
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSyntheses, structures and properties of group 12 element (Zn, Cd, Hg) coordination polymers with a mixed-functional phosphonate-biphenyl-carboxylate linker†
Christian
Heering
a,
Biju
Francis‡
a,
Bahareh
Nateghi
a,
Gamall
Makhloufi
a,
Steffen
Lüdeke
 b and
Christoph
Janiak
*a
b and
Christoph
Janiak
*a
aInstitut für Anorganische Chemie und Strukturchemie, Universität Düsseldorf, Universitätsstr. 1, 40225 Düsseldorf, Germany. E-mail: janiak@hhu.de
bInstitut für Pharmazeutische Wissenschaften, Universität Freiburg, Albertstr. 25, 79104 Freiburg, Germany
First published on 25th May 2016
Abstract
The new phosphonate-carboxylate ligand from 4-phosphono-biphenyl-4′-carboxylic acid (H2O3P–(C6H4)2–CO2H, H3BPPA) is based on the rigid biphenyl system and is studied toward the coordination behavior of group 12 elements zinc, cadmium and mercury. The crystalline products from hydrothermal syntheses highlight the versatile and different coordination modes with the (partially) deprotonated H3BPPA ligand to give coordination polymeric 3D-[Zn5(μ3-OH)4(μ4-O3P–(C6H4)2–CO2-μ2)2]n (5), 2D-[Zn(μ6-O3P–(C6H4)2–CO2H)]n (6), 3D-[Cd3(μ5-O3P–(C6H4)2–CO2-μ2)(μ6-O3P–(C6H4)2–CO2-μ3)]n (7) and 2D-[Hg(μ3-HO3P–(C6H4)2–CO2H)]n (8). The cobalt complex, 2D-[Co(μ4-O3P–(C6H4)2–CO2H)]n (9) is isostructural to 6. Through additional classic strong carbonyl O–H⋯O hydrogen bonding the dimensionality of the 2D coordination networks increases to 3D supramolecular frameworks. The carboxy-phosphonate ligand shows five different coordination modes which can be described as μ4-O3P–CO2-μ2 (5), μ6-O3P– (6), μ5-O3P–CO2-μ2, μ5-O3P–CO2-μ3 (7), and μ3-O3P– (8), that is, the ligand bridges altogether between 3 to 8 metal atoms with the phosphonate group alone connecting already 3 to 6 metal atoms. Layers of metal–oxygen polyhedra are interconnected via the biphenyl linker, which either coordinates metal atoms with both donor groups or the –COOH end forms tail-to-tail hydrogen bonds to create 3D or 2D coordination networks, respectively. In the flat {MOx} layers in 6 and 7 the Zn and Cd metal nodes represent a honeycomb and an mcm net, respectively. The coordination polyhedra of the Cd atoms in compound 7 were analyzed towards a trigonal-prismatic coordination environment. The complexes are hydrolytically very stable due to their hydrothermal preparation from aqueous solution at 180–200 °C. The compounds could be stored in water or air for months without apparent decomposition. Compounds 5 and 7, where the ligand is fully deprotonated, start to decompose at ∼400 °C. The fluorescence emission spectrum of the ligand, 4, shows an intense peak at 365 nm (λex = 316 nm). The fluorescence emission of the metal complexes 5, 7 and 9 is shifted towards larger wavelengths with values of 417 nm, 415 nm and 410 nm, respectively (λex = 354 nm for 5, λex = 350 nm for 7, λex = 400 nm for 8, λex = 360 nm for 9). In addition, the crystal structures of the H3BPPA ligand precursors 4-iodo-4′-biphenylcarboxylic acid methyl ester, and 4-diethylphosphono-4′-biphenylcarboxylic acid methyl ester are described here for the first time.
Introduction
Metal–organic frameworks, MOFs, and coordination polymers became a rapidly growing research area in recent years, because of possible applications,1,2 for example in catalysis,3 sensing by fluorescence changes,4,5 gas storage6,7 and separation.8,9 However, the water or moisture stability of such networks is often low.10One approach to increase hydrothermal stability is the use of three- or four-valent, highly charged metal ions such as Cr3+, Al3+, Fe3+, Ti4+ or Zr4+ in MIL-MOF compounds11–14 (MIL = Materials of Institute Lavoisier) or UiO-MOFs15 (UiO University of Oslo).16–18 Carboxylate linkers were also replaced by azolates with improved stability of analogous or isoreticular compounds.19,20 Yet, the focus has seldom been set on very strongly coordinating (organo)phosphonate groups,21,22 which could significantly raise the stability of such metal–ligand coordination compounds.
Organophosphonic acids, which have a pKa1 of 2.0 for the first and a pKa2 of 6.59 for the second proton, are more acidic ligands than pyrazoles (pKa = 2.49). Organophosphonates generate strong metal–ligand coordinative bonds in thermodynamically stable complexes with high stability constants. Metal organophosphonate compounds are organic–inorganic hybrid materials,23 can be porous networks,24–27 and as such be placed in between zeolite-like28,29 and metal–organic framework materials.30 Metal organophosphonates are stable in water or aqueous environment31 and can be reversibly hydrated and dehydrated.27,32–36 The use of metal phosphonates in catalysis, luminescence,37 ion or proton exchange or conductivity38,39 and in separation is discussed and investigated.40 Cobalt and iron organophosphonates exhibit magnetic properties.41–48
Organophosphonates can contain additional functional groups such as carboxylate, hydroxyl or amino in the organo-moiety which presents a tunable functionality with a wide variety of structural motifs and properties.49
The controlled growth of crystalline metal–organophosphonate complexes is not a straightforward procedure.28 This is probably due to the strong metal–ligand bond, which is formed with little reversibility to correct for crystal defects. Therefore, we started to investigate carboxyl-phosphonate ligands,50 which can be seen as intermediates between pure carboxylates and pure phosphonates, and share synergies of both ligand classes. Metal complexes with mixed-functional 4-phosphono-benzoic acid (H3PPA) (Scheme 1 and S1 in ESI†) with barium,51 cobalt,52 copper,52 europium,53 lead,54 lithium,55 silver,56 strontium,57 thorium,58 titanium,59 uranium58 and zinc55,60,61 are known. Carboxy-phosphonates can form porous or 3D metal–ligand networks.62–64 Weng et al. described a 3D zinc carboxy-phosphonate, ZnPC-2, as a material for CO2 adsorption.65 As in MOFs, di- or tri-substituted rigid phosphonato-aryl-carboxylate linkers are a rational choice for rigid networks.
 | ||
| Scheme 1 4-Phosphono-benzoic acid (H3PPA) and 4-phosphono-4′-biphenylcarboxylic acid (H3BPPA, 4). | ||
Here we present the synthesis of the new mixed-functional linker 4-phosphono-4′-biphenylcarboxylic acid, H3BPPA (4) (Schemes 1 and 2) and four crystal structures of coordination networks with the group 12 metal ions zinc, cadmium and mercury.
 | ||
| Scheme 2 Reaction sequence for the synthesis of H3BPPA (4) from 4-biphenyl carboxylic acid. | ||
The concept of a mixed-functional rigid linker with carboxylate and phosphonate coordinating groups was used to obtain hydrothermally stable coordination networks. Up to now, no carboxy-phosphonate complexes with mercury have been known or published to the best of our knowledge.
Results and discussion
Linker synthesis
The linker H3BPPA has been synthesized for the first time here (Scheme 2), following a known procedure by Merkushev et al. to synthesize 4′-iodo-biphenyl-4-carboxylic acid (1) through the iodination of 4-biphenyl carboxylic acid.66 Compound 1 was transformed into the carboxy methyl ester 2, followed by the nickel(II) catalyzed conversion to a phosphonate ester (3), which after hydrolysis gave H3BPPA (4).Crystals of the 4-iodo-4′-biphenylcarboxylic acid methyl ester (2) were obtained by slow evaporation of a chloroform solution. The crystal structure of compound 2 shows a nearly in-plane conformation of both aryl rings and the carboxy methyl ester group (Fig. 1a). The dihedral angle between both aryl rings is 1.4(3)° and between the plane of the –COOMe group and its aryl ring it is 8.1(6)°. In 4-biphenyl carboxylic acid the dihedral angle between the –COOH group and the aryl ring is 29.05° and the dihedral angle between both aryl rings is 2.89°.67 This implies a better π-conjugation in the 4-iodo-derivative 2. Compound 2 crystallizes in the non-centrosymmetric orthorhombic space group Pca21. The non-centrosymmetric packing (Flack parameter 0.080(17))68 originates from the identical orientation of the iodo or carboxyl groups, respectively, along the crystallographic c axis at an angle of ±21.7(8)° (Fig. 1b).
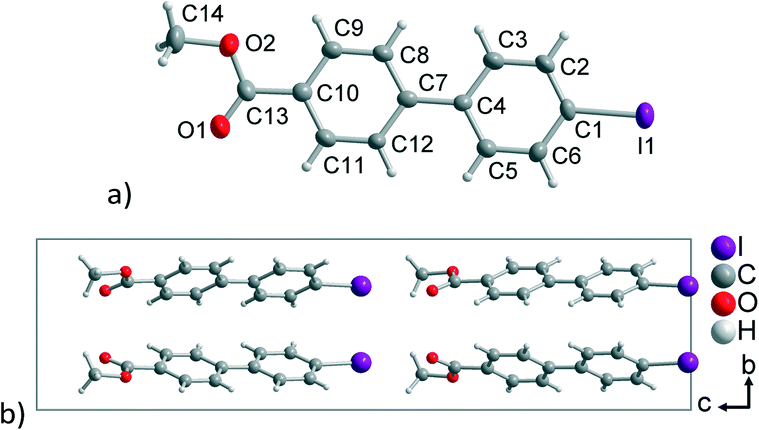 | ||
| Fig. 1 (a) Asymmetric unit (50% thermal ellipsoids) and (b) projection of the unit cell packing of 2. Selected distances (Å) and angles (°): C1–I1 2.082(9), C13–O1 1.202(13), C13–O2 1.303(12), O1–C13–C10 123.4(9), O1–C13–O2 123.6(10). | ||
Crystals of 4-diethylphosphono-4′-biphenylcarboxylic acid methyl ester (3) were derived from mesitylene solution. In the crystal structure of compound 3 (Fig. 2) the carboxy methyl ester and aryl ring form a dihedral angle of 13.7(2)°, the two aryl rings a dihedral angle of 35.8(2)°.
 | ||
| Fig. 2 (a) Asymmetric unit (50% thermal ellipsoids) and (b) crystal packing in 3. Selected distances (Å) and angles (°): P1–O1 1.560(2), P1–O2 1.462(2), P1–O3 1.567(2), C13–O5 1.195(3), C13–O4 1.341(3), O1–P1–O2 116.11(13), O2–P1–O3 113.98(13), O1–P1–O3 102.44(13). | ||
The thermal stability of the phosphono-carboxylic acid 4 (mp > 350 °C, decomp. > 400 °C, see TGA in Fig. S18 in ESI†) is higher than that of biphenyl-4,4′-dicarboxylic acid, which has a melting point of 310 °C.69
Metal compound syntheses and structures
Due to poor solubility of the phosphono-carboxylic acid in most common solvents, the reactions between ligand and metal salts were carried out under hydrothermal conditions at high temperatures (up to 180 °C) in water. All five metal complexes 5–9 share the common coordination motif of layers of metal ions, bridged by either carboxylate and phosphonate or the phosphonate group only (Scheme 3). | ||
| Scheme 3 Coordination modes of the BPPA ligand in the structures of 5–9. The metal atom (M) can be Zn, Cd, Hg or Co. | ||
The zinc compound [Zn5(μ3-OH)4(μ4-O3P–(C6H4)2–CO2-μ2)2] (5) was obtained from a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio of Zn(OAc)2 and H3BPPA in water in the presence of oxalic acid. Oxalic acid was added as a buffer to give stable pH values. There was little effect on the yield or product composition when the reactant stoichiometry was adjusted to the ratio found in the X-ray structure in subsequent repeated reactions.
