. Structurally, when the tetramer is viewed perpendicular to the lipid surface, it bears a striking resemblance to the crystal of β-haematin viewed perpendicular to the bc-plane (Fig. 4b). Thus, its orientation relative to the surface of the lipid corresponds to that of Hz lying with its (100) face parallel to the lipid–water interface. This particular orientation of crystals relative to the lipid surface has been observed experimentally for β-haematin produced at a monomyristoylglycerol–water interface21 as well as in P. falciparum for Hz crystals aligned with the inner DV membrane.15 A noteworthy observation therefore is that propagation of aggregation in the tetramer is expected to occur parallel to the lipid surface which corresponds to the two fastest growing directions reported for the Hz crystal, namely along the b- and c-axes.23 Further structural analysis of the tetramer revealed that at various stages during the dynamics simulation, a propionic acid side chain of one ferrihaem molecule is positioned near the iron centre of another (approx. 3.2–3.4 Å). In this respect, the structure is reminiscent of that previously proposed as the precursor to the μ-propionato ferrihaem dimer and consequently the putative precursor to Hz (Fig. 4a, inset).10 It should be noted, however, that the interaction observed here occurs between π–π dimers (inter-dimer) as opposed to an intra-dimer interaction in the putative Hz μ-propionato precursor. The π–π aggregate may thus represent the initial stage of Hz formation. Indeed, such a proposal has also been intimated by Klonis et al. where, following re-analysis of the Hz crystal structure, these authors suggested that the crystal can rather be considered as an array of ferrihaem π–π dimers which are stabilized by iron–carboxylate bonds.47
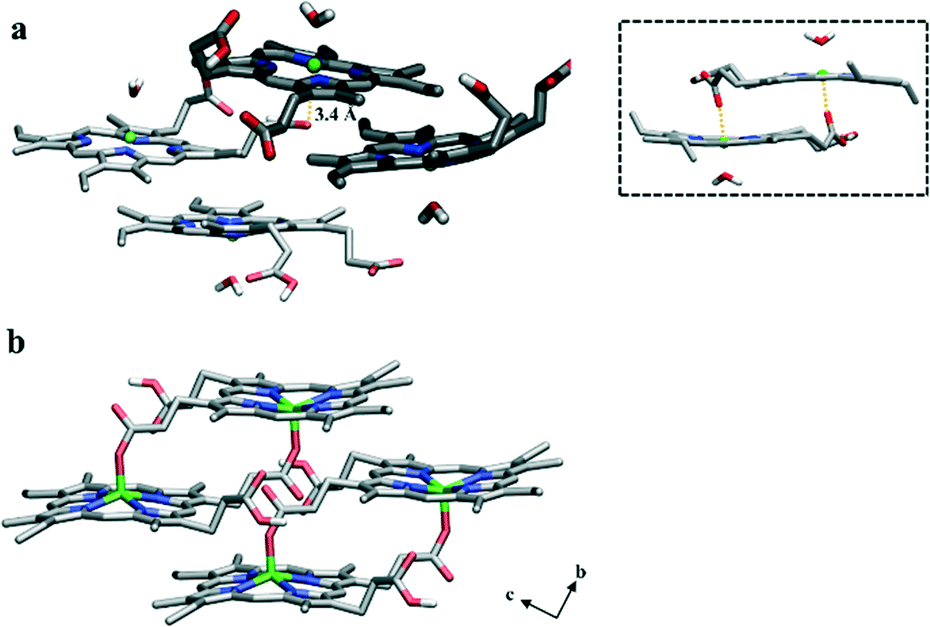 | ||
| Fig. 4 Molecular structures of ferrihaem. (a) The π–π tetramer aggregate observed during MD simulation at a DOG–water interface viewed perpendicular to the lipid surface. The two dimers that were initially placed in positions within the water and lipid sub-phases are shaded dark and light, respectively. An inter-dimer interaction between a propionic acid side chain and an Fe centre is observed and the distance of 3.4 Å is indicated by the yellow dotted line. Inset: Vacuum-minimized structure of the π–π dimer previously proposed as the precursor to the μ-propionato dimer.10 (b) The reported crystal structure of β-haematin viewed perpendicular to the bc-plane.1 This orientation corresponds to the (100) face of Hz crystals which has been shown to lie parallel to lipid surfaces. Close structural congruency between (a) and (b) is evident. Carbon, nitrogen, oxygen and iron atoms are coloured grey, blue, red and green, respectively. For clarity, non-polar hydrogen atoms have been omitted. | ||
Iron–carboxylate bond formation
