
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards design strategies for anion–π interactions in crystal engineering
Antonio
Bauzá
a,
Tiddo J.
Mooibroek
*b and
Antonio
Frontera
 *a
*a
aDepartament de Quimica, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, Spain. E-mail: toni.frontera@uib.es
bSchool of Chemistry of the University of Bristol, Cantock's Close, BS8 1TS, Bristol, UK. E-mail: chtjm@bristol.ac.uk
First published on 8th October 2015
Abstract
For well over half a century part of the scientific community has been committed to understanding and predicting how molecules recognize each other. This subject of unceasing interest, ‘supramolecular chemistry’, relies on the understanding of noncovalent interactions. Among the noncovalent forces, the anion–π interaction has attracted increasing attention ever since its inception about two decades ago. This highlight article first summarizes some of the fundamental aspects of this interaction leading to several design strategies. In the main body we highlight some relevant examples that illustrate the viability of these strategies and the importance of anion–π interactions in crystal engineering.
 Antonio Bauzá | Mr. Antonio Bauzá received his degree in Chemistry from the Universitat de les Illes Balears (Spain) in 2011. After obtaining his master's thesis degree in 2012, he started his PhD working with Prof. A. Frontera. His thesis is devoted to the study from a theoretical perspective of the so-called unconventional noncovalent interactions that involve elements from group IV to VIII. |
 Tiddo J. Mooibroek | Dr. T. J. Mooibroek obtained his PhD working with Prof. E. Drent and Prof. E. Bouwman on the P2Pd-catalyzed reductive carbonylation of nitroaromatics (Leiden University, the Netherlands). He then joined the group of Prof. A. P. Davis to help develop artificial carbohydrate receptors (Bristol University, United Kingdom). He currently works as a research fellow in Bristol on the development of glucose-responsive insulin with Dr. T. S. Høeg-Jensen (Novo Nordisk) and Prof. A. P. Davis (Bristol University, United Kingdom). |
 Antonio Frontera | Prof. A. Frontera received his PhD from the Universitat de les Illes Balears (Spain) in 1994. During this period, he combined theory and experiment to propose a plausible mechanism for the direct lithiation of polyphenolic compounds. Moreover, he studied the mechanism of diotropic reactions. After two years of postdoctoral research in the laboratory of Prof. W. L. Jorgensen (Yale University, USA) in 1995–1996 devoted to the OPLS force field parametrization of carbohydrates, he came back to Spain as a research scientist at the Universitat de les Illes Balears, where he is currently a professor working on the study of noncovalent interactions. |
1. Introduction
Supramolecular chemistry is a highly multidisciplinary field of research. Its development has had a profound impact on the growing efficacy and success of constructing molecular assemblies of different sizes.1,2 Supramolecular chemists depend on the understanding of the noncovalent forces that govern molecular recognition phenomena. The design strategy of constructing interaction-complementary receptors for a target host molecule has resulted in host–guest systems with high affinities (Kass ≥104 M−1),3–5 even in very competitive media such as water (i.e., small, polar and protic).6 In the solid state the interaction-complementary approach is even more pronounced, as competing solvent molecules are mostly absent. As a result, the manner in which crystalline materials are organized can be seen as a reflection of strong and weak intermolecular forces balancing themselves towards a stable state. Strong and highly directional interactions such as hydrogen bonding and other σ-hole bonding7–15 together with less directional forces like ion pairing are indeed commonly used to engineer crystals.Interactions involving π-systems (e.g. aromatic rings, nucleobases) are very relevant for molecular interactions. For instance C–H/π, π–π and cation–π interactions are important in protein structure and enzyme catalysis.16–19 During the past two decades the anion–π interaction has been established as the attractive interaction between an anion and an electron-deficient π-system.20–25 The birth of this increasingly established interaction can be pinpointed to several publications.26–30 In 1996 Woollins et al. described a close contact between Cl− and the aromatic ring [S4N3]+ in the crystal packing of thiotrithiazylium chloride and named this a “π-facial interaction”.26 Other early publications are from Schneider et al.27,28 and Yamabe et al.29,30 Recently, this interaction has been described as a subclass of π-hole bonding interactions.15b,31 Various theoretical32–35 and experimental36–40 investigations showcase that anion–π interactions can be of structural and functional significance. Indeed, the relevance of the anion–π interaction has been noticed in biochemical processes and in fields like environmental chemistry, medicine and supramolecular chemistry.20,41–45
This highlight article is not just a bibliographic review of the literature related to anion–π interactions since several surveys have been written for that purpose.20,24,46–49 Instead, we showcase herein that a fundamental understanding of the interaction leads to design strategies that can be exploited in crystal engineering.
2. Fundamental aspects of anion–π interactions
2.1 Physical nature
The physical nature of the anion–π interaction has been studied and rationalized using high-level ab initio calculations and several partition energy schemes.50,51 The general assumption is that ion-induced polarization and electrostatic terms are the main contributors to the anion–π interaction.52–54The molecular polarizability of a compound is denoted as α‖. The π-cloud on an aromatic compound is readily polarized by an anion, leading to an ‘induced dipole’ (see Fig. 1A). The anion can then interact with this induced dipole. This polarization contribution to the total interaction energy of an anion–π contact can be substantial.50–53 Actually, the electrostatic repulsion anticipated in the benzene⋯Cl− pair is largely overruled by the favourable induced dipole interaction, resulting in a nearly negligible overall repulsive interaction energy of +2.4 kcal mol−1.55 Induced polarization interactions can also rationalize the dual binding mode exhibited by arenes with a marginal permanent quadrupole moment perpendicular to the ring plane (Qzz in Buckinghams, B).56,57 That is, molecules with very small Qzz values such as 1,3,5-trifluorobenzene (Qzz = 0.57 B) and s-triazine (Qzz = 0.90 B) are able to interact with both anions and cations because the electrostatic term is negligible and the interaction is thus dominated by the polarization term (always favourable).