:2 molar ratio of Zn(OAc)2 and H3BPPA in water in the presence of oxalic acid. Oxalic acid was added as a buffer to give stable pH values. There was little effect on the yield or product composition when the reactant stoichiometry was adjusted to the ratio found in the X-ray structure in subsequent repeated reactions.
The structure of 5 consists of Zn(II) ions that are coordinated by hydroxide ions and either carboxylate (–COO−) or phosphonate groups (–PO32−). The asymmetric unit contains three crystallographically different zinc atoms (one of them, Zn3 is half occupied on the special position of a C2 rotation axis, bisecting the O1–Zn–O1vi angle, Fig. 3a), two triply bridging (μ3) hydroxide ligands and one fully deprotonated BPPA3− ligand. Thereby, a {Zn2O4} tetrahedron, a {Zn1O5} trigonal bipyramid and a {Zn3O6} octahedron are formed (Fig. 3 and Fig. S27 in ESI†). For the five-coordinated Zn1 atom the difference between the two largest angles yields τ = (172.9 − 128.6)°/60° = 0.74, which is closer to the value for a trigonal bipyramid (τ = 1) than for a square pyramid (τ = 0).70 The two oxygens from the –COO− group bridge two Zn ions and the –PO32− group is coordinated to four Zn ions (Fig. 3a). Overall the BPPA3− ligand bridges between six Zn atoms. The {ZnOx} polyhedra are edge- and corner-sharing (Fig. 3c and S28 in ESI†). The {ZnOx} (x = 4–6) polyhedra with their bridging μ3-OH−, μ4-PO32− and μ2-COO− groups are arranged in layers (parallel to the bc plane) (Fig. 3b and c) and these layers are connected by the biphenyl part of the ligand to a 3D network (Fig. 3d).
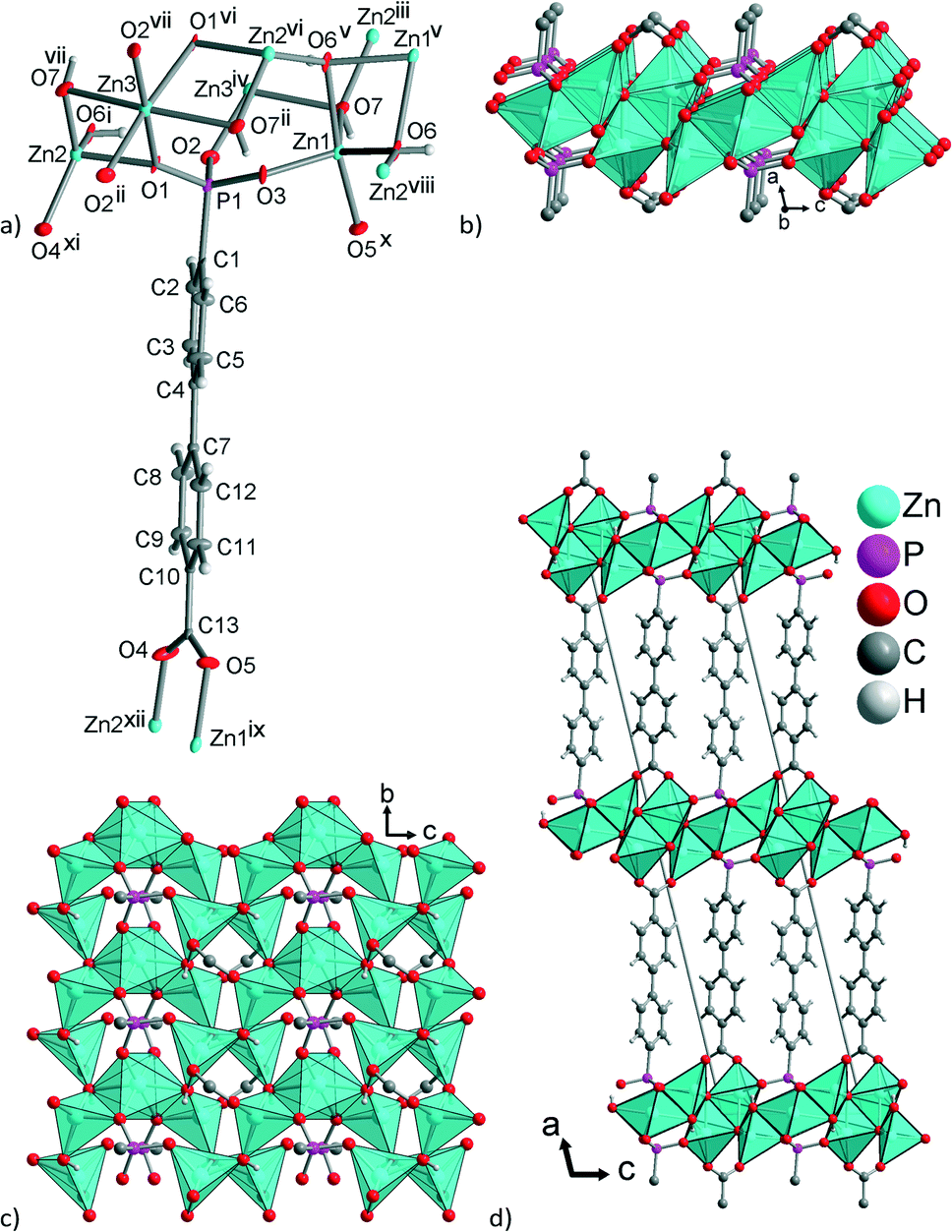 | ||
| Fig. 3 (a) Expanded asymmetric unit of 5 (70% thermal ellipsoids, except for C with 50% and H with arbitrary radii) and (b, c) layers of zinc ions, bridged by hydroxido, carboxylate and phosphonate groups, {Zn5(μ3-OH)4(μ4-O3PC–)2(–CO2-μ2)2} with the {ZnOx} polyhedra presented as such. (d) Projection of the three-dimensional packing on the ac plane. Symmetry transformations: i = x, y, 1 + z; ii = x, 1 + y, z; iii = x, y, −1 + z; iv = x, −1 + y, z; v = −x, y, −z; vi = −x, y, 1 − z; vii = −x, 1 + y, 1 − z; viii = −x, −1 + y, 1 − z; ix = 1/2 − x, 1/2 + y, 1 − z; x = 1/2 − x, −1/2 + y, 1 − z; xi = 1/2 − x, 1/2 + y, 2 − z; xii = 1/2 − x, −1/2 + y, 2 − z. Further figures also showing the O–H⋯O H-bonds are given in ESI† as Fig. S27–S29. Selected distances and angles are given in Table 1. | ||
The phosphorus atom in 5 has four different substituents, thus, is asymmetric and R-configured in the investigated single crystal. Only P atoms of the same handedness are assembled in the layers of 5 giving the non-centrosymmetric point group C2 (Flack parameter 0.030(3)).68 Circular dichroism (CD) spectra of 5 performed with KBr pellets (0.05 wt% of 5 according to a previously published protocol for solid state CD)71 (Fig. S33 in ESI†) do not contain any significant spectral features that would indicate enantiomeric excess of either one of the two possible enantiomers of 5. Therefore, as previously observed for other cases of spontaneous resolution,72,73 the overall crystal ensemble is racemic.
Zn compounds with 4-phosphonate-biphenyl-3′,5′-dicarboxylate as a linker and templating amine bases are known74 but do not show the same layer motif. A zinc complex with the shorter 4-phosphonate-benzoate ligand PPA3− has a zeolite-like structure, in which zigzag chains exist and {Zn4O} tetrahedra are coordinated by PPA3−.75
The zinc compound [Zn(μ6-O3P–(C6H4)2–CO2H)] (6) was obtained from a 1:1 molar ratio of Zn(NO3)2·4H2O and H3BPPA in water without any modifier. No deprotonating or buffer agent was added. The structure of 6 consists of Zn(II) ions that are coordinated by the HBPPA2− ligand with only the phosphono group deprotonated (–PO32−) and the carboxyl group still protonated (–COOH). The asymmetric unit contains a unique zinc atom and the ligand HBPPA2− on special positions with half-occupancy. This leads to a crystallographically induced disorder of the ligand over two split-positions (Fig. 4a).
 | ||
| Fig. 4 (a) Expanded asymmetric unit of the zinc compound 6 (isostructural with the cobalt compound 9) (50% thermal ellipsoids) and (b, c) layers of zinc ions, bridged by phosphonate groups, {Zn(μ6-O3PC–)} with the {ZnO6} octahedra shown as polyhedra. (d) Section of the three-dimensional supramolecular packing through the tail-to-tail hydrogen-bonding arrangement (orange dashed lines) of the carboxylic acid groups. Symmetry transformations: i = −x + 1, y, −z + 1; ii = −x + 3/2, −y + 3/2, −z + 1; iii = x − 1/2, y + 1/2, z; iv = −x + 1/2, −y + 3/2, −z + 1; v = −x + 1, −y + 1, −z + 1; vi = x, −y + 1, z; vii = x + 1/2, y − 1/2, z; viii = −1 + x, y, z; ix = 1 − x, y, 1 − z; x = −1/2 + x, −1/2 + y, z; xi = −1/2 + x, 3/2 − y, z; xii = 1/2 + x, 3/2 − y, z; xiii = 2 − x, 1 − y, −z. Selected distances and angles are given in Table 2. | ||
In the cobalt structure with the shorter 4-phosphono benzoic acid ligand,52 which is analogous to compound 9 (see below), the same disorder of the phenyl ring and of the –COOH was observed. The Zn atom is coordinated by six oxygen atoms in a distorted octahedron with four shorter bonds in the equatorial plane and two longer axial bonds. The oxygen atoms of μ6-PO32− coordinate to six zinc ions with each O atom bridging two zinc ions (Fig. 4a). The distorted {ZnO6} octahedra are edge-sharing and form a flat honeycomb layer parallel to the ab plane (Fig. 4b and c). The 2D-{Zn(μ6-O3PC–)} layers are connected by the biphenyl-carboxylic acid part of the ligand to a supramolecular 3D network through the carboxylic acid groups which are oriented towards each other with the typical tail-to-tail arrangement, also known as R22(8)-motif in the Etter-notation (Fig. 4d and Table S5 in ESI†).76
Similarly compound [Co(μ6-O3P–(C6H4)2–CO2H)] (9) was prepared from an equimolar ratio of CoCl2 and H3BPPA in water. The single-crystal data set of 9 was not of sufficient quality to present a publishable structure determination. Yet, the isostructural nature of 6 and 9 could be established and also confirmed by comparison of the powder X-ray diffractograms (see Fig. S24 in ESI†).