To probe ferrihaem iron–carboxylate bond formation in a lipid environment, DFT calculations were conducted. To reduce computational cost, calculations were performed on a truncated system which comprised a single H2O–Fe(III) porphine core, H2O–FeP, and either an acetic acid or acetate molecule which represented a propionic acid and propionate side chain, respectively. Since n-octanol has similar polarity to that measured for neutral glycerolipids,19 an implicit n-octanol solvent continuum model was used to mimic a lipid environment. DFT calculations revealed that formation of a five coordinate FeP–acetic acid product has a very small energy gain relative to H2O–FeP and acetic acid reactants (Fig. 5a). In contrast, coordination of an acetate anion to FeP produces a highly stable product relative to the H2O–FeP and acetate reactants (Fig. 5b). This is unsurprising given that this five coordinate FeP–acetate species is analogous to β-haematin, which is very stable. These observations are in accord with FeP bond dissociation energies reported for a variety of neutral and anionic ligands.39 Further calculations indicate that the formation of either a FeP–acetic acid or FeP–acetate species is less likely to proceed via a dissociative pathway since the four coordinate FeP intermediate is higher in energy than the reactants. Formation of a six coordinate intermediate is more favourable, on the other hand, particularly in the case of the acetate system. This finding suggests that iron–carboxylate bond formation could proceed through an associative pathway, although formation via a dissociative mechanism cannot be discounted given the small energy barrier.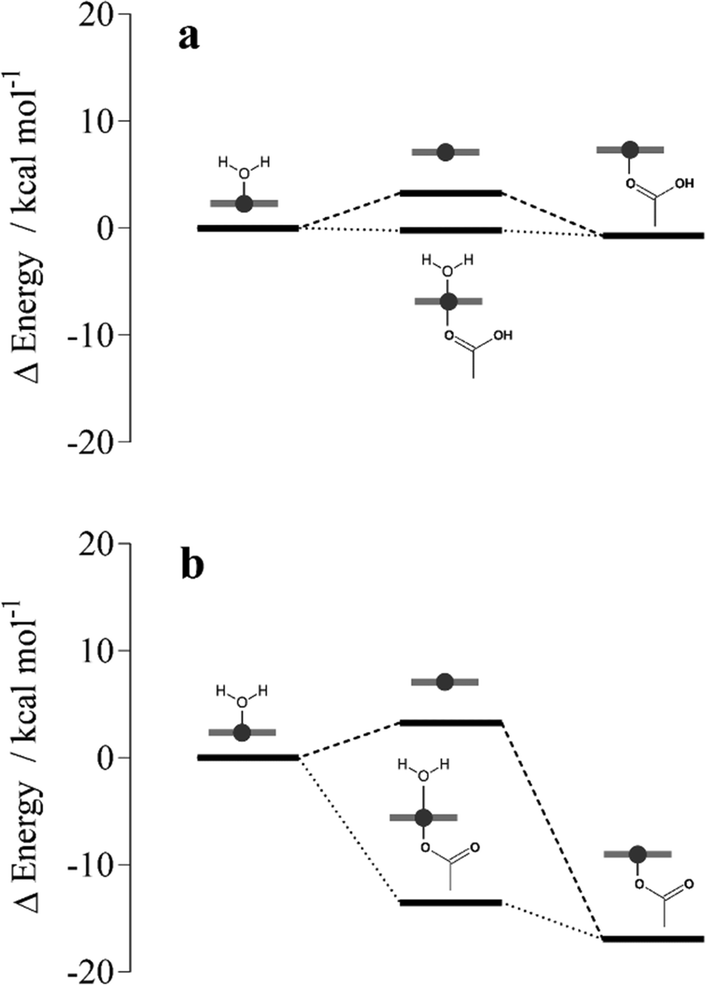 | ||
| Fig. 5 DFT-calculated energy level diagram of associative (dotted lines) and dissociative pathways (dashed lines) for the coordination of (a) acetic acid and (b) acetate to Fe(III) porphine in implicit n-octanol. Energies are reported relative to those computed for reactants H2O–Fe(III) porphine and acetate/acetic acid molecules (see Table S5† for values). Fe(III) porphine species are depicted schematically in grey. | ||
On the basis of these calculations, the feasibility of iron coordination occurring in an observed conformation of the π–π aggregate was next investigated. DFT geometry optimisations in implicit n-octanol were performed on H2O–FeP with an acetic acid or acetate molecule initially positioned at 3.4 Å. This distance is representative of that observed in MD simulations between an iron(III) atom and a propionic acid side chain (Fig. 4a). While greater distances were observed between iron centres and propionate groups, the MD simulations performed are unable to model proton transfer and thus it is plausible that the ionised form of a propionic acid side chain (i.e. a propionate) may also exist at 3.4 Å from the iron centre. Geometry optimisations of H2O–FeP and an acetic acid molecule initially placed at 3.4 Å revealed that coordination did not occur. Rather, the acetic acid molecule moved further away to a distance of 3.6 Å. In contrast, the acetate anion coordinated following geometry optimisation to give a H2O–FeP–acetate intermediate species. These findings thus indicate that iron coordination in the tetramer aggregate is indeed feasible, but likely only occurs in the case of the propionate and not the propionic acid side chain. In order for reciprocal propionate coordination to occur in the tetramer aggregate presented in Fig. 4a, the axial water ligand of the ferrihaem molecule with propionic acid 3.4 Å from the iron centre of its neighbour would first need to dissociate. Based on the fairly low energy barrier calculated from DFT for water ligand dissociation (3.3 kcal mol−1), this structural requirement is unlikely to hinder reciprocal coordination in the tetramer. These observations provide further support for the role of the π–π aggregate in nucleating Hz, assuming that nucleation occurs from an aggregate that resembles the final product.