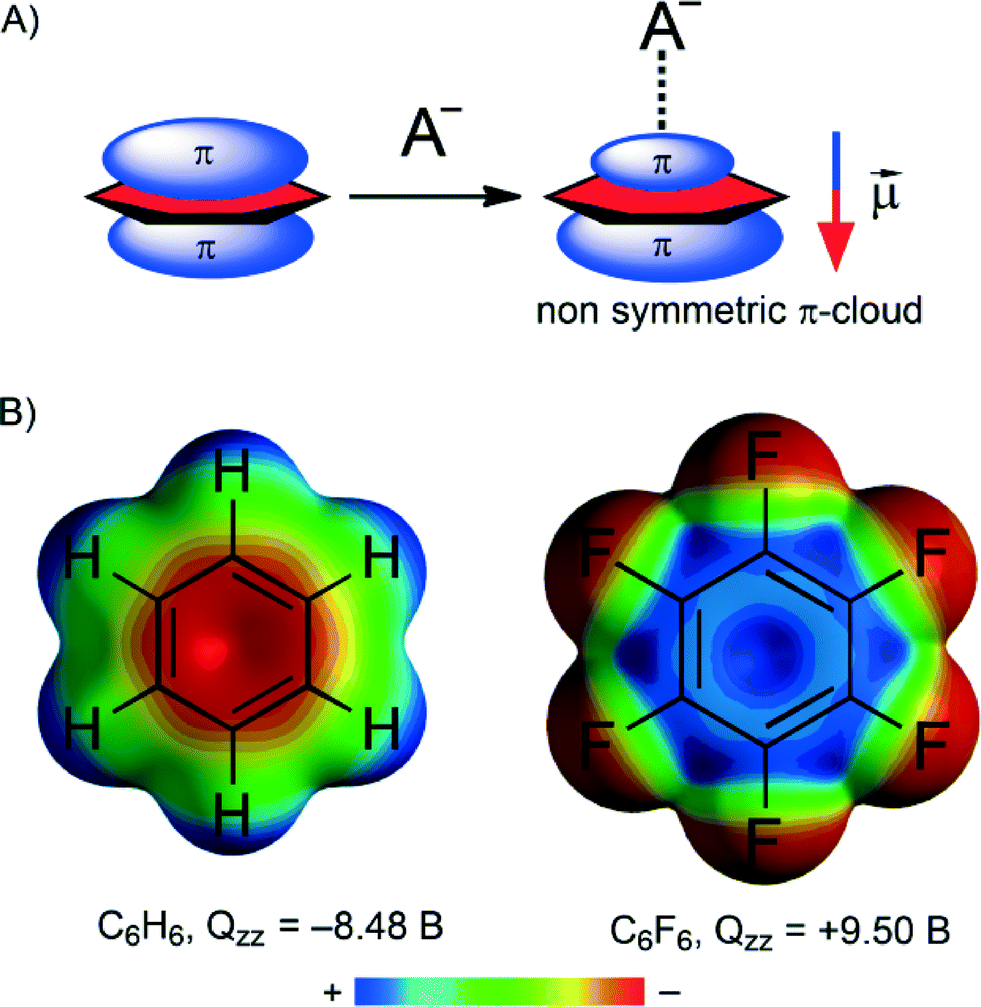 | ||
| Fig. 1 Illustrations to aid in the understanding of the physical nature of anion–π interactions: (A) schematic representation of the ion-induced dipole. (B) Molecular electrostatic potential (MEP) of benzene and hexafluorobenzene; MEP surfaces computed at the B3LYP/6-31G* level of theory. | ||
The electrostatic contribution to an anion–π interaction correlates well with the permanent quadrupole moment perpendicular to the ring plane (Qzz in Buckinghams, B). This physical property is a measure of the electron deficiency of an aromatic system. As one might expect, a more electron-poor aromatic tends to display stronger binding to anions. The magnitude and sign of Qzz depend on the substitution pattern on a given aromatic. For example (see Fig. 1B), the negative Qzz found for benzene (Qzz = −8.48 B) predicts a repulsive interaction with anions. If, however, H is replaced by F (i.e. hexafluorobenzene) the Qzz becomes positive (9.50 B) and the interaction becomes attractive.
The anion–π interaction has also been examined using molecular orbital analysis and compared to the related cation–π interaction.58,59 Interestingly, a totally different picture emerged: the atomic orbitals of a cation do not participate in the molecular orbitals of the cation–π complex, while conversely, the atomic orbitals of an anion contribute significantly to the molecular orbitals of the anion–π complex.58,59
2.2. Energies and directionality
The usefulness of an intermolecular interaction for molecular engineers relies in part on its strength and directionality. For the solid state (where competing solvent molecules are mainly absent), energies computed using high-level theoretical models provide a reliable estimate of the actual enthalpic component of an anion–π interaction. Various such inquiries have revealed that a single anion–π interaction varies in strength from very weak (−3.6 kcal mol−1 for 1,4-difluorobenzene⋯Cl− complex) to strong (−24.0 kcal mol−1 for 1,3,5-trinitrobenzene⋯Cl−) compared to hydrogen bonding (typically −2–−25 kcal mol−1).60In light of such estimated binding enthalpies it can be expected that anion–π interactions are directional. The geometry that is intuitively preferred can be seen as a ‘T-shaped’ geometry, where the anion is located above/below the centre of an aromatic ring at a ring plane–anion angle of 90°. However, the rather wide electropositive region above/below an aromatic implies that lateral deviation from this geometry has a small energy penalty. The size of the electropositive potential may thus impede on the directionality of anion–π interactions.