The cadmium compound [Cd3(μ6-O3P–(C6H4)2–CO2-μ2)(μ6-O3P–(C6H4)2–CO2-μ3)] (7) was obtained from a 3:2 molar ratio of Cd(OAc)2 and H3BPPA in water in the presence of oxalic acid (added to allow stable pH values).
In the crystal structure of 7, the asymmetric unit consists of three different Cd atoms and two fully deprotonated BPPA3− ligands. The Cd atoms are coordinated by six oxygen atoms from both μ2- and μ3-COO− and μ5-PO32− groups (Fig. 5a).
 | ||
| Fig. 5 (a) Expanded asymmetric unit of the cadmium compound 7 (Cd and P 70%, C and O 50% thermal ellipsoids) and (b, c) layers of cadmium ions, bridged by phosphonate and carboxylate groups, {Cd3(μ5-O3PC–)2(–CO2-μ2/μ3)2} with the {CdO6} trigonal prisms depicted as polyhedra. (d) Projection of the three-dimensional packing on the ac plane. Symmetry transformations: i = 1/2 − x, y, 1/2 + z; ii = 1/2 − x, y, −1/2 + z; iii = −x, 2 − y, 1/2 + z; iv = −x, 2 − y, −1/2 + z; v = −1/2 + x, 1 − y, z; vi = 1/2 + x, 1 − y, z; vii = x, −1 + y, z; ix = 1/2 + x, 2 − y, z; x = −1/2 + x, 2 − y, z; xi = −x, 1 − y, −1/2 + z; xii = −x, 1 − y, 1/2 + z; xiii = 1/2 − x, −1 + y, −1/2 + z; xiv = 1/2 − x, 1 + y, 1/2 + z. Selected distances are given in Table 3. Angles are listed in Table S6 in ESI.† | ||
Significant distortion from classical octahedral towards trigonal-prismatic (TP) geometry around Cd is clearly revealed in the structure of 7 (see Cd-polyhedra in Table S12 in ESI†). An idealized octahedron has two exactly parallel, staggered equilateral triangles and all edges of side s (Scheme 4). Consideration of the angular parameters θ, ρ and ω (Scheme 5) in the comparison between octahedral and trigonal prismatic limiting structures77,78 indicates that the Cd polyhedra in compound 7 can be regarded as distorted trigonal prisms (see average values in Scheme 5 from the data in Table S12 in ESI†).
 | ||
| Scheme 4 Octahedron and trigonal prism with enhanced triangular faces and relevant parameters for the assessment of their relationship. ϕ is the twist angle between the enhanced triangles around the C3 axis. | ||
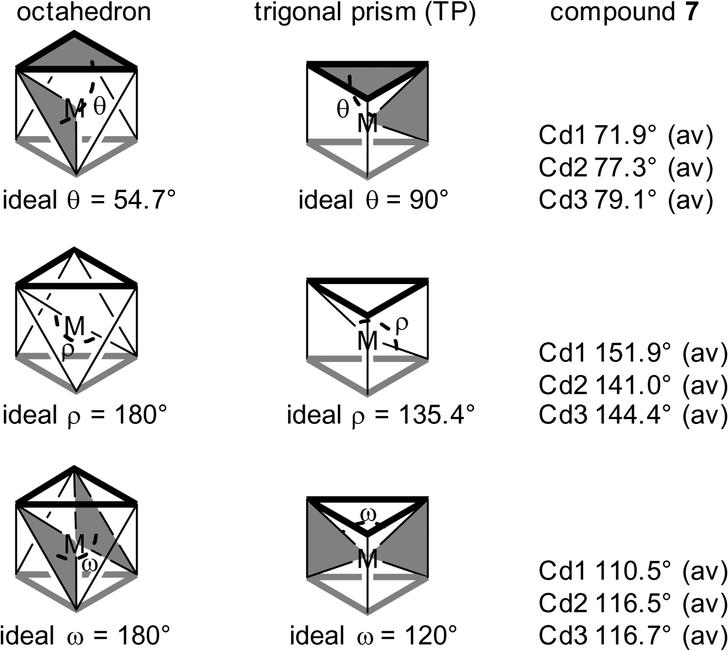 | ||
| Scheme 5 Angular parameters involved in the comparison between the octahedral and trigonal-prismatic limiting structures. θ = angle between the mean plane of the two O3 triangles and the chelate planes defined by the metal and each pair of near eclipsed O vertices. ρ = angle between the metal and trans O-donor sites. ω = angle between the triangular faces defined by the metal and near eclipsed O atoms. The angle for Cdi is the average of three angles, which are listed in Table S12 in ESI.† | ||
The –PO32− oxygens bridge between five Cd atoms, while the –COO− groups bridge either two or three Cd atoms (Fig. 5a, Scheme 3 and Fig. S31 in ESI†). Overall, the BPPA3− ligand bridges seven or eight Cd atoms. The edge- and vertex-sharing {CdO6} trigonal prisms with their bridging μ5-PO32− and μ2/μ3-COO− groups are arranged in flat layers (parallel to the ab plane) (Fig. 5b and c) and these layers are connected by the biphenyl part of the ligand to a 3D network (Fig. 5d).
Compound 7 crystallizes in the non-centrosymmetric orthorhombic space group Pca21. The non-centrosymmetric packing (Flack parameter 0.007(9))68 originates from the identical orientation of the phosphonato ends (or the carboxylato ends, respectively) of the 2−O3P–(C6H4)2–CO2− linkers along the crystallographic c axis (Fig. 5d).
Five edge- and vertex-sharing {CdO6} polyhedra form a pentagon, which assembles into a 2D mcm-net (Fig. 6a).79 In its regular form this pentagon tiling consists of two kinds of vertices (53 and 54, connecting 3 or 4 pentagons, respectively), two kinds of edges and one kind of face.
 | ||
| Fig. 6 Left: Layer of {CdO6} polyhedra in 7 with the Cd atoms connected by black lines to illustrate the pentagonal mcm tiling. Right: mcm net from http://rcsr.net/layers/mcm.79 | ||
The mercury(I) compound with the sum formula [Hg(μ3-HO3P–(C6H4)2–CO2H)] (8) was obtained from an equimolar ratio of mercury(II) acetate, Hg(OAc)2, and H3BPPA in water in the presence of oxalic acid to allow stable pH values. In 8, the mercury oxidation state assignment depends on the presence or absence of the calculated proton on the phosphonato group, −HO3P.
The phosphonate group of the ligand remains singly protonated (or is only singly deprotonated), that is, –PO3H− with the O3 atom carrying the H atom. The carboxyl group remains protonated as is clearly evidenced by the tail-to-tail hydrogen-bonding arrangement. The H3 atom on O3 of –PO3H− could be found and refined with Uiso(H) = 1.5Ueq(O) and the DFIX restraint 0.90 ± 0.05 Å.
Bond valence sum calculations80,81 done on the Hg atom of 8 gave a value of 0.83 when assuming a valence of 1 and a value of 1.00 when assuming a valence of 2. The calculated values support the valence of 1.
In the structure of 8, two mercury ions form a handle with C2-symmetry-related Hg atoms at a Hg–Hg distance of 2.5116(14) Å, presumably the well-known dinuclear Hg22+ dication. A possible Hg24+ cation is rare. The di-mercury(II) complex bis(μ-P,N-1-benzyl-2-imidazolyl-diphenylphosphine)(μ-O,O′-diperchloratedimercury(II) diperchlorate (C44H38N4O16P2Cl4Hg2) is one example,82 but the Hg–Hg distance with 3.071(1) Å is longer than in 8. The short Hg–Hg distance of ∼2.5 Å falls in the distance range of 2.46–2.60 Å observed for a Hg(I)22+ cation.83
Hence, we assume a Hg(I) oxidation state with single-deprotonated H3BPPA ligand. The asymmetric unit contains one unique Hg atom and one H2BPPA− ligand. Each Hg atom is coordinated by three oxygens from three different −HO3P-groups (Fig. 7a) with a severely distorted tripodal HgO3 coordination, which is tilted with respect to the Hg–Hg bond (Fig. 7b and Fig. S32, ESI†). There is one short Hg1–O1 contact of 2.118(13) Å and two considerably longer contacts Hg1–O2iii = 2.566(16) Å and Hg1–O3iv = 2.729(15) Å (Table 4 and Fig. 7a). This coordination environment agrees with a classical L–Hg(I)–Hg(I)–L species. For Hg(II), a higher coordination number of four to eight is expected, whereas a coordination number of three for H(II) is very rare.84 More importantly, no direct Hg–Hg bonds are found in coordination polymers of mercury(II).84 For Hg22+, there are several examples of complexes in the Cambridge Structure Database in which the overall coordination number of a Hg(I) atom is four.85 Furthermore, an in situ reduction of Hg2+ to Hg22+ was also observed by Su et al.85c
 | ||
| Fig. 7 (a) Expanded asymmetric unit of 8 (Hg and P 70%, C and O 50% thermal ellipsoids; H bonds are orange dashed lines) and (b, c) layers of Hg22+ handles, bridged by phosphonate groups, {Hg2(μ3-HO3PC–)2} with the {HgO3}/{Hg2O6} depicted as polyhedra. (d) Section of the supramolecular three-dimensional packing through the tail-to-tail hydrogen-bonding arrangement (orange dashed lines) of the carboxylic acid groups. Symmetry transformations: i = 1 − x, y, 1/2 − z; ii = x, −y, 1/2 + z; iii = x, −y, −1/2 + z; iv = x, 1 − y, −1/2 + z; v = 1 − x, −y, 1 − z; vi = x, 1 + y, z; vii = 1 − x, 1 + y, 1/2 − z; viii = 1 − x, 1 − y, 1 − z; ix = 1 − x, −1 + y, 3/2 − z; x = x, 2 − y, −1/2 + z; xi = x, −1 + y, z; xii = 1 − x, 2 − y, 1 − z. Selected distances and angles are given in Table 4; hydrogen bonding interactions in Table S8 in ESI.† | ||
| a Symmetry transformations: i = x, y, 1 + z; ii = x, 1 + y, z; iii = x, y, −1 + z; iv = x, −1 + y, z; v = −x, y, −z; vi = −x, y, 1 − z; vii = −x, 1 + y, 1 − z; viii = −x, −1 + y, 1 − z; ix = 1/2 − x, 1/2 + y, 1 − z; x = 1/2 − x, −1/2 + y, 1 − z; xi = 1/2 − x, 1/2 + y, 2 − z; xii = 1/2 − x, −1/2 + y, 2 − z. | |||
|---|---|---|---|
| Zn1–O3 | 1.940(4) | Zn2–O4xi | 1.909(5) |
| Zn1–O5x | 2.010(5) | Zn2–O6i | 1.970(4) |
| Zn1–O6 | 2.007(4) | Zn2–O7vii | 1.974(4) |
| Zn1–O6v | 2.256(5) | Zn3–O1 | 2.133(4) |
| Zn1–O7 | 2.014(5) | Zn3–O2ii | 2.013(5) |
| Zn2–O1 | 1.991(4) | Zn3–O7ii | 2.264(4) |
| O3–Zn1–O6v | 110.82(18) | O4xi–Zn2–O1 | 103.41(19) |
| O3–Zn1–O5x | 96.31(18) | O6i–Zn2–O1 | 104.64(19) |
| O6–Zn1–O5x | 99.97(18) | O7vii–Zn2–O1 | 89.09(18) |
| O3–Zn1–O7 | 116.73(18) | O2ii–Zn3–O2vii | 97.0(3) |
| O6–Zn1–O7 | 128.57(19) | O2ii–Zn3–O1 | 86.30(16) |
| O5x–Zn1–O7 | 93.40(19) | O2vii–Zn3–O1 | 175.21(18) |
| O3–Zn1–O6v | 90.79(17) | O1vi–Zn3–O1 | 90.6(2) |
| O6–Zn1–O6v | 77.4(2) | O2ii–Zn3–O7ii | 98.01(16) |