Ferrihaem tautomerism
While the results presented in this work have focused on ferrihaem species which have an axial water ligand, one propionate and one propionic acid side chain (H2O–ferrihaem), it is important to note that a tautomer also exists. This tautomeric form can arise from proton transfer between an axial water ligand and a propionate side chain to give a species with an axial hydroxide ligand and two propionic acid side chains, HO–ferrihaem. Based on the absence of a propionate group and the fact that hydroxide binds more strongly to iron than water,48 this latter tautomer would not be expected to lead to iron coordination. Indeed, DFT calculations of HO–FeP confirm that no energetically favourable interaction with acetic acid or even acetate is expected (Table S5†). This suggests that HO–ferrihaem would not be capable of forming Hz and consequently ferrihaem tautomerism must have a pronounced effect on Hz formation.In an attempt to probe ferrihaem tautomerism in a non-aqueous environment, UV-visible spectroscopy was employed. Ideally, investigations would preferably have been conducted in a solvent such as n-octanol since this is a good mimic of a lipid environment. The poor solubility of ferrihaem in anhydrous n-octanol, however, necessitated the used of rigorously dried methanol instead. The UV-visible spectrum recorded of ferrihaem in anhydrous methanol displayed a moderately intense Soret band with a pronounced shoulder in the low wavelength region as well as several less intense peaks between 480 and 600 nm (Fig. 6a). These spectroscopic features are characteristic of HO–ferrihaem. This was confirmed by adding aliquots of water buffered at pH 10 to the system which produced almost no change to the spectrum (Fig. 6a). It was found that tautomerism could be induced through the addition of slightly acidified water (pH 6.5) to ferrihaem in anhydrous methanol. This produced marked changes to the spectrum which included intensifying of the Soret band as well as minor shifts in the long wavelength region (Fig. 6b). These observed spectroscopic features are consistent with the formation of the H2O–ferrihaem tautomer.37 The ability of water to influence ferrihaem tautomerism may arise as a result of solvation. In the absence of water, formation of the HO–ferrihaem tautomer is preferred, presumably because its two uncharged propionic acid side chains are more hydrophobic than the single propionate group in the zwitterionic H2O–ferrihaem tautomer. Evidence for this is seen from MD simulations which show that propionic acid groups are less solvated by water than their respective propionates. Addition of water to an anhydrous organic environment soon shifts the tautomeric equilibrium in favour of the H2O–ferrihaem species, where aqueous solvation of the hydrophilic propionate side chain and axial water ligand is now feasible.