A convenient source of information regarding the actual directionality of intermolecular interactions is contained within the ever-growing Cambridge Structure Database (CSD). It is indeed commonly assumed that a proper statistical analysis of the CSD can unveil the geometric preferences of a given supramolecular interaction. This assumption stems from the intuition that even weak intermolecular forces must somehow manifest themselves in the way molecules pack in crystals.
Some CSD surveys have shown the close proximity of electron-rich/anionic entities above/below electron-deficient (hetero) aromatics.20,61–63 The directional character of anion–π (but also lone-pair–π)64 interactions has only recently been demonstrated by a thorough CSD evaluation of interactions between anionic/electron-rich atoms and a pentafluorophenyl vs. a phenyl ring.64 The method used in these inquiries takes account of the non-spherical volume of the arene and corrects for a random scattering of the data. The reasoning behind the method is akin to the ‘cone-correction method’65 that is known to be essential for a proper evaluation of the geometric preferences of hydrogen bonding.60 These studies clearly showed that the predicted ‘T-shaped’ binding mode is preferred for pentafluorophenyl rings, while a clustering of data near the phenyl's H-atoms was observed.64
2.3. Towards design strategies
From the above it is clear that to engineer anion–π interactions, the π–binding units should have a large molecular polarizability (α‖) and a large and positive quadrupole moment (Qzz).The polarizability can be somewhat tuned by the size of the aromatic; larger conjugated systems (e.g. naphthalene) are more polarizable than smaller π-systems (e.g. benzene).
Enlarging the permanent quadrupole moment of an arene can be achieved in several ways. Attachment of strong electron-withdrawing groups like -NO2 and -CN leads to large positive Qzz values (e.g.Fig. 2A). For benzene derivatives this strategy is somewhat limited because it is difficult to install more than three such groups. Using polycyclic aromatic compounds enables one to attach more electron-withdrawing groups, with the added advantage of increased molecular polarizability (see e.g.Fig. 2A). An alternative way of rendering an arene ring electron deficient is to have it partake in a cation–π interaction. The induced positive potential on the ring can then interact favourably with anions to give strong ‘cation–π–anion complexes’ in which the arene effectively bridges the ion pair.66–72 These ternary complexes exhibit large binding energies and shorter equilibrium distances than those in the corresponding binary ion–π complexes. Alternatively, the ring itself can contain a positive charge, such as in tropylium or protonated azines.
 | ||
| Fig. 2 Illustrations of design strategies to obtain strong anion–π interactions: (A) MEP of 1,3,5-trinitrobenzene and 1,3,5,7-tetranitronaphtalene, values of MEP, quadrupole moments (Qzz) and molecular polarizabilities (αzz). (B) MEP of s-triazine, [s-triazine⋯3HF], [s-triazine⋯3CuICl] and 1,3,5-triazine 1,3,5-trioxide. MEP surfaces computed at the B3LYP/6-31G* level of theory. MEP values over the ring centre in kcal mol−1. | ||
N-Substituted heteroaromatics can be particularly useful to generate anion–π interactions. While some (e.g. s-triazine, Fig. 2B) already have a positive electrostatic potential over the ring centre, the MEP value can be greatly enhanced when a N-atom is bound to some positively polarized entity or when it is oxidized to an N-oxide. As illustrated in Fig. 2B for s-triazine, hydrogen bonding, coordination to a metal ion and oxidation all lead to a larger positive MEP value over the ring centre.48,73–77 Particularly useful about N-heterocyclic arenes is that their coordination chemistry can be exploited to construct larger molecular assemblies. Another design factor is the additivity of the anion–π interaction. For example, for a series of complexes between Cl−/Br− and some s-triazines it was computed that the ternary “sandwich” complexes were twice as stable as the corresponding binary complexes.78 Thus, designing systems that can encapsulate an anion using multiple anion–π contacts seems advantageous.
3. Highlighted structures
3.1 General remarks and overview
We have chosen to highlight several structures where anion–π interactions were intended and/or closely studied by the authors of the original manuscripts. For ease of reference we use the CSD reference codes in graphical renderings of these examples. In addition to reported examples, we examined recent entries (post November 2010) in the CSD for particularly evident anion–π interactions, that is, structures where the van der Waals shell of an atom belonging to an anion overlaps with all six atoms of a six-membered aromatic ring composed of C and/or N.† Nearly all of these close contacts have been overlooked by the authors of the original manuscripts, highlighting just how prevalent yet non-canonical anion–π interactions are. An asterisk (*) has been added to their CSD reference codes to demarcate them from the structures where anion–π interactions were clearly intended and/or studied. Occasionally we also discuss some theoretical studies that are related to the structure highlighted and/or some interesting experimental findings reported by the authors.Shown in Fig. 3 is an overview of the aromatics used in the crystal structures we mention in this work. The selected structures are grouped under three main headings based on the type of aromatic used, namely: formally charge-neutral arenes rendered electron deficient by covalently attached groups (A), arenes that are coordinated to a metal ion (B), and arenes that bear a formal positive charge. For completion we also indicate in what coordination environment the metal ions are.
 | ||
| Fig. 3 Overview of examples (including CSD reference codes) highlighted in this paper. Three classes have been distinguished where the interacting aromatic is formally charge neutral (A), coordinated to a metal ion (B) or where the aromatic bears a formal positive charge (C). The part of the aromatic involved in an anion–π interaction is highlighted in blue. The red asterisk (*) accompanying some CSD reference codes denote that the structure was found by the CSD search† and was not intended/studied by the authors of the original work. | ||
3.2 Charge-neutral aromatics
Pyrazine-N,N′-dioxide (PDO) is formally charge-neutral, yet has a substantial electron-deficient centre due to the high degree of polarization of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. This has been exploited by using PDO as a linking component in octacyanotungstenate-based networks.79 The hybrid networks are co-organized by two types of interactions, as indicated in Fig. 4: anion–π interactions between the pyrazine ring centre and a single NCN atom of [W(CN)8]3−, and an interaction where four NCN atoms point towards the Npyrazine atoms.