| O5x–Zn1–O6v | 172.90(17) | O2ii–Zn3–O7vii | 90.37(16) |
| O7–Zn1–O6v | 83.34(17) | O1–Zn3–O7ii | 92.64(16) |
| O4xi–Zn2–O6i | 110.3(2) | O1vi–Zn3–O7ii | 78.43(15) |
| O4xi–Zn2–O7vii | 126.2(2) | O2ii–Zn3–O7vii | 90.37(16) |
| O6i–Zn2–O7vii | 116.62(19) | O2vii–Zn3–O7vii | 98.01(16) |
| O1–Zn3–O7vii | 78.32(15) | ||
| Zn1–O6–Zn2iii | 127.5(2) | O1vi–Zn3–O7vii | 92.64(16) |
| Zn2–O1–Zn3 | 97.19(17) | O7ii–Zn–O7vii | 167.4(2) |
| Zn1–O7–Zn3iv | 110.38(19) | ||
| a Symmetry transformations: i = −x + 1, y, −z + 1; ii = −x + 3/2, −y + 3/2, −z + 1; iii = x − 1/2, y + 1/2, z; iv = −x + 1/2, −y + 3/2, −z + 1; vii = x + 1/2, y − 1/2, z; xii = 1/2 + x, 3/2 − y, z. | |||
|---|---|---|---|
| Zn–O1 | 1.931(4) | Zn–O1xii | 2.620(4) |
| Zn–O2iii | 1.973(6) | ||
| O1–Zn–O1i | 93.2(3) | O1iv–Zn–O1xii | 171.1(3) |
| O1–Zn–O2ii | 162.8(3) | O1–Zn–O1xii | 82.8(4) |
| O1–Zn–O2iii | 93.7(2) | Zn–O1–Zniv | 76.6(2) |
| O2ii–Zn–O2iii | 84.2(4) | Znii–O2–Znvii | 95.8(4) |
| O1i–Zn–O1xii | 103.4(3) | ||
| a For angles see Table S6 in ESI. Symmetry transformations: i = 1/2 − x, y, 1/2 + z; ii = 1/2 − x, y, −1/2 + z; iii = −x, 2 − y, 1/2 + z; v = −1/2 + x, 1 − y, z; vi = 1/2 + x, 1 − y, z; vii = x, −1 + y, z; ix = 1/2 + x, 2 − y, z; x = −1/2 + x, 2 − y, z; xii = −x, 1 − y, 1/2 + z; xiii = 1/2 − x, −1 + y, −1/2 + z; xiv = 1/2 − x, 1 + y, 1/2 + z. | |||
|---|---|---|---|
| Cd1–O1 | 2.370(7) | Cd2–O8v | 2.273(8) |
| Cd1–O2 | 2.294(7) | Cd2–O9xii | 2.387(9) |
| Cd1–O3x | 2.359(8) | Cd2–O10xii | 2.458(9) |
| Cd1–O4iii | 2.177(9) | Cd3–O3 | 2.328(8) |
| Cd1–O7vii | 2.263(7) | Cd3–O5i | 2.132(9) |
| Cd1–O9xiv | 2.406(9) | Cd3–O6 | 2.324(8) |
| Cd2–O1x | 2.286(8) | Cd3–O7vi | 2.201(7) |
| Cd2–O2 | 2.226(7) | Cd3–O8 | 2.328(8) |
| Cd2–O6 | 2.289(8) | Cd3–O10i | 2.541(10) |
| a Symmetry transformations: i = 1 − x, y, 1/2 − z; ii = x, −y, 1/2 + z; iii = x, −y, −1/2 + z; iv = x, 1 − y, −1/2 + z. | |||
|---|---|---|---|
| Hg1–O1 | 2.118(13) | Hg1–O3iv | 2.729(15) |
| Hg1–O2iii | 2.566(16) | Hg1–Hg1i | 2.5116(14) |
| O1–Hg1–Hg1i | 171.7(4) | P1–O1–Hg1 | 133.6(9) |
| O1–Hg1–O2iii | 78.3(5) | P1–O2–Hg1ii | 120.1(8) |
| Hg1i–Hg1–O2iii | 108.9(3) | O3iv–Hg1–Hg1i | 101.3(3) |
| O3iv–Hg1–O2iii | 97.0(5) | ||
The phosphonate groups of six ligands (three on each Hg) are bridging the Hg–Hg handles to a flat 2D-{Hg2(μ3-HO3P–)} layer (Fig. 7b and c). These 2D-layers are connected by the biphenyl-carboxylic acid part of the ligand to a supramolecular 3D network through the carboxylic acid groups which are oriented towards each other in the tail-to-tail arrangement (R22(8)-motif in the Etter-notation)76 (Fig. 7d and S32, Table S8, ESI†).
A mixed-valent mercury complex with L-alanine, [Hg12(L-ala)8(NO3)8]·2H2O showed a coordination environment for Hg similar to the one found for 8.86 A compound of mercury with a phosphono-carboxylate linker has been unknown.
There are seven entries in the Cambridge Structure Database, CSD, with a –P–O–Hg coordination, from which only two (AFIWES87 and SARSAF88) contain phosphonates coordinated to Hg(II) and of which none has Hg–Hg handles.
All metal-linker compound 5–9 share the layer motif for the {MOx} polyhedra. The driving force appears to be the separation of polar and nonpolar parts of molecules, which is a common packing motif.89–91
CH-π contacts between neighbouring biphenyl systems in 5–8 are listed in Tables S9–S11 in ESI† with angles and distances of significant interactions.
A listing of the dihedral angles in the BPPA linker in complexes 5–8 is given in Table S13 in the ESI.† With dihedral angles ranging from 0.1(6)° (5) to 2.0(2)° (8), the aryl systems are nearly co-planar (0°) to each other, favouring the π-electron delocalisation. The aryl-COO(H) angles vary from 2.0(2)° (6, 7) to 16.3(9)° (5), which is explained by the different state of protonation and coordination of the –COO group. Still, a close to planar geometry of the ligand is observed in all complexes.
Hydrothermal stability
The noteworthy water stability of the complexes 5–9 has been investigated by long-time exposure of the complexes in water. IR and powder X-ray diffraction, PXRD, did not indicate any structural changes within months. Furthermore, the hydrothermal synthesis conditions (water at 180–200 °C) would not allow the formation of water-unstable structures.The thermogravimetric behavior in Fig. 8 (detailed Fig. S19–S23 in ESI†) reveals a high thermal stability for 5 and 7. The thermally stable ligand (m.p. >350 °C) contributes to the overall stability of the compounds through the strong metal-phosphonate coordination. The mass loss of ∼5% in 4 (see Fig. S18 in ESI†) can be explained by a dehydration (−1H2O, 18 g mol−1, 6%) through the condensation of phosphonic acid groups to give anhydrides.92
 | ||
| Fig. 8 Thermogravimetric analysis (TGA) curves of compounds 4–9. | ||
Compounds 5 and 7, where the ligand is fully deprotonated without a remaining –COOH group, start to decompose at ∼400 °C. The isostructural compounds 6 and 9, where only the phosphono group is deprotonated (–PO32−) and the carboxyl group still protonated (–COOH), show a steady weight loss starting at ∼150–200 °C, presumably first by a condensation (−1H2O, 18 g mol−1: 5.3% in 6 and 9) followed by a decarboxylation (−1CO2, 44 g mol−1: 13% in 6; 13.5% in 9). The mercury compound 8 undergoes a rapid mass loss, starting already at ∼100 °C, which can also be observed in other Hg complexes.93 A decarboxylation is observed in the temperature range from 120 to 180 °C (−1CO2, 45 g mol−1, 9.5%). Hg complexes vaporize completely because of the high volatility of elemental Hg above 500 °C. Elemental Hg is formed by decomposition of intermediate HgO to its elements.
Luminescence
Many organic molecules are only fluorescent in solution because in their solid-state they are subject to self-quenching with concomitant low quantum yield of their luminescence.94 Immobilization/rigidification of organic molecules as linkers or as guest molecules in a solid framework like a coordination polymer95 or MOF with d10 metal ions such as Zn2+ and Cd2+ will reduce non-radiative relaxation caused by free rotation and vibration and, therefore, lead to stronger emissions.96,97The emission spectrum of 4 (Fig. 9) shows a broad band with a maximum at around 365 nm when excited at 316 nm. The spectrum is comparable to that of dihydroxybiphenyls, such as 4,4′-dihydroxybiphenyl (λmax = 354 nm) and 2,2′-dihydroxybiphenyl (λmax = 356 nm) and many other previously reported substituted biphenyl systems (λmax = 318–420 nm).98
 | ||
| Fig. 9 Solid-state luminescence spectra of complexes 5 (λex = 354 nm), 7 (λex = 350 nm), 8 (λex = 345 nm) and 9 (λex = 360 nm) in comparison with the spectrum of the ligand, 4 (λex = 316 nm). The spikes in the emission spectra are probably due to the lamp fluctuations or random pickup by the photomultiplier. | ||
It is well documented that the luminescence observed in MOFs or coordination polymers of transition metal ions without unpaired electrons (such as Cd2+ and Zn2+) are typically ligand centered.99,100 The emission spectra of the Zn compound 5 (λex = 354 nm), the Cd compound 7 (λex = 350 nm), the Hg compound 8 (λex = 345 nm) and the Co compound 9 (λex = 360 nm) also exhibit broad emission bands in the visible range at 410–420 nm suggesting that the luminescence in all compounds originate from the electronic transitions of the ligand. The emission spectra of the complexes are red shifted compared to that of the ligand, 4, due to the different coordination and crystal packing interactions. The positions of the emission bands may be explained by different dihedral angles of the aromatic rings in the ligand and the complexes. Smaller dihedral angles result in enhanced π electron interactions, which in turn shift the emission bands to lower energies.101 Although the dihedral angle in the ligand 4 is not known, in the metal compounds it is consistently very close to 0°: 0.1(6)° (5), 1.5(1)° (6), 0.5(2)° (7) and 2.0(2)° (8). Even though the dihedral angles in the excited states cannot be determined, it may be assumed that due to the crystal in rigid structures the conformations do not change significantly. To the best of our knowledge, no luminescent phosphonate–carboxylate compounds with Zn2+, Hg22+ and Cd2+ ions have been reported so far.