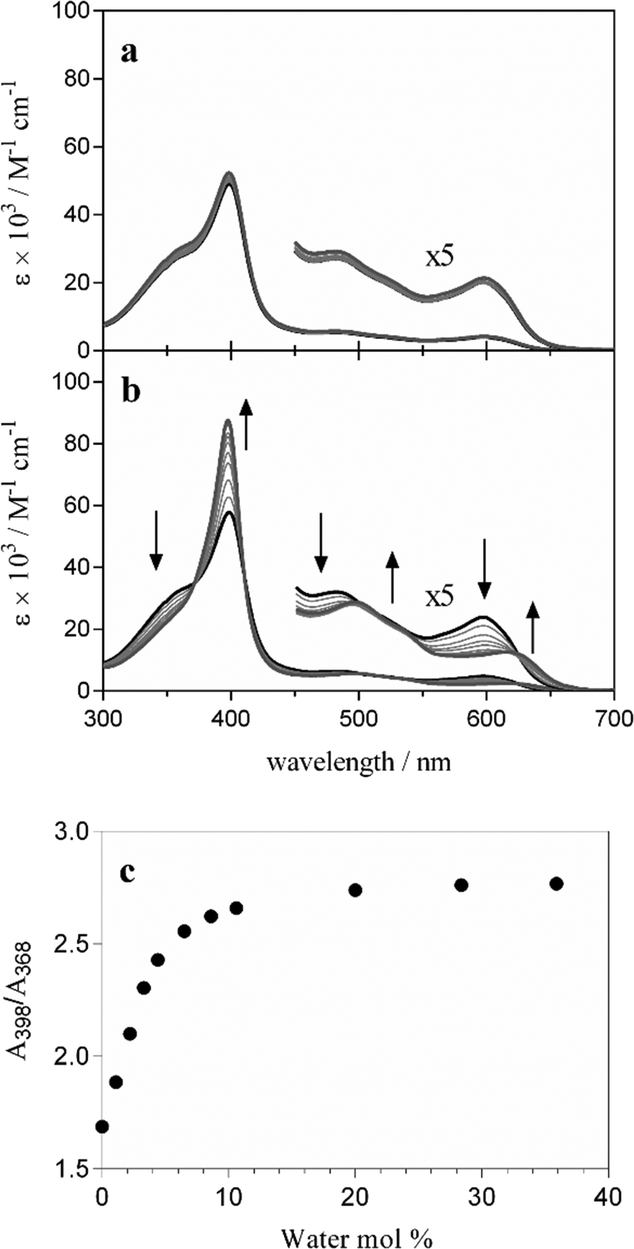 | ||
| Fig. 6 Effects of water on ferrihaem speciation in anhydrous methanol. Spectroscopic changes observed upon addition of water buffered at (a) pH 10 and (b) pH 6.5. The spectrum of ferrihaem in anhydrous methanol is represented by the thick black line and that obtained after addition of a 36% (mol) aqueous solution is given by the thick grey line. Arrows indicate the direction of absorbance change in (b). (c) Ratio of the Soret peak maximum (398 nm) to the isosbestic point on the absorbance shoulder (368 nm) in (b) as a function of aqueous buffer added to anhydrous methanol indicating the tautomerism of HO–ferrihaem to H2O–ferrihaem. 50% conversion occurs at 2.7% (mol) water. | ||
It can thus be concluded that the presence of water is essential in facilitating Hz formation since it promotes the formation of H2O–ferrihaem over HO–ferrihaem. The former tautomer is the only species capable of producing Hz because it contains a labile axial ligand and a propionate side chain, both of which are necessary for iron coordination. This conclusion is supported by experimental observations of β-haematin formation in n-octanol.49 Under anhydrous conditions, n-octanol is unable to support β-haematin formation, presumably because HO–ferrihaem is the predominant species. On the other hand, β-haematin is rapidly formed from n-octanol saturated with aqueous citric acid buffer (pH 4.8). Based on our findings, the water present in the latter system likely promotes the H2O–ferrihaem tautomer necessary for Hz formation. Indeed, the measured water content in this system23 is much greater than the approximately 10% (mol) required for essentially complete tautomerism to occur in methanol (Fig. 6c). Similarly, based on the water content measured in a mixture of glycerolipids,23 it can be concluded that H2O–ferrihaem is almost certainly the dominant tautomer present at such lipid–water interfaces which can mediate β-haematin formation. These findings highlight that the influence of water on ferrihaem tautomerism is an important consideration for Hz biocrystallisation.