O bonds. This has been exploited by using PDO as a linking component in octacyanotungstenate-based networks.79 The hybrid networks are co-organized by two types of interactions, as indicated in Fig. 4: anion–π interactions between the pyrazine ring centre and a single NCN atom of [W(CN)8]3−, and an interaction where four NCN atoms point towards the Npyrazine atoms.
 | ||
| Fig. 4 Fragment of the X-ray crystal structure containing pyrazine-N,N′-dioxide moieties. The relevant anion–π interactions are indicated by dashed lines. The CSD reference code is indicated. H-atoms omitted for clarity. | ||
The electron-deficient pentafluorophenyl (PFP) group is also a good building block for generating anion–π interactions.64 Giese et al.80–82 have exploited this experimentally by focusing on purely organic salts where the positive charge is located some distance away from the PFP group. From their work it is evident that the position of an anion above the PFP π-system can be tuned. Indeed, in the solid-state structures of [N-(pentafluorobenzyl)pyridinium]+[X]− salts, the way in which the anion is encapsulated by C–H⋯X hydrogen bonds and anion–π contacts depends on the anion.81
In this particular system, both π-acidic rings (C6F5 and pyridinium moieties) are involved in the anion–π contacts (see Fig. 5). For X = I3− and PF6−, there are cooperative anion–π interactions between the two electron-deficient arenes and the anion located between them (Fig. 5A), while for X = Br− or BF4− (Fig. 5B), this cooperativity is absent.
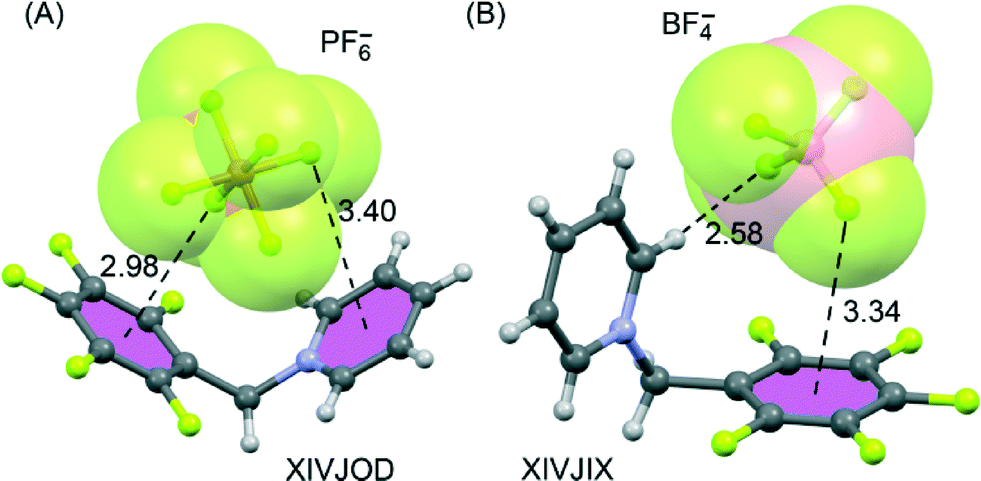 | ||
| Fig. 5 X-ray crystal structures of pentafluorophenyl derivatives involving hexafluorophosphate (A) and tetrafluoroborate (B). The CSD reference codes are indicated. Distances in Å. | ||
In a related investigation82 they used simple triphenyl(pentafluorobenzyl)-phosphonium and bis(pentafluorobenzyl)-phosphonium salts. The X-ray structures provided crucial data on the influence of anion size on the molecular structure of cations containing two adjacent electron-deficient rings (see Fig. 6).
 | ||
| Fig. 6 X-ray crystal structures of pentafluorophenyl derivatives involving bromide (A) and hexafluorophosphate (B). The CSD reference codes are indicated. Distances in Å. H-atoms omitted for clarity. | ||
In particular, whereas bromide anions interact by means of anion–π interactions in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mode with the pentafluorobenzene unit Z-configured (Fig. 6A), the bulkier anions iodide, tetrafluoroborate, and hexafluorophosphate (Fig. 6B) result in a 1:2 tweezer-like anti-configuration in which the anion interacts simultaneously with two pentafluorobenzene units. Apparently, when the spatial separation of the two electron-deficient rings matches the size of the anion, the formation of two concurrent anion–π interactions induce a conformational change from the anti-form observed for the smaller anion to the tweezer-like syn-form for the greater one.
:1 mode with the pentafluorobenzene unit Z-configured (Fig. 6A), the bulkier anions iodide, tetrafluoroborate, and hexafluorophosphate (Fig. 6B) result in a 1:2 tweezer-like anti-configuration in which the anion interacts simultaneously with two pentafluorobenzene units. Apparently, when the spatial separation of the two electron-deficient rings matches the size of the anion, the formation of two concurrent anion–π interactions induce a conformational change from the anti-form observed for the smaller anion to the tweezer-like syn-form for the greater one.
The naphthalenediimide (NDI) core seems to readily engage in anion–π interactions, likely because NDI is both electron deficient and easily polarized (large, hence large α‖). Several groups used NDI to construct fascinating assemblies in the solid state and several works deserve special attention. Liu et al.83 have generated a panchromatic hybrid crystal of iodoplumbate nanowires and J-aggregated NDIs. The organic–inorganic hybrid is constituted by anionic iodoplumbate nanowires that strongly bind to the NDIs by means of charge-assisted anion–π interactions (see Fig. 7).