Conclusions
We have presented the crystal structures of five complexes with the new BPPA3− or HBPPA2− ligand. The new mixed-functional linker assumes (five) different metal–oxygen(ligand) coordination modes depending on the metal. Yet, all metal-linker compounds 5–9 share the common motif of {MOx} polyhedra arranged in a mostly flat layer, which are connected by the biphenyl part, either directly or through tail-to-tail carboxyl groups. This way, polar and nonpolar parts of the molecules are separated, which is a common packing motif. In the flat {MOx} layers in 6 and 7 the Zn and Cd metal nodes represent a honeycomb and an mcm net, respectively. Between the biphenyl groups, CH-π contacts are present (see ESI†). The coordination polyhedra of the Cd atoms in compound 7 were analyzed towards a trigonal-prismatic coordination environment. When the linker is fully deprotonated to BPPA3− the metal–ligand networks feature a high thermal stability of ∼400 °C. When the carboxyl group is still protonated (–COOH in HBPPA2−) a decarboxylation starts at ∼150–200 °C, yet further decomposition only occurs above 400 °C. All compounds are very resistant against hydrolysis as they were synthesized in water at up to 200 °C and were stored in either water or air. The fluorescence emission of the H3BPPA ligand is red shifted by about 50 nm upon deprotonation and metal complexation. It was our aim to demonstrate, that the combination of the phosphonato, –PO32− and carboxylato, –COO− function leads to hydrothermally stable network compounds.Experimental section
Materials and methods
All reactants were commercially available and used without further purification. Doubly de-ionized (DI) water was used.Elemental (CHN) analyses were performed on a Perkin Elmer CHN 2400. 1H, 13C and 31P NMR spectra were measured with a Bruker Avance DRX-200, Bruker Avance DRX-500 or Bruker Avance DRX-600. High-resolution electron-spray ionization mass spectra (HR-ESI-MS) were collected with a UHR-QTOF maXis 4G from Bruker Daltonics. Electron impact (EI) mass spectra were collected on a TSQ 7000 by Finnigan MAT. FT-IR spectra were recorded in ATR mode (Platinum ATR-QL, Diamond) on a Bruker TENSOR 37 IR spectrometer in the range of 4000–500 cm−1. The absorbance bands have been described by the following terms: strong (s), medium (m), weak (w) and broad (br). Solid-state fluorescence spectra (2D) were obtained with a FluoroMax spectrometer from Horiba at room temperature. The samples were ground to a powder and pressed to fit securely in the sample holder (the mercury compound 8 has not been measured because of its toxicity). The reflection angle was set to be 60°. For thermogravimetric analysis (TGA) a Netzsch TG 209 F3 Tarsus was used, equipped with Al-crucible and a measurement range from 20 to 700 °C with a heating rate of 10 K min−1 under inert atmosphere (N2). Programmable ovens with heating and cooling ramps were from Memmert.
Melting points were determined using a Büchi Melting Point B540. For powder X-ray diffraction patterns (PXRD) a Bruker D2 Phaser powder diffractometer was used with flat panel low background sample holder, CuKα radiation (λ = 1.5418 Å) at 30 kV, 10 mA, a scan speed of 0.2 s per step and a step size of 0.02° (2θ).
Circular dichroism (CD) spectra of 5 were recorded on a J-810 spectropolarimeter (JASCO) with KBr pellets (0.05 wt% of 5). To correct for linear anisotropy artifacts the KBr pellets were rotated during a measurement by the use of a motor-driven sample holder (20 rpm). The detector integration time was set to 16 s (scan rate: 20 nm min−1). Each pellet was measured on the front and the reverse side. The resulting spectra (see Fig. S33 in ESI†) were averaged.
Single crystal X-ray diffractometry
Suitable crystals were carefully selected under a polarizing microscope, covered in protective oil and mounted on a 0.05 mm cryoloop. Data collection. Bruker Kappa APEX2 CCD X-ray diffractometer with microfocus tube, Mo-Kα radiation (λ = 0.71073 Å), multi-layer mirror system, ω- and θ-scans. Data collection with APEX II,102 cell refinement with SMART, data reduction with SAINT103 and experimental absorption correction with SADABS.104Structure analysis and refinement: The structures were solved by direct methods, SHELXS-97, refinement was done by full-matrix least squares on F2 using the SHELX-97 program suite.105 Non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were positioned geometrically and refined using riding models [AFIX 43 for aromatic CH with C–H = 0.93 Å, AFIX 23 for CH2 with C–H = 0.97 and Uiso(H) = 1.2Ueq(C), AFIX 137 for CH3 with C–H = 0.96 Å and Uiso(H) = 1.5Ueq(C); AFIX 83 for –COOH and Uiso(H) = 1.5Ueq(O)]. Hydrogen atoms H1 and H7 on the OH groups in 5 have been freely refined in position and Uiso. Crystals of 5–9 were of very small size. It is known that very small crystals diffract weaker than larger crystals,106 resulting in lower data quality and subsequent problems during refinement. Some of the checkcif alerts are due to the small crystal size, e.g., poor data/parameter ratio (6, 7). In 6 the disorder of the biphenyl moiety (treated with the PART command in SHELX) and –COOH group, which can have two different orientations, is causing level B alerts. The large residual electron density near the Hg atoms in 8 gave level A and B alerts. Nine of the 13 largest peaks of the residual electron density (including the three largest) are within 2 Å of the Hg atom in 8. Crystal data and details on the structure refinement are given in Tables 5 and 6. Graphics were drawn with DIAMOND.107 Analyses on the supramolecular C–H⋯O-, C–H⋯π- and π–π-stacking interactions were done with PLATON for Windows.108 The crystallographic data have been deposited with the Cambridge Crystallographic Data Center (CCDC-numbers 1465329–1465334 for 3, 6, 8, 5, 2, 7, respectively).| Compound | 2 | 3 |
|---|---|---|
| a Largest difference peak and hole. b R 1= [∑(‖Fo| − |Fc‖)/∑|Fo|]; wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2. c Goodness-of-fit, S = [∑[w(Fo2 − Fc2)2]/(n − p)]1/2. d w = 1/[σ2(Fo2) + (aP)2 + bP] where P = (max(Fo2 or 0) + 2Fc2)/3. e Ref. 68. | ||
| Chemical formula | C14H11IO2 | C18H21O5P |
| M r | 338.13 | 348.32 |
| Crystal system | Orthorhombic | Triclinic |
| Space group | Pca21 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Temperature (K) | 150 | 296 |
| a (Å) | 6.0355 (4) | 8.094(2) |
| b (Å) | 7.2980(4) | 8.186(2) |
| c (Å) | 27.8833(18) | 13.626(4) |
| α (°) | 90 | 104.044(15) |
| β (°) | 90 | 92.510(15) |
| γ (°) | 90 | 99.071(14) |
| V (Å3) | 1228.18(13) | 861.7(4) |
| Z | 4 | 2 |
| μ (mm−1) | 2.593 | 0.18 |
| Crystal size (mm) | 0.10 × 0.10 × 0.05 | 0.90 × 0.20 × 0.20 |
| T min, Tmax | 0.724, 0.745 | 0.620, 0.744 |
| No. measured, indep. and obs. [I > 2σ(I)] reflect. | 10290, 2382, 2325 |
11291, 3248, 2650 |
| R int | 0.049 | 0.053 |
| (sin θ/λ)max (Å−1) | 0.624 | 0.613 |
| R 1 [F2 > 2σ(F2)], wR2 (F2),bSc | 0.030, 0.099, 1.24 | 0.060, 0.175, 1.07 |
| No. of reflect./restraints/parameters | 2382/1/155 | 3248/0/220 |
| Weight. scheme w; a/bd | 0.050/0.6335 | 0.0925/0.6193 |
| Max./min. Δρ (e Å−3)a | 0.81, −0.82 | 0.53, −0.47 |
| Absolute structure (Flack) parametere | 0.080(17) | — |
| Compound | 5 | 6 | 7 | 8 |
|---|---|---|---|---|
| a Largest difference peak and hole. b R 1 = [∑(‖Fo| − |Fc‖)/∑|Fo|]; wR2 = [∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]]1/2. c Goodness-of-fit, S = [∑[w(Fo2 − Fc2)2]/(n − p)]1/2. d w = 1/[σ2(Fo2) + (aP)2 + bP] where P = (max(Fo2 or 0) + 2Fc2)/3. e Ref. 68. | ||||
| Chemical formula | C26H20O14P2Zn5 | C13H9O5PZn | C26H16Cd3O10P2 | C13H10HgO5P |
| M r | 945.31 | 341.54 | 887.53 | 477.77 |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic | Monoclinic |
| Space group | C2 | C2/m | Pca21 | C2/c |
| Temperature (K) | 173 | 173 | 173 | 173 |
| a (Å) | 32.8388(12) | 5.2848(12) | 9.3961(7) | 59.003(11) |
| b (Å) | 5.3763(19) | 8.1031(18) | 9.5450(7) | 5.4907(9) |
| c (Å) | 7.913(3) | 27.371(6) | 27.3230(19) | 7.8375(12) |