Conclusions
Based on the findings from experimental and computational investigation, a mechanism describing the initial stages of Hz nucleation in P. falciparum is proposed (Fig. 7):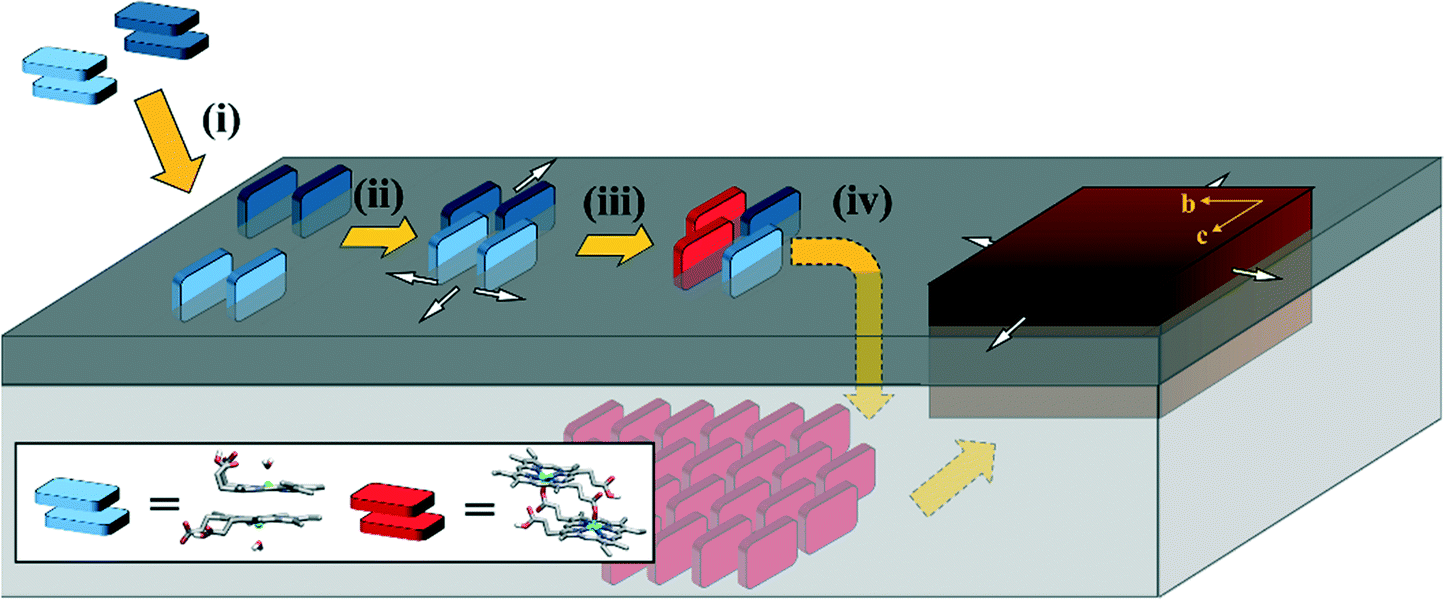 | ||
| Fig. 7 Proposed mechanism of the first stages of Hz nucleation at a lipid–water interface. Major aspects involving (i) ferrihaem partitioning from an aqueous medium into a lipid, (ii) ferrihaem aggregation within the lipid, (iii) iron coordination, and (iv) crystal growth are discussed in the text. For the sake of clarity, ferrihaem dimers supplied from the aqueous phase for crystal growth in step (iv) have been omitted. Lipid headgroups and tails are represented by dark and light grey blocks, respectively. The aqueous phase above the lipid headgroups is not shown and the ferrihaem π–π dimers are shaded in dark and light blue to highlight the inter-dimer nature of the reciprocal coordination reaction forming the μ-propionato dimer. White arrows indicate the proposed directions of π–π aggregate growth which correspond to the directions of fastest growth of the Hz crystal (brown block). | ||
Partitioning (i)
The neutral ferrihaem π–π dimer (consisting of two ferrihaem molecules each with an axial water ligand, one propionic acid and one propionate group) is the dominant form in an aqueous solution at pH 5. This amphiphilic species readily partitions into lipids near the hydrated headgroup region. Here the hydrophobic methyl and vinyl substituents are accommodated, while also facilitating favourable hydrogen bonds between solvent molecules and the axial water ligand, propionate and propionic acid side chains. Solvation of the first two hydrophilic groups by water at the interface ensures that the H2O–ferrihaem tautomer of the π–π dimer is maintained preferentially over the HO–ferrihaem species. The latter form of ferrihaem is unable to produce Hz since the more stable and less labile axial hydroxide ligand cannot be displaced by a propionate side chain.Aggregation (ii)
The spontaneous partitioning of H2O–ferrihaem π–π dimers into lipids causes an increased local concentration of ferrihaem within the lipid. This leads to aggregation at the lipid–water interface which is not a random conglomeration of molecules but rather consists of ferrihaem π–π dimers that are oriented with porphyrin rings perpendicular to the plane of the lipid interface. Growth of the aggregate likely occurs in the plane of the lipid surface. Glycerolipids seem highly efficient at promoting this process. Phospholipids on the other hand appear to have a lower propensity to promote ferrihaem aggregation, which coincides with their less efficient mediation of β-haematin crystallisation. This process of oriented aggregation is crucial to nucleating Hz crystals.Coordination (iii)