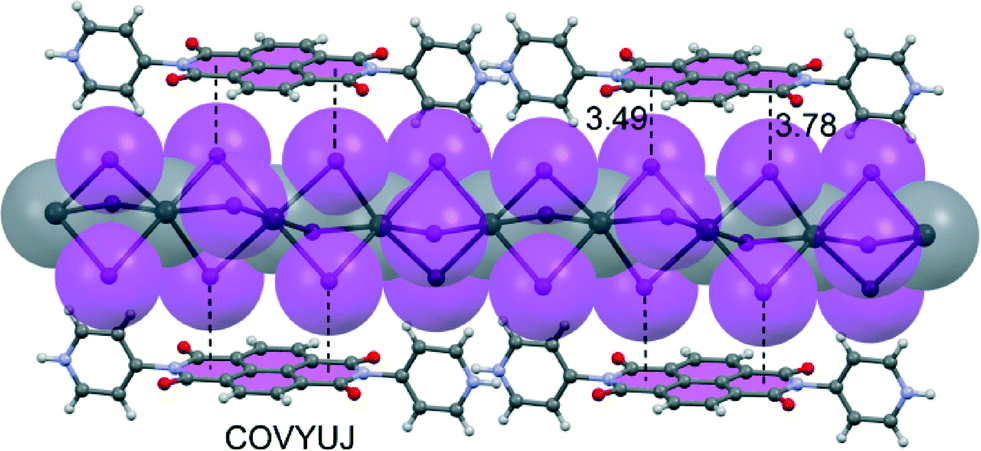 | ||
| Fig. 7 X-ray structure of the hybrid crystal of iodoplumbate nanowire and J-aggregated NDIs. The CSD reference code is indicated. Distances in Å. | ||
The material has photo-induced long-lived charge-separated states (also when irradiated with sunlight), making this study interesting for research in the area of solar cells and photocatalysis. The solid-state UV-vis diffuse reflectance spectral analysis revealed that the crystal is a panchromatic hybrid with a broad absorption band from 200 nm to 800 nm, making this material much ‘darker’ than other iodoplumbate nanowires.84,85 It is worthy to note that the long-lived charge separation in this hybrid material is basically due to the intense and panchromatic absorption and the close contacts between organic and inorganic units through anion–π interactions that aid the electron transfer (see Fig. 7).
Kumar et al.86 attached two phosphonium groups to an NDI unit (hence ‘NDI(PPh3)2’) and were able to characterize the radical cation [(NDI(PPh3)2˙+)BPh4−] and its extraordinary π-acidic precursor [(NDI(PPh3)22+)·2BF4−]. In this latter structure (see Fig. 8) the NDI unit is tightly sandwiched in between two BF4− anions through anion–π contacts.
 | ||
| Fig. 8 Fragments of the X-ray crystal structures containing NDI π-acidic moieties. The relevant anion–π interactions are indicated by dashed lines. The CSD reference code is indicated. Distances in Å. H-atoms omitted for clarity. | ||
A remarkable finding of their study is the extraordinary stability of both complexes, which were air-stable and resisted conventional silica column chromatography. What is more, the straightforward electron transfer (even in nonpolar solvents) strongly tunes the optical properties of the radical ion. This implies the usefulness of such materials in research towards switchable panchromatic materials, phosphorous-based stable radical ions and, in general, spin-based research.
NDIs have also been applied to encapsulate/stabilize polyoxoxmetalates (POMs) by anion–π interactions, as shown in Fig. 9, where [O40PW14 ]6− is encapsulated by three N,N′-di(4-pyridyl)-1,4,5,8-naphthalenediimide units.87 This enquiry demonstrated for the first time that anion–π interactions are appropriate for the stabilization and immobilization of functional POM anions.
 | ||
| Fig. 9 Fragment of the X-ray crystal structures containing POM and NDI moieties. The relevant anion–π interactions are indicated by dashed lines. The CSD reference code is indicated. Distance in Å. | ||
More recently, the combination of NDIs, POMs, Zn(II) and several co-ligands has been used88 to synthesize a very rare radical-doped POM-based host–guest crystalline material. Interestingly, it has fast-responsive reversible photochromic properties and photocontrolled tunable luminescence. In addition, this material is able to photocatalytically oxidize benzylic alcohols to aldehydes using air as a catalyst re-oxidant. Of particular importance is that the trapped POM anions interact with functional NDIs via directional anion–π contacts. The anion–π interactions stabilize and immobilize the functional POM anions and also promote the charge transfer and exchange among components, leading to interesting properties of the crystalline material (i.e. reversible photochromism, photocontrolled tunable luminescence, and photocatalytic activity).
Stoddart et al.89 reported on a macrocycle linking three NDIs together using 1,2-diaminocyclohexane, leading to molecular triangular prisms able to encapsulate anions (see Fig. 10). They have used both theory and experiment to demonstrate orbital interactions and electron transfer effects between the anion and the π-acidic surface of the NDIs in their synthesized redox-active prisms. The presence of three NDI moieties symmetrically distributed leads to a large number of individually accessible redox states in the triangular prisms, opening the door to potential applications in molecular electronics. The electron-deficient interior of the molecular prism is perfect for studying anion–π interactions. This ability is evidenced by the trapping of linear I3− anions inside the receptor cavity, causing a significant change in the packing of the prisms in the extended solid-state architecture. The encapsulation of I3− anions provokes π–π stacking of the chiral prisms into supramolecular helices, providing an extraordinary example of anion-induced self-assembly. In addition, the chirality provided by the six stereogenic centres of the cyclohexane corners in the occupied prisms dictates the either right- or left-handedness of their packing in the solid state.