| β (°) | 103.531(11) | 91.766(12) | 90 | 93.644(9) |
| V (Å3) | 1358.2(9) | 1171.6(5) | 2450.5(3) | 2534.0(7) |
| Z | 2 | 4 | 4 | 8 |
| μ (mm−1) | 4.55 | 2.25 | 2.77 | 12.29 |
| Crystal size (mm) | 0.10 × 0.01 × 0.01 | 0.03 × 0.03 × 0.03 | 0.10 × 0.07 × 0.05 | 0.01 × 0.01 × 0.01 |
| T min, Tmax | 0.502, 0.748 | 0.724, 0.745 | 0.685, 0.747 | 0.515, 0.746 |
| No. measured, indep. and obs. [I > 2σ(I)] reflect. | 8001, 3360, 2930 | 5207, 1106, 912 | 48627, 4301, 4281 |
11056, 2121, 1633 |
| R int | 0.060 | 0.054 | 0.043 | 0.089 |
| (sin θ/λ)max (Å−1) | 0.703 | 0.594 | 0.595 | 0.594 |
| R 1 [F2 > 2σ(F2)], wR2 (F2),bSc | 0.041, 0.085, 1.01 | 0.068, 0.163, 1.05 | 0.032, 0.077, 1.13 | 0.070, 0.176, 1.03 |
| No. of reflections/restraints/parameters | 3360/1/220 | 1106/0/129 | 4301/1/310 | 2121/0/155 |
| Weight. scheme w; a/bd | 0.0302/0 | 0.0504/40.479198 | 0.008/37.947201 | 0.0831/363.958771 |
| Max./min. Δρ (e Å−3)a | 1.67, −1.09 | 3.63, −1.72 | 1.73, −2.15 | 8.07, −3.81 |
| Absolute structure (Flack) parametere | 0.030(3) | — | 0.007(9) | — |
Syntheses
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1 = 2976 (w, br), 2820 (w, br), 2668 (w), 2546 (w), 1827 (vs), 1677 (m), 1605 (m), 1572 (m), 1549 (m), 1514 (m), 1474 (m), 1424 (m), 1386 (m), 1321 (m), 1298 (m), 1280 (m), 1196 (s), 1177(m), 1123 (m), 1104 (m), 1062 (m), 1019 (m), 1010 (m), 996 (m), 954 (m), 871 (m), 818 (m), 767 (m), 744 (vs), 710 (m), 698 (m), 661 (m), 623 (m). 1H-NMR (200 MHz; DMSO-d6) δ/ppm = 7.53 (d, 2H, J = 8.0 Hz), 7.79 (d, 2H, J = 8.0 Hz), 7.84 (d, 2H, J = 8.0 Hz), 8.01 (d, 2H, J = 8.0 Hz). Spectra are given in Fig. S1 and S2 in the ESI.†
:v = 1:1). Then 6.2 g (45.0 mmol) of K2CO3 were added to the suspension under stirring. A portion of 6.4 g (45.0 mmol) of methyliodide was added dropwise over one hour resulting in the formation of a yellowish, oily layer. The layers were separated, the upper DMF layer was discarded, the solvent from the yellowish CCl4 was removed in vacuum affording an off-white powder (5.0 g; 98% yield). Alternatively: In a 250 mL flask 2.5 g (7.9 mmol) of 1 were suspended in 100 mL of dry methanol. Conc. sulphuric acid (5 mL) was added and the suspension heated to reflux for 4 h. The solid was filtered and washed with plenty of water (>100 mL) to remove the acid and methanol. The product was dried to afford 2.4 g (90%) of an off-white powder. Mp = 188 °C (lit.: 189 °C).110 FT-IR (ATR) /cm−1 = 3399 (w, br), 2998 (w), 2944 (w), 2842 (w), 1936 (m), 1902 (m), 1781 (m), 1708 (s), 1602 (m), 1576 (m), 1474 (m), 1453 (m), 1431 (m), 1387 (m), 1330 (m), 1287 (m), 1269 (s), 1209 (m), 1186 (m), 1136 (m), 1110 (s), 1066 (m), 1018 (m), 995 (m), 949 (m), 860, 819 (s), 764 (vs), 697 (m), 662 (m), 622 (m), 588 (m). 1H-NMR (200 MHz, CDCl3) δ/ppm = 3.92 (s, 3H), 7.34 (d, 2H, J = 8.6 Hz), 7.59 (d, 2H, J = 8.6 Hz), 7.77 (d, 2H, J = 8.6 Hz), 7.08 (d, 2H, J = 8.6 Hz). Spectra are given in Fig. S3 and S4 in ESI.†
/cm−1 = 3785 (w), 2969 (w), 2911 (w), 1637 (w), 1593 (w), 1521 (w), 1428 (m), 1329 (m), 1250 (s), 1198 (m), 1091 (m), 1032 (m), 1010 (m), 962 (m), 824 (w), 768 (m), 686 (m), 611 (vs). 1H-NMR (500 MHz, DMSO-d6) δ/ppm = 1.22 (t, 6H), 3.87 (s, 3H), 4.01 (q, 4H), 7.33–8.35 (m, 8H). 13C-NMR (125.7 MHz, DMSO-d6) δ/ppm = 16.5 (–CH3), 39.8 (–CH2–), 52.5 (O–CH3), 62.1 (O–CH3), 127.6 (d), 130.1 (d), 132.1 (d), 142.5, 143.3 (aromatic C), 176.5 (–COOH). 31P{H}-NMR (121.5 MHz, DMSO-d6) δ/ppm = 17.53. Spectra are given in Fig. S5–S8 in ESI.† EI-MS(+) at 100 °C (M = C18H21O5P (348.11) m/z (%) 348 (15) [M], 320 (28) [M–C2H4]+, 292 (41) [M–C2H5–C2H4]+, 261 (22) [M–C2H4–C2H3O2]+, 152 (65) [M–PO3(C2H5)2–CO2CH3 = C12H8]+. Calcd. for C18H21O5P·H2O (366.35 g mol−1): C 59.01, H 6.33%. Found C 58.79, H 5.99%.
/cm−1 = 3324 (w, b), 2955 (w, br), 2849 (w, br), 2677 (m), 2557 (m), 2166 (m), 1935 (m), 1808 (m), 1677 (vs), 1605 (m), 1574 (w), 1550 (w), 1429 (m), 1391 (m), 1302 (m), 1214 (m), 1179 (m), 1145 (m), 1102 (m), 1002 (vs), 946 (m), 868 (m), 831 (m), 799 (m), 771 (m), 755 (m), 722 (m), 689 (m), 577 (vs). 1H-NMR (200 MHz, DMSO-d6) δ/ppm = 7.70–7.90 (m, 6H), 8.04 (d, 2H, J = 7.70 Hz), 8.2–9.0 (broad signal, P–OH) 13C-NMR (125.7 MHz, DMSO-d6) δ/ppm = 127.1, 127.2, 127.5, 127.6, 129.5, 130.5, 131.7, 133.6, 134.8, 138.3, 141.7, 143.9 (aromatic C), 167.5 (–COOH). 31P-NMR (121.5 MHz, DMSO-d6) δ/ppm = 10.91. Spectra are given in Fig. S9–S12 in ESI.† EI-MS(+) at 260 °C, M = C13H11O5P, (278): m/z (%) 278 (100) [M], 198 (22) [M–PO3H]+, 152 (40) [M–PO3H2–CO2H = C12H8]+. Calcd. for C13H11O5P (278.2 g mol−1): C 56.13, H 3.99%. Found C 56.17, H 4.10%.
/cm−1 = 3460 (w), 3069 (w), 1691 (w), 1586 (w), 1553 (w), 1519 (w), 1423 (m), 1167 (m), 1143 (m), 1073 (vs), 962 (vs), 925 (s), 869 (m), 830 (s), 775 (vs), 728 (m), 700 (m), 595 (vs) (Fig. S13, ESI†). Calc. for C26H20O14P2Zn5 (945.29 g mol−1): C 33.04, H 2.13%. Found C 33.71, H 2.39%.
/cm−1 = 2988 (w, br), 2875 (w, br), 2669 (w, b), 2543 (w, br), 1681 (m), 1606 (w), 1427 (w), 1275 (w), 1179 (m), 1146 (s), 1059 (vs), 958 (vs), 828 (m), 756 (m), 691 (w), 599 (vs), 562 (s) (Fig. S14, ESI†). Calc. for C13H9O5PZn (341.56 g mol−1): C 45.71, H 2.66%. Found C 45.69, H 2.54%.
/cm−1 = 3402 (m, b), 3284 (m), 2855 (w, b), 2546 (w), 1684 (vs), 1604 (m), 1425 (s), 1126 (m), 1089 (s), 1031 (m), 948 (s), 823 (s), 765 (s), 719 (s), 687 (m), 578 (s) (Fig. S15, ESI†). Calc. for C26H16Cd3O10P2 (887.59 g mol−1): C 35.18, H 1.82%. Found C, 35.26, H 2.0%.
/cm−1 = 2976 (w, br), 2820 (w, br), 2668 (w), 2546 (w), 1827 (vs), 1677 (m), 1605 (m), 1572 (m), 1549 (m), 1514 (m), 1474 (m), 1424 (m), 1386 (m), 1321 (m), 1298 (m), 1280 (m), 1196 (s), 1177(m), 1123 (m), 1104 (m), 1062 (m), 1019 (m), 1010 (m), 996 (m), 954 (m), 871 (m), 818 (m), 767 (m), 744 (vs), 710 (m), 698 (m), 661 (m), 623 (m). 1H-NMR (200 MHz; DMSO-d6) δ/ppm = 7.53 (d, 2H, J = 8.0 Hz), 7.79 (d, 2H, J = 8.0 Hz), 7.84 (d, 2H, J = 8.0 Hz), 8.01 (d, 2H, J = 8.0 Hz). Spectra are given in Fig. S1 and S2 in the ESI.†
:v = 1:1). Then 6.2 g (45.0 mmol) of K2CO3 were added to the suspension under stirring. A portion of 6.4 g (45.0 mmol) of methyliodide was added dropwise over one hour resulting in the formation of a yellowish, oily layer. The layers were separated, the upper DMF layer was discarded, the solvent from the yellowish CCl4 was removed in vacuum affording an off-white powder (5.0 g; 98% yield). Alternatively: In a 250 mL flask 2.5 g (7.9 mmol) of 1 were suspended in 100 mL of dry methanol. Conc. sulphuric acid (5 mL) was added and the suspension heated to reflux for 4 h. The solid was filtered and washed with plenty of water (>100 mL) to remove the acid and methanol. The product was dried to afford 2.4 g (90%) of an off-white powder. Mp = 188 °C (lit.: 189 °C).110 FT-IR (ATR) /cm−1 = 3399 (w, br), 2998 (w), 2944 (w), 2842 (w), 1936 (m), 1902 (m), 1781 (m), 1708 (s), 1602 (m), 1576 (m), 1474 (m), 1453 (m), 1431 (m), 1387 (m), 1330 (m), 1287 (m), 1269 (s), 1209 (m), 1186 (m), 1136 (m), 1110 (s), 1066 (m), 1018 (m), 995 (m), 949 (m), 860, 819 (s), 764 (vs), 697 (m), 662 (m), 622 (m), 588 (m). 1H-NMR (200 MHz, CDCl3) δ/ppm = 3.92 (s, 3H), 7.34 (d, 2H, J = 8.6 Hz), 7.59 (d, 2H, J = 8.6 Hz), 7.77 (d, 2H, J = 8.6 Hz), 7.08 (d, 2H, J = 8.6 Hz). Spectra are given in Fig. S3 and S4 in ESI.†
/cm−1 = 3785 (w), 2969 (w), 2911 (w), 1637 (w), 1593 (w), 1521 (w), 1428 (m), 1329 (m), 1250 (s), 1198 (m), 1091 (m), 1032 (m), 1010 (m), 962 (m), 824 (w), 768 (m), 686 (m), 611 (vs). 1H-NMR (500 MHz, DMSO-d6) δ/ppm = 1.22 (t, 6H), 3.87 (s, 3H), 4.01 (q, 4H), 7.33–8.35 (m, 8H). 13C-NMR (125.7 MHz, DMSO-d6) δ/ppm = 16.5 (–CH3), 39.8 (–CH2–), 52.5 (O–CH3), 62.1 (O–CH3), 127.6 (d), 130.1 (d), 132.1 (d), 142.5, 143.3 (aromatic C), 176.5 (–COOH). 31P{H}-NMR (121.5 MHz, DMSO-d6) δ/ppm = 17.53. Spectra are given in Fig. S5–S8 in ESI.† EI-MS(+) at 100 °C (M = C18H21O5P (348.11) m/z (%) 348 (15) [M], 320 (28) [M–C2H4]+, 292 (41) [M–C2H5–C2H4]+, 261 (22) [M–C2H4–C2H3O2]+, 152 (65) [M–PO3(C2H5)2–CO2CH3 = C12H8]+. Calcd. for C18H21O5P·H2O (366.35 g mol−1): C 59.01, H 6.33%. Found C 58.79, H 5.99%.