Dimer aggregation leads to inter-dimer interactions between ferrihaem propionate/propionic acid side chains and Fe(III) centres which facilitates iron coordination. Propionic acid side chains in proximity to an iron atom will not coordinate unless proton transfer to form a propionate is possible. Iron coordination by a propionate group is strongly favoured and upon reciprocal propionate coordination, the μ-propionato dimer is formed.Crystal growth (iv)
Once formed, the μ-propionato ferrihaem dimer penetrates deeper into the interior of the lipid. This occurs because the hydrophilic axial water ligand and a propionate side chain present in its π–π dimer precursor state have been released and coordinated to iron, respectively, leading to an overall more hydrophobic molecule. Within the non-polar lipid interior, there is a natural propensity for neighbouring μ-propionato dimers to hydrogen bond due to reduced competition from water molecules or polar lipid headgroups. The subsequent growth of Hz crystals likely follows the modified vapour–liquid–solid crystallisation mechanism described by Vekilov et al.46
This proposed mechanism applies to any aqueous–lipid or analogous biomimetic system that can promote β-haematin formation; however, based on TLC analysis, Hz biocrystallisation in P. falciparum likely occurs at a glycerolipid–water interface. This interface may exist as a lipid raft in the parasite DV membrane or as separate droplets within this same organelle. Currently, the precise identity and composition of these Hz-nucleating glycerolipids are still uncertain, but they form the focus of future studies. Further work on modelling Hz crystal growth using MD simulations is also currently underway.
Acknowledgements
Financial support for this work was provided by the National Research Foundation (NRF) (grant no. 88165 and grant no. CPRR13082330465). Any opinion, findings and conclusions or recommendations expressed in this material are those of the authors and therefore the NRF does not accept any liability in regard thereto. We thank the Centre for High Performance Computing in Cape Town and the Scientific Computing Research Unit at the University of Cape Town for the use of their computing resources as well as the Electron Beam Unit at Stellenbosch University and the Centre for Imaging & Analysis at the University of Cape Town for microscopy services. We also thank Dr. MA Ambele for providing transmission electron micrographs, Ms. NA Dare (Centre for Supramolecular Chemistry Research) for performing powder X-ray diffraction experiments and Dr. GA Venter at the University of Cape Town for helpful discussions.Notes and references
- S. Pagola, P. W. Stephens, D. S. Bohle, A. D. Kosar and S. K. Madsen, Nature, 2000, 404, 307–310 CrossRef CAS PubMed.
- M. F. Oliveira, J. C. P. D'Avila, C. R. Torres, P. L. Oliveira, A. J. Tempone, F. D. Rumjanek, C. M. S. Braga, J. R. Silva, M. Dansa-Petretski, M. A. Oliveira, W. de Souza and S. T. Ferreira, Mol. Biochem. Parasitol., 2000, 111, 217–221 CrossRef CAS PubMed.
- M. F. Oliveira, S. W. Kycia, A. Gomez, A. J. Kosar, D. S. Bohle, E. Hempelmann, D. Menezes, M. A. Vannier-Santos, P. L. Oliveira and S. T. Ferreira, FEBS Lett., 2005, 579, 6010–6016 CrossRef CAS PubMed.