 | ||
| Fig. 10 The X-ray crystal structure of the NDI-based triangular prism. The CSD reference code is indicated. H-atoms are omitted for clarity. | ||
Another family of neutral anion receptors that exploited the additivity of anion–π interactions is based on a tetraoxacalix[2]arene[2]triazine macrocyclic host reported by Wang et al.90 They combined several techniques (electrospray ionization mass spectrometry, fluorescence titration and X-ray crystallography) to demonstrate that tetraoxacalix[2]arene[2]triazine forms 1:1 complexes with several polyatomic and monoatomic anions. The solid-state structures with polyatomic anions like NO3−, BF4−, PF6− and SCN− reveal that two opposing s-triazine rings act as a pair of tweezers to interact with the anions through cooperative anion–π interactions (see Fig. 11A). In monoatomic (smaller) anions like Cl− and Br−, a water molecule is also involved in the anion binding and the tweezers interact with the anion–water pair through anion–π and lp–π interactions (see Fig. 11B).91 Interestingly, Wang et al. also altered the tetraoxacalix[2]arene[2]triazine host by introducing hydroxyl groups in the meta-position of the arene rings. In the NMe4Br co-crystal of this novel macrocycle (see Fig. 11C),92 the introduced –OH groups act like water in the unsubstituted version (Fig. 11B), hydrogen bonding to Br− and interacting with an s-triazine ring by a lone-pair–π interaction.
 | ||
| Fig. 11 X-ray crystal structures of tetraoxacalix[2]arene[2]triazine hosts involving thiocyanate (A), chloride (B) and bromide (C) anions. The CSD reference codes are indicated. Distances in Å. H-atoms omitted for clarity. | ||
1,4,5,8,9,12-Hexaazatriphenylene (HAT) is a large (and thus easily polarizable) electron-deficient aromatic known to generate anion–π interaction. For example, K. Dunbar et al. constructed a HAT derivative bearing six (electron-withdrawing) cyano groups.93 This neutral yet strongly π-acidic molecule co-crystallized with N(n-Pr)4Br from benzene, leading to a packing where HAT interacts with four anions simultaneously (three anions by one side and one by the opposite side, see Fig. 12). Furthermore, Ballester's group94 has studied anion–π complexes of HAT(CN)6 both theoretically and experimentally and demonstrated that the formation of anion–π interactions, charge-transfer or electron transfer adducts strongly depends on the type of anion.
 | ||
| Fig. 12 X-ray structure of [HAT(CN)6]2[Br−]3. The CCDC reference code is indicated. | ||
Finally, it is worth pointing out some theoretical studies with the bicycle pyridazino[4,5-d]pyridazine and larger polycyclic analogues thereof.76,77 These inquiries demonstrated that the anion–π interaction strengthens as the number of fused rings increases. Interestingly, when such extended heteroaromatics were hydrogen bonded to water, even stronger complexes were predicted to form with anions.
3.3 Aromatics coordinated to metal ions
Dunbar's group has greatly contributed to highlighting the crucial role of the anion–π interaction in the formation of self-assembled metallacycles.95–97 Taking advantage of the structural versatility of metal ions and the directionality of metal–ligand interactions98–103 they have recently described104 the spontaneous assembly of elegant supramolecular architectures with unusual properties and applications or intriguing host–guest behaviour. They have provided unambiguous evidence that anion–π interactions are the main driving force in the templation process leading to the formation of Fe(II) metallacycles with π-acidic cavities, using 3,6-bis(2-pyridyl)-1,2,4,5-tetrazine as chelating ligand in the solid state. The anions play a decisive role in the formation and size of the supramolecular polygons, establishing multiple anion–π interactions with the electron-deficient walls, as exemplified in Fig. 13 for TIWHEO.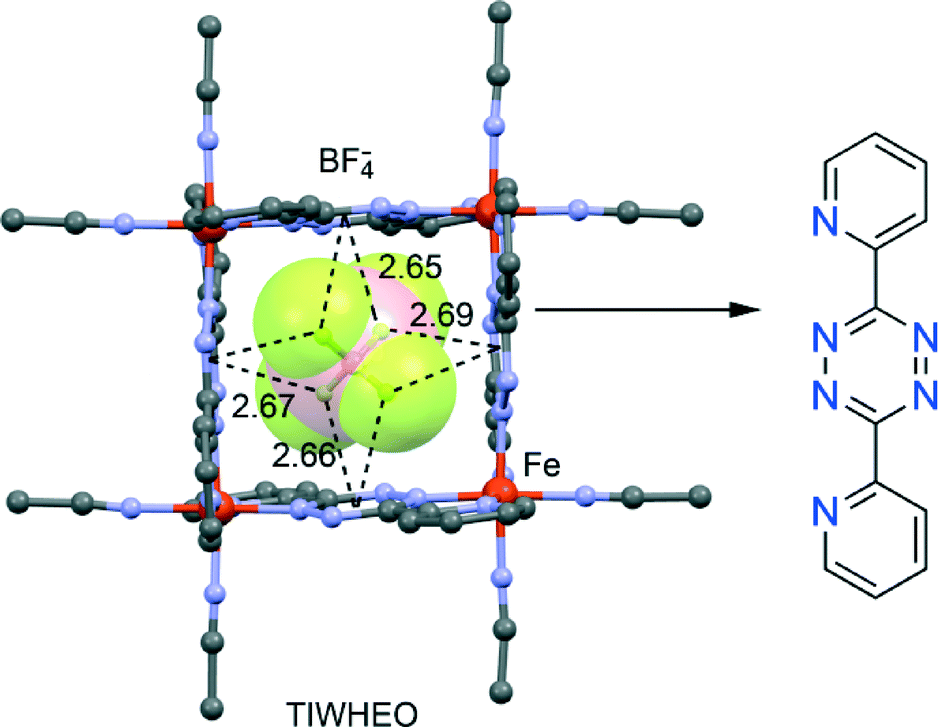 | ||
| Fig. 13 Fragment of the X-ray crystal structures containing 3,6-bis(2-pyridyl)-1,2,4,5-tetrazine ligands. The relevant anion–π interactions are indicated by dashed lines. The CSD reference code is also indicated. H-atoms omitted for clarity. Distances in Å. | ||
Safin et al.105 recently reported anion-induced self-assemblies using AgI salts and the two multidentate ligands 3,6-bis(2′-pyrimidyl)-1,2,4,5-tetrazine and 2,4,6-tris(2-pyrimidyl)-1,3,5-triazine. Interestingly, the selective use of PF6−, ClO4− and OTf− anions leads to the formation of different solid-state architectures, providing clear evidence of the pivotal role played by different anions (size and shape) and their interactions with the ligand. Two structures where anion–π directed assembly is most evident are depicted in Fig. 14A and B.