/cm−1 = 3324 (w, b), 2955 (w, br), 2849 (w, br), 2677 (m), 2557 (m), 2166 (m), 1935 (m), 1808 (m), 1677 (vs), 1605 (m), 1574 (w), 1550 (w), 1429 (m), 1391 (m), 1302 (m), 1214 (m), 1179 (m), 1145 (m), 1102 (m), 1002 (vs), 946 (m), 868 (m), 831 (m), 799 (m), 771 (m), 755 (m), 722 (m), 689 (m), 577 (vs). 1H-NMR (200 MHz, DMSO-d6) δ/ppm = 7.70–7.90 (m, 6H), 8.04 (d, 2H, J = 7.70 Hz), 8.2–9.0 (broad signal, P–OH) 13C-NMR (125.7 MHz, DMSO-d6) δ/ppm = 127.1, 127.2, 127.5, 127.6, 129.5, 130.5, 131.7, 133.6, 134.8, 138.3, 141.7, 143.9 (aromatic C), 167.5 (–COOH). 31P-NMR (121.5 MHz, DMSO-d6) δ/ppm = 10.91. Spectra are given in Fig. S9–S12 in ESI.† EI-MS(+) at 260 °C, M = C13H11O5P, (278): m/z (%) 278 (100) [M], 198 (22) [M–PO3H]+, 152 (40) [M–PO3H2–CO2H = C12H8]+. Calcd. for C13H11O5P (278.2 g mol−1): C 56.13, H 3.99%. Found C 56.17, H 4.10%.
/cm−1 = 3460 (w), 3069 (w), 1691 (w), 1586 (w), 1553 (w), 1519 (w), 1423 (m), 1167 (m), 1143 (m), 1073 (vs), 962 (vs), 925 (s), 869 (m), 830 (s), 775 (vs), 728 (m), 700 (m), 595 (vs) (Fig. S13, ESI†). Calc. for C26H20O14P2Zn5 (945.29 g mol−1): C 33.04, H 2.13%. Found C 33.71, H 2.39%.
/cm−1 = 2988 (w, br), 2875 (w, br), 2669 (w, b), 2543 (w, br), 1681 (m), 1606 (w), 1427 (w), 1275 (w), 1179 (m), 1146 (s), 1059 (vs), 958 (vs), 828 (m), 756 (m), 691 (w), 599 (vs), 562 (s) (Fig. S14, ESI†). Calc. for C13H9O5PZn (341.56 g mol−1): C 45.71, H 2.66%. Found C 45.69, H 2.54%.
/cm−1 = 3402 (m, b), 3284 (m), 2855 (w, b), 2546 (w), 1684 (vs), 1604 (m), 1425 (s), 1126 (m), 1089 (s), 1031 (m), 948 (s), 823 (s), 765 (s), 719 (s), 687 (m), 578 (s) (Fig. S15, ESI†). Calc. for C26H16Cd3O10P2 (887.59 g mol−1): C 35.18, H 1.82%. Found C, 35.26, H 2.0%.
Inside a Teflon-lined stainless-steel autoclave 11.0 mg (0.034 mmol) of Hg(OAc)2 and 3.6 mg (0.029 mmol) of oxalic acid dihydrate were weighed and dissolved in 4 mL of DI water. A portion of 10.0 mg (0.036 mmol) of H3BPPA was added and the suspension stirred to homogeneity. The reactor was heated to 120 °C within 24 h and kept at this temperature for 72 h. After cooling to room temperature within 24 h the solids were filtered and washed with water (3 × 10 mL). Colorless block-shaped crystals of very small size (0.01 × 0.01 × 0.01 mm) were isolated, which were used for X-ray structure determination. Crystal yield 8.0 mg (50%). Mp > 300 °C. FT-IR (ATR) /cm−1 = 2969 (m, br) 2849 (m, br), 2674 (w), 2547 (w), 2323 (w), 1807 (w), 1682 (vs), 1604 (m), 1573 (w), 1426 (m), 1392 (m), 1280 (m), 1139 (m), 1070 (m), 824 (s), 765 (s), 752 (m), 720 (m), 582 (s) (Fig. S16, ESI†). Calc. for C13H10HgO5P (477.78 g mol−1): C 32.68, H 2.11%. Found C 30.84, H 1.91%.
/cm−1 = 3445 (w, b), 2961 (w, br), 2777 (w, br), 1681 (m), 1602 (w), 1419 (m), 1390 (m), 1138 (m), 944 (vs), 828 (s), 768 (s), 753 (m), 723 (m), 579 (m) (Fig. S17, ESI†). Calc. for C13H9CoO5P (335.12 g mol−1): C 46.59, H 2.71%. Found: C 46.16, H 3.24%.
Acknowledgements
The work was supported by the German Science Foundation (DFG) through grant Ja466/25-1 and in part by BMBF project OptiMat 03SF0492C. We thank Mrs. Marija Marolt for the CD measurements.Notes and references
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed
; M. Gaab, N. Trukhan, S. Maurer, R. Gummaraju and U. Müller, Microporous Mesoporous Mater., 2012, 157, 131–136 CrossRef
- S. Li and F. Huo, Nanoscale, 2015, 7, 7482–7501 RSC
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkovc and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC
- Y. Xu, M. Wu, Y. Liu, X.-Z. Feng, X.-B. Yin, X.-W. He and Y.-K. Zhang, Chem. – Eur. J., 2013, 19, 2276–2283 CrossRef CAS PubMed
- B. Gole, A. K. Bar and P. S. Mukherjee, Chem. – Eur. J., 2014, 20, 13321–13336 CrossRef CAS PubMed
- R. B. Getman, Y.-S. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2012, 112, 703–723 CrossRef CAS PubMed
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed
- S. Qiu, M. Xue and G. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 RSC
- F. Jeremias, D. Fröhlich, C. Janiak and S. Henninger, New J. Chem., 2014, 4, 24073–24082 Search PubMed
- G. Férey, Dalton Trans., 2009, 4400–4415 RSC
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, S. Surble, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296–6301 CrossRef CAS PubMed
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surble and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed
- G. Akiyama, R. Matsuda and S. Kitagawa, Chem. Lett., 2010, 39, 360–361 CrossRef CAS
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed
- J. Canivet, A. Fateeva, Y. Guo, B. Coasnec and D. Farrusseng, Chem. Soc. Rev., 2014, 43, 5594–5617 RSC
- F. Jeremias, V. Lozan, S. Henninger and C. Janiak, Dalton Trans., 2013, 42, 15967–15973 RSC
- C. Heering, I. Boldog, V. Vasylyeva, J. Sanchiz and C. Janiak, CrystEngComm, 2013, 15, 9757–9768 RSC
- C. Montoro, F. Linares, E. Quartapelle Procopio, I. Senkovska, S. Kaskel, S. Galli, N. Masciocchi, E. Barea and J. A. R. Navarro, J. Am. Chem. Soc., 2011, 133, 11888–11891 CrossRef CAS PubMed
- P. O. Adelani, L. J. Jouffret, J. E. S. Szymanowski and P. C. Burns, Inorg. Chem., 2012, 51, 12032–12040 CrossRef CAS PubMed
- P. O. Adelani, G. E. Sigmon and P. C. Burns, Inorg. Chem., 2013, 52, 6245–6247 CrossRef CAS PubMed
- Reviews: K. Maeda, Microporous Mesoporous Mater., 2004, 73, 47–55 CrossRef CAS
- S. Kirumakki, J. Huang, A. Subbiah, J. Yao, A. Rowland, B. Smith, A. Mukherjee, S. Samarajeewa and A. Clearfield, J. Mater. Chem., 2009, 19, 2593–2603 RSC
- F. Zhai, Q. Zheng, Z. Chen, Y. Ling, X. Liu, L. Wenig and Y. Zhou, CrystEngComm, 2013, 15, 2040–2043 RSC
- S. Kirumakki, J. Huang, A. Subbiah, J. Yao, A. Rowland, B. Smith, A. Mukherjee, S. Samarajeewa and A. Clearfield, J. Mater. Chem., 2008, 19, 2593–2603 RSC
- X. Zhao, J. G. Bell, S.-F. Tang, L. Li and K. M. Thomas, J. Mater. Chem. A, 2016, 4, 1353–1365 CAS
-
A. Clearfield and K. Demadis, Metal Phosphonate Chemistry: From Synthesis to Applications, Royal Society of Chemistry, Oxford, 2012, ch. 2–4, pp. 45–128 Search PubMed
- M. Deng, X. Liu, Q. Zheng, Z. Chen, C. Fang, B. Yue and H. He, CrystEngComm, 2013, 15, 7056–7061 RSC
- K. J. Gagnon, H. P. Perry and A. Clearfield, Chem. Rev., 2012, 112, 1034–1054 CrossRef CAS PubMed
- J. M. Taylor, R. Vaidhyanathan, S. S. Iremonger and G. K. H. Shimizu, J. Am. Chem. Soc., 2012, 134, 14338–14340 CrossRef CAS PubMed
- T. L. Kinnibrugh, A. A. Ayi, V. I. Bakhmutov, J. Zon and A. Clearfield, Cryst. Growth Des., 2013, 13, 2973–2981 CAS
- T. Zheng, J. M. Clemente-Juan, J. Ma, L. Dong, S.-S. Bao, J. Huang, A. Coronado and L.-M. Zheng, Chem. – Eur. J., 2013, 19, 16394–16402 CrossRef CAS PubMed
- M. Taddei, F. Costantino, A. Lenco, A. Comotti, P. V. Dau and S. M. Cohen, Chem. Commun., 2013, 49, 1315–1317 RSC
- R. El Osta, M. Frigoli, J. Marrot, N. Guillou, H. Chevreau, R. I. Walton and F. Millange, Chem. Commun., 2012, 48, 10639–10641 RSC
-
A. Clearfield and K. D. Demadis, Metal Phosphonate Chemistry, RSC Publishing, Cambridge, 2011 Search PubMed
- A. R. Patterson, W. Schmitt and R. C. Evans, J. Phys. Chem. C, 2014, 118, 10291–10301 CAS
- L. Jiménez-García, A. Kaltbeitzel, W. Pisula, J. S. Gutmann, M. Klapper and K. Müllen, Angew. Chem., Int. Ed., 2009, 48, 9951–9953 CrossRef PubMed
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed
- G. K. H. Shimizu, R. Vaidhyanathan and J. M. Taylor, Chem. Soc. Rev., 2009, 38, 1430–1449 RSC
- Review: T. Rojo, J. L. Mesa, J. Lago, B. Bazan, J. L. Pizarro and M. I. Arriortua, J. Mater. Chem., 2009, 19, 3793–3818 RSC
- Cobalt compounds:
(a) S.-Z. Hou, D.-K. Cao, X.-G. Liu, Y.-Z. Li and L.-M. Zheng, Dalton Trans., 2009, 2746–2750 RSC
- Iron compounds: H. A. Habib, B. Gil-Hernández, K. Abu-Shandi, J. Sanchiz and C. Janiak, Polyhedron, 2010, 29, 2537–2545 CrossRef CAS
- J.-S. Feng, S.-S. Bao, M. Ren, Z.-S. Cai and L.-M. Zheng, Chem. – Eur. J., 2015, 21, 17336–17343 CrossRef CAS PubMed