- J. M. Pisciotta, E. L. Ponder, B. Fried and D. Sullivan, Int. J. Parasitol., 2005, 35, 1037–1042 CrossRef CAS PubMed.
- M. Lvova, M. Zhukova, E. Kiseleva, O. Mayboroda, P. Hensbergen, E. Kizilova, A. Ogienko, V. Besprozvannykh, B. Sripa, A. Katokhin and V. Mordvinov, Int. J. Parasitol., 2016, 46, 147–156 CrossRef CAS PubMed.
- D. Jani, R. Nagarkatti, W. Beatty, R. Angel, C. Slebodnick, J. Andersen, S. Kumar and D. Rathore, PLoS Pathog., 2008, 4, e1000053 Search PubMed.
- M. Chugh, V. Sundararaman, S. Kumar, V. S. Reddy, W. A. Siddiqui, K. D. Stuart and P. Malhotra, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5392–5397 CrossRef CAS PubMed.
- K. Bendrat, B. J. Berger and A. Cerami, Nature, 1995, 378, 138–139 CrossRef CAS PubMed.
- C. D. Fitch, G. Z. Cai, Y. F. Chen and J. D. Shoemaker, Biochim. Biophys. Acta, 1999, 1454, 31–37 CrossRef CAS.
- T. J. Egan, J. Y.-J. Chen, K. A. de Villiers, T. E. Mabotha, K. J. Naidoo, K. K. Ncokazi, S. J. Langford, D. McNaughton, S. Pandiancherri and B. R. Wood, FEBS Lett., 2006, 580, 5105–5110 CrossRef CAS PubMed.
- A. Dorn, S. R. Vippagunta, H. Matile, A. Bubendorf, J. L. Vennerstrom and R. G. Ridley, Biochem. Pharmacol., 1998, 55, 737–747 CrossRef CAS PubMed.
- J. M. Pisciotta, I. Coppens, A. K. Tripathi, P. F. Scholl, J. Schuman, S. Bajad, V. Shulaev and D. J. Sullivan, Biochem. J., 2007, 402, 197–204 CrossRef CAS PubMed.
- N. T. Huy, Y. Shima, A. Maeda, T. T. Men, K. Hirayama, A. Hirase, A. Miyazawa and K. Kamei, PLoS One, 2013, 8, e70025 CAS.
- R. Stiebler, D. Majerowicz, J. Knudsen, K. C. Gondim, D. W. Wright, T. J. Egan and M. F. Oliveira, PLoS One, 2014, 9, e88976 Search PubMed.
- S. Kapishnikov, A. Weiner, E. Shimoni, P. Guttman, G. Schneider, N. Dahan-Pasternak, R. Dzikowski, L. Leiserowtiz and M. Elbaum, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11188–11193 CrossRef CAS PubMed.
- J. B. R. Corrêa Soares, C. M. Maya-Monteiro, P. R. B. Bittencourt-Cunha, G. C. Atella, F. A. Lara, J. C. P. D'Avila, D. Menezes, M. A. Vannier-Santos, P. L. Oliveira, T. J. Egan and M. F. Oliveira, FEBS Lett., 2007, 581, 1742–1750 CrossRef PubMed.
- C. D. Fitch, Life Sci., 2004, 74, 1957–1972 CrossRef CAS PubMed.
- K. E. Jackson, N. Klonis, D. J. P. Ferguson, A. Adisa, C. Dogovski and L. Tilley, Mol. Microbiol., 2004, 54, 109–122 CrossRef CAS PubMed.
- A. N. Hoang, K. K. Ncokazi, K. A. de Villiers, D. W. Wright and T. J. Egan, Dalton Trans., 2010, 39, 1235–1344 RSC.
- M. A. Ambele and T. J. Egan, Malar. J., 2012, 11, 337 CrossRef CAS PubMed.
- K. A. de Villiers, M. Osipova, T. E. Mabotha, I. Solomonov, Y. Feldman, K. Kjaer, I. Weissbuch, T. J. Egan and L. Leiserowtiz, Cryst. Growth Des., 2009, 9, 626–632 CAS.
- M. A. Ketchum, K. N. Olafson, E. V. Petrova, J. D. Rimer and P. G. Vekilov, J. Chem. Phys., 2013, 139, 121911 CrossRef PubMed.