 | ||
| Fig. 14 Fragments of the X-ray crystal structures containing pyrazine-N,N′-dioxide moieties and hexafluorophosphate (A) or perchlorate (B) anions. The relevant anion–π interactions are indicated by dashed lines. The CSD reference codes are indicated. Distances in Å. | ||
In TUNDIR, the PF6− anion interacts with three central tetrazine rings of the 3,6-bis(2′-pyrimidyl)-1,2,4,5-tetrazine ligand, and in TUNDAJ the ClO4− is sandwiched between two central rings of the 2,4,6-tris(2-pyrimidyl)-1,3,5-triazine ligand.
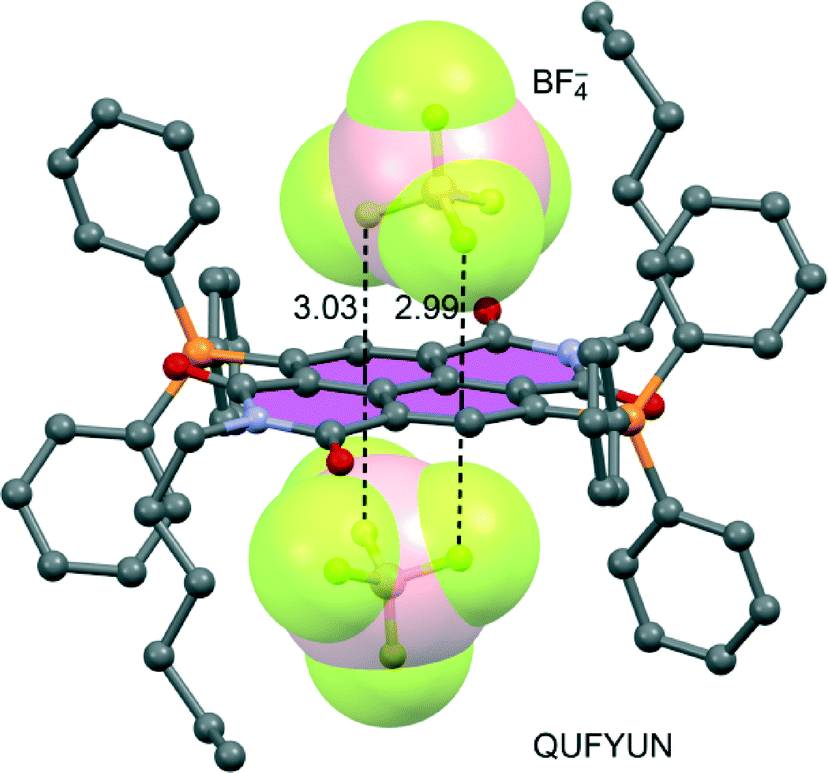
Another clear example of this strategy has been reported by Domasevitch's group.74 In particular, several X-ray crystal structures of s-tetrazine μ4-coordinated to AgI have been reported, exhibiting very close contacts between anions and the s-tetrazine ring, indicating strong anion–π interactions. Computational models of their structures corroborated that strong anion–π interactions are present between nitrate anions and the tetrazine ring. The polycyclic heteroaromatic arene pyridazino[4,5-d]pyridazine mentioned earlier has been studied as an anion–π donor when coordinated to metal ions by Gural'skiy et al.,74 clearly revealing the formation of several anion–π interactions. Qin et al.106 have reported the aqueous synthesis of an octanuclear macrocycle that includes fluorescent carbazole-based dipyrazole ligands, which coordinate to dipalladium corners. The X-ray diffraction analysis (see Fig. 15) reveals that this hybrid metallomacrocycle traps BF4− anions in the dipalladium–phenantroline clips through short anion–π contacts between BF4− anions and coordinated phenanthroline ligands.