- X.-J. Yang, S.-S. Bao, M. Ren, N. Hoshino, T. Akutagawa and L.-M. Zheng, Chem. Commun., 2014, 50, 3979–3981 RSC
- Y.-Z. Zheng and R. E. P. Winpenny, Sci. China: Chem., 2012, 55, 910–913 CrossRef CAS
- Z.-Y. Du, H.-R. Wen, C.-M. Liu, Y.-H. Sun, Y.-B. Lu and Y.-R. Xie, Cryst. Growth Des., 2010, 10, 3721–3726 CAS
- Y.-Y. Zhang, M.-H. Zeng, Y. Qi, S.-Y. Sang and Z.-M. Liu, Inorg. Chem. Commun., 2007, 10, 33–36 CrossRef CAS
- M. Menelaou, M. Dakanali, C. P. Raptopoulou, C. Drouza, N. Lalioti and A. Salifoglou, Polyhedron, 2009, 28, 3331–3339 CrossRef CAS
- C. Heering, B. Nateghi and C. Janiak, Crystals, 2016, 6, 22 CrossRef
- J. Svoboda, V. Zima, L. Beneš, K. Melánová, M. Trchová and M. Vlček, Solid State Sci., 2008, 10, 1533–1542 CrossRef CAS
- A.-M. Pütz, L. M. Carrella and E. Rentschler, Dalton Trans., 2013, 42, 16194–16199 RSC
- J.-M. Rueff, N. Barrier, S. Boudin, V. Dorcet, V. Caignaert, P. Boullay, G. B. Hix and P.-A. Jaffrès, Dalton Trans., 2009, 10614–10620 RSC
- J.-M. Rueff, O. Perez, A. Leclaire, H. Couthon-Gourvès and P.-A. Jaffrès, Eur. J. Inorg. Chem., 2009, 4870–4876 CrossRef CAS
- J.-T. Li, L.-R. Guo, Y. Shen and L.-M. Zheng, CrystEngComm, 2009, 11, 1674–1678 RSC
- J.-M. Rueff, O. Perez, V. Caignaert, G. Hix, M. Berchel, F. Quentel and P.-A. Jaffrès, Inorg. Chem., 2015, 54, 2152–2159 CrossRef CAS PubMed
- V. Zima, J. Svoboda, L. Beneš, K. Melánová, M. Trchová and J. Dybal, J. Solid State Chem., 2007, 180, 929–939 CrossRef CAS
- P. O. Adelani and T. E. Albrecht-Schmitt, Inorg. Chem., 2010, 49, 5701–5705 CrossRef CAS PubMed
- K. Melánová, J. Klevcov, L. Beneš, J. Svoboda and V. Zima, J. Phys. Chem. Solids, 2012, 73, 1452–1455 CrossRef
- J.-T. Li, D.-K. Cao, T. Akutagawa and L.-M. Zheng, Dalton Trans., 2010, 39, 8606–8608 RSC
- Z. Chen, Y. Zhou, L. Weng and D. Zhao, Cryst. Growth Des., 2008, 8, 4045–4053 CAS
- Y. Ling, M. Deng, Z. Chen, B. Xia, X. Liu, Y. Yang, Y. Zhou and L. Wenig, Chem. Commun., 2013, 49, 78–80 RSC
- B. A. Breeze, M. Shanmugam, F. Tuna and R. E. P. Winpenny, Chem. Commun., 2007, 5185–5187 RSC
- J.-M. Rueff, O. Perez, A. Leclaire, H. Couthon-Gourvès and P.-A. Jaffrès, Eur. J. Inorg. Chem., 2009, 4870–4876 CrossRef CAS
- T.-B. Liao, Y. Ling, Z.-X. Chen, Y.-M. Zhou and L.-H. Weng, Chem. Commun., 2010, 46, 1100–1102 RSC
- E. B. Merkushev and N. D. Yudina, Zh. Org. Khim., 1982, 17, 2598–2601 Search PubMed
- C. P. Brock, J. R. Blackburn and K. L. Haller, Acta Crystallogr., Sect. B: Struct. Sci., 1984, 40, 493–498 CrossRef
- H. D. Flack, M. Sadki, A. L. Thompson and D. J. Watkin, Acta Crystallogr., Sect. A: Found. Crystallogr., 2011, 67, 21–34 Search PubMed
- R. B. DeVasher, L. R. Moore and K. H. Shaughnessy, J. Org. Chem., 2004, 69, 7919–7927 CrossRef CAS PubMed
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC
- A.-C. Chamayou, G. Makhloufi, L. A. Nafie, C. Janiak and S. Lüdeke, Inorg. Chem., 2015, 54, 2193–2203 CrossRef CAS PubMed
- B. Gil-Hernández, J. K. Maclaren, H. A. Höppe, J. Pasan, J. Sanchiz and C. Janiak, CrystEngComm, 2012, 14, 2635–2644 RSC
- M. Enamullah, A. Sharmin, M. Hasegawa, T. Hoshi, A.-C. Chamayou and C. Janiak, Eur. J. Inorg. Chem., 2006, 2146–2154 CrossRef CAS
- W. Dan, X. Liu, M. Deng, Y. Ling, Z. Chen and Y. Zhou, Inorg. Chem. Commun., 2013, 37, 93–96 CrossRef CAS
- Z. Chen, Y. Zhou, L. Weng, C. Yuan and D. Zhao, Chem. – Asian J., 2007, 2, 1549–1554 CrossRef CAS PubMed
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS
- E. I. Stiefel and G. F. Brown, Inorg. Chem., 1972, 11, 434 CrossRef CAS
- S. Banerjee, A. Ghosh, B. Wu, P.-G. Lassahn and C. Janiak, Polyhedron, 2005, 24, 593–599 CrossRef CAS
- The three letter symbols, proposed by M. O'Keeffe, can be retrieved with examples and further information from the Reticular Chemistry Structure Resource database, http://rcsr.anu.edu.au/
- Bond valences (s) calculated from the Hg–O bond lengths (R) according to s = exp(R0 − R)/B and R0 = 1.900 for Hg(I)–O or 1.972 for Hg(II)–O, B = 0.37; program VALENCE as implemented in PLATON.
- I. D. Brown and R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1973, 29, 266 CrossRef CAS
- R. Galassi, F. Bachechi and A. Burini, J. Mol. Struct., 2006, 791, 82–88 CrossRef CAS
-
S. T. Liddle, in Molecular Metal–Metal Bonds: Compounds, Synthesis, Properties, Wiley-VCH Verlag, Weinheim, Germany, 2015, ch. 12.2.1.1 Search PubMed
- A. Morsali and M. Y. Masoomi, Coord. Chem. Rev., 2009, 253, 1882–1905 CrossRef CAS
-
(a) K. Brodersen, R. Dölling and G. Liehr, Z. Anorg. Allg. Chem., 1980, 464, 17–29 CrossRef CAS
- C. D. L. Saunders, N. Burford, U. Werner-Zwanziger and R. McDonald, Inorg. Chem., 2008, 47, 3693–3699 CrossRef CAS PubMed
- C. R. Samanamu, E. N. Zamora, J.-L. Montchamp and A. F. Richards, J. Solid State Chem., 2008, 181, 1462–1471 CrossRef CAS
- B. Kamenar, A. Hergold-Brundic and M. Bruvo, Z. Kristallogr., 1988, 184, 103 CrossRef CAS
- T. Dorn, A.-C. Chamayou and C. Janiak, New J. Chem., 2006, 30, 156–167 RSC
- J. K. Maclaren, J. Sanchiz, P. Gili and C. Janiak, New J. Chem., 2012, 36, 1596–1609 RSC
- M. Enamullah, V. Vasylyeva and C. Janiak, Inorg. Chim. Acta, 2013, 408, 109–119 CrossRef CAS
- S. Vairam and S. Govindarajan, Thermochim. Acta, 2004, 414, 263–270 CrossRef CAS
- L. W. Collins, E. K. Gibson and W. W. Wendlandt, Thermochim. Acta, 1975, 11, 177–185 CrossRef CAS
- T. Gunnlaughson, M. Glynn, G. M. Tocci, P. E. Kruger and F. M. Pfeffer, Coord. Chem. Rev., 2006, 250, 3094–3117 CrossRef CAS
- H. A. Habib, A. Hoffmann, H. A. Höppe, G. Steinfeld and C. Janiak, Inorg. Chem., 2009, 48, 2166–2180 CrossRef CAS PubMed
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed
- J. W. Bridges, P. J. Creaven and R. T. Williams, Biochem. J., 1965, 96, 872–878 CrossRef CAS PubMed
- Q. Gong, Z. Hu, B. J. Deibert, T. J. Emge, S. J. Teat, D. Banerjee, B. Mussman, N. D. Rudd and J. Li, J. Am. Chem. Soc., 2014, 136, 16724–16727 CrossRef CAS PubMed
- A. Majumder, G. M. Rosair, A. Mallick, N. Chattopadhyay and S. Mitra, Polyhedron, 2006, 25, 1753–1762 CrossRef CAS
-
(a) W. W. Lestari, P. Lonnecke, M. B. Sarosi, H. C. Streit, M. Adlung, C. Wickleder, M. Handke, W.-D. Einicke, R. Glaser and E. Hey-Hawkins, CrystEngComm, 2013, 15, 3874–3884 RSC
-
APEXII, Data collection program for the APEXII CCD area-detector system, Bruker Analytical X-ray Systems, Madison, Wisconsin, USA, 2010 Search PubMed
-
SMART, Data Collection Program for the CCD Area-Detector System, SAINT: Data Reduction and Frame Integration Program for the CCD Area-Detector System, Bruker Analytical X-ray Systems, Madison, Wisconsin, USA, 1997 Search PubMed
-
G.-M. Sheldrick, Program SADABS: Area-detector absorption correction, University of Göttingen, Germany, 1996 Search PubMed
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed
- J. Newman, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 27–31 CrossRef PubMed
-
K. Brandenburg, DIAMOND (Version 3.2), Crystal and Molecular Structure Visualization, Crystal Impact – K. Brandenburg & H. Putz, Gbr: Bonn, Germany, 2009 Search PubMed
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed
- H. O. Wirth, W. Kern and E. Schmitz, Makromol. Chem., 1963, 68(1), 66–69 CrossRef
- R. Pummerer and L. Seligsberger, Ber. Dtsch. Chem. Ges., 1931, 64, 2477–2486 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available: NMR data for 1–4, IR data for 1–9, additional structure graphics, bond lengths and angles, TGA and PXRD data for 4–9, photos of crystals (5, 7, 8, 9), geometrical calculations for 5–8, and CD spectra of 5. CCDC 1465329–1465334. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce00587j |
| ‡ Permanent address: CSIR-Network of Inst. for Solar Energy, National Institute for Interdisciplinary Science & Technology (NIIST), Thiruvananthapuram-695 019, India. |
| This journal is © The Royal Society of Chemistry 2016 |