- K. N. Olafson, M. A. Ketchum, J. D. Rimer and P. G. Vekilov, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4946–4951 CrossRef CAS PubMed.
- W. Trager and J. B. Jensen, Science, 1976, 193, 673–675 CAS.
- J. F. Meisner, T. W. Chuprevich and S. L. Inhorn, Nat. New Biol., 1973, 245, 145–147 CrossRef PubMed.
- M. A. Ambele, B. T. Sewell, F. R. Cummings, P. J. Smith and T. J. Egan, Cryst. Growth Des., 2013, 13, 4442–4452 CAS.
- T. J. Egan, W. W. Mavuso and K. K. Ncokazi, Biochemistry, 2001, 40, 204–213 CrossRef CAS PubMed.
- J. Gildenhuys, T. le Roux, T. J. Egan and K. A. de Villiers, J. Am. Chem. Soc., 2013, 135, 1037–1047 CrossRef CAS PubMed.
- Purification of Laboratory Chemicals, ed. D. D. Perrin and L. F. Armarego, Pergamon Press, Oxford, 3rd edn, 1988 Search PubMed.
- B. R. Brooks, C. L. Brooks III, A. D. MacKerell Jr., L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, H. F. I. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- E. L. Wu, X. Cheng, S. Jo, K. C. Song, E. M. Dávila-Contreras, Y. Qi, J. Lee, V. Monje-Galvan, R. M. Venable, J. B. Klauda and W. Im, J. Comput. Chem., 2014, 35, 1997–2004 CrossRef CAS PubMed.
- S. Jo, J. B. Lim, J. B. Klauda and W. Im, Biophys. J., 2009, 97, 50–58 CrossRef CAS PubMed.
- S. Jo, T. Kim and W. Im, PLoS One, 2007, 2, e880 Search PubMed.
- D. Kuter, V. Streltsov, N. Davydova, G. A. Venter, K. J. Naidoo and T. J. Egan, Inorg. Chem., 2014, 53, 10811–10824 CrossRef CAS PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- D. Kuter, G. A. Venter, K. J. Naidoo and T. J. Egan, Inorg. Chem., 2012, 51, 10233–10250 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision C.01), Gaussian, Inc, Wallingford, CT, 2010 Search PubMed.
- M. C. Durrant, Dalton Trans., 2014, 43, 9754–9765 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- T. White, S. Bursten, D. Federighi, R. A. Lewis and E. Nudelman, Anal. Biochem., 1998, 258, 109–117 CrossRef CAS PubMed.
- E. D. Guerra, D. S. Bohle and M. Cerruti, Langmuir, 2016, 32, 4479–4484 CrossRef CAS PubMed.
- C. Asher, K. A. de Villiers and T. J. Egan, Inorg. Chem., 2009, 48, 7994–8003 CrossRef CAS PubMed.
- C. Y. Botté, Y. Yamaryo-Botté, T. W. T. Rupasinghe, K. A. Mullin, J. I. MacRae, T. P. Spurck, M. Kalanon, M. J. Shears, R. L. Coppel, P. K. Crellin, E. Maréchal, M. J. McConville and G. I. McFadden, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7506–7511 CrossRef PubMed.
- S. Gulati, E. H. Ekland, K. V. Ruggles, R. B. Chan, B. Jayabalasingham, B. Zhou, P. Y. Mantel, M. C. S. Lee, N. Spottiswoode, O. Coburn-Flynn, D. Hjelmqvist, T. S. Worgall, M. Marti, G. Di Paolo and D. A. Fidock, Cell Host Microbe, 2015, 18, 371–381 CAS.
- P. G. Vekilov, J. D. Rimer, K. N. Olafson and M. A. Ketchum, CrystEngComm, 2015, 17, 7790–7800 RSC.
- N. Klonis, R. Dilanian, E. Hanssen, C. Darmanin, V. Streltsov, S. Deed, H. Quiney and L. Tilley, Biochemistry, 2010, 49, 6804–6811 CrossRef CAS PubMed.
- J. E. Wertz and J. R. Bolton, Electron spin resonance: Elementary theory and practical applications, McGraw-Hill, New York, 1986 Search PubMed.
- K. N. Olafson, J. D. Rimer and P. G. Vekilov, Cryst. Growth Des., 2014, 14, 2123–2127 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ce00866f |
| This journal is © The Royal Society of Chemistry 2016 |