 | ||
| Fig. 15 Fragment of the X-ray crystal structure showing the anions trapped in the dipalladium clips through anion–π interactions. The relevant anion–π interactions are indicated by dashed lines. The CSD reference code is indicated. Distance in Å. | ||
A recent experimental and theoretical study107 has evidenced the formation of anion–π/π–π/π–anion assemblies in the solid-state structures of several copper(II) complexes with a tetradentate Schiff base (see Fig. 16 for a selected example). Similar anion–π/π–π/π–anion assemblies in the solid state have also been found in N6-decyladenine hydrochloride salts and bisadenine derivatives.108
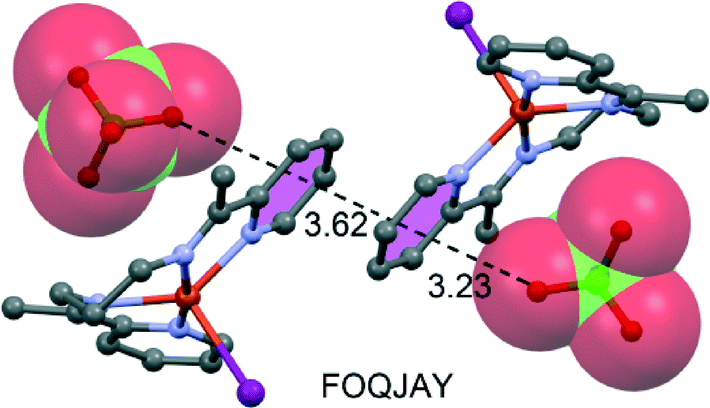 | ||
| Fig. 16 Fragment of the X-ray crystal structure exhibiting anion–π/π–π/π–anion assembly. The CSD reference code is indicated. Distances in Å. | ||
The interplay between π–π and anion–π interactions has also been investigated by others by combining theoretical studies and searches in the CSD.109–111 These studies too revealed that the mutual influence between ion–π and π–π interactions can lead to strong cooperativity effects.
Atwood and co-workers reported on calixarenes and cyclotriveratrylene macrocycles that have their arene rings μ6-coordinated to transition metal ions (Ru, Ir, Rh).69–72 This renders these molecules (which normally act as a host for cations) into an electron-deficient bowl to accommodate anions, as illustrated in Fig. 17.
 | ||
| Fig. 17 Fragments of the X-ray crystal structures containing quinolizinium and tropylium moieties. The relevant anion–π interactions are indicated by dashed lines. The CSD reference codes are indicated. | ||
In addition to the examples highlighted above, we next summarize some structures we found by our CSD inquiry† where very strong anion–π contacts have been overlooked by the authors of the original manuscripts. Graphical illustrations of these examples are collected in Fig. 18A–D, and in all cases the van der Waals shell of an atom belonging to an anion overlaps with all six atoms of the interacting arene, representing η6 anion–π interactions. In one example, UTEGIK (see Fig. 18A), a PF6− anion interacts with phthalazine with an exceptionally short F⋯ring centroid distance of 2.64 Å; the sum of the van der Waals radii of F (1.47) and C (1.70) equals 3.17 Å. The shortest F⋯C distance of 2.908 Å is 0.262 Å shorter than the benchmark of overlapping van der Waals shells. Another example is REJGEK as depicted in Fig. 18B, where the electrochemically active Rh(III) coordination complex exhibits an η6 anion–π interaction between the phenyl aromatic ring and one chlorine atom of the anion. The Cl−⋯ring centroid distance of 3.09 Å is again exceptionally short when compared to the sum of the van der Waals radii of 3.45 Å for Cl (1.75) and C (1.70). Kiplinger and co-workers recently investigated the coordination behaviour of bulky β-diketiminate ligands toward UCl4.112 The reaction of two equivalents of the ligand with UCl4 led to an interesting cationic diuranium complex (see Fig. 18C). The X-ray structure demonstrates that the β-diketiminate ligand is symmetrically bound to the U and an interesting combination of anion–π and halogen bonding interactions is crucial to rationalize the solid-state architecture of this compound. That is, the Cl− counterion is located almost perfectly over the ring centre of the aromatic ring of one complex and simultaneously establishes several Cl⋯Cl interactions with the coordinated U chlorido ligands belonging to another complex, generating a remarkably supramolecular assembly (see Fig. 18C). Finally, Li et al.113 reported on the self-assembly of enantiopure 2,5-bis(4,5-pinene-2-pyridyl)pyrazine ligands with copper(II) nitrate, leading to second-order nonlinear optically (NLO) active square Cu(II) enantiomeric pairs (one enantiomer is shown in Fig. 18D). The X-ray structure presents four crystallographically equivalent Cu(II) ions and four chiral ligands forming a molecular square. Each Cu(II) ion in these squares forms an octahedron by coordinating to four nitrogen atoms from the ligands and two oxygen atoms of two monodentate NO3− anions. Remarkably, each pyrazine ring forms extraordinary short anion–π interactions with the monodentate NO3− anion of the neighbouring complex (only the naked anions are shown in Fig. 18D for clarity), thereby gluing together the individual squares.
 | ||
| Fig. 18 (A–D) Illustrations of examples where an η6 anion–π interaction involving a metal-coordinated heteroaromatic was found in the CSD,† which has escaped the attention of the authors of the original manuscripts. The CSD reference codes are given. Distances in Å. The sum of van der Waals radii [Σr(vdW)] values is also given. In A, B and D structures the H-atoms have been omitted for clarity. | ||
At this point it is worth mentioning that in some of the examples shown above one may argue that the anion simply tries to snug as close to the cation as sterically feasible. The π-system is them thought to be in the way and it is indeed difficult to differentiate between the contribution of the π-system and that of the positive charge. In this respect, several theoretical studies have showed that the contribution of the anion–π interaction is usually large and naturally assisted by the increased electron deficiency induced by coordination.114 Moreover, since the anion-π interaction is directional while ion pairing is not, the final position of the (poly)anion is most likely determined by the anion–π interaction.
3.4 Positively charged or protonated aromatics
The geometric and energetic features of anion–π+ complexes of several aromatic cations (tropylium, quinolizinylium) have been investigated theoretically and by analysis of the CSD.115–118 Two selected examples retrieved from the literature (KASVOP10119 and CADZUC120) are shown in Fig. 19A and B. | ||
| Fig. 19 Fragments of the X-ray crystal structures containing quinolizinium and tropylium moieties. The relevant anion–π interactions are indicated by dashed lines (distances in Å). The CSD reference codes are indicated. | ||
The importance of the anion–π+ interaction in protonated purine and pyrimidine bases has been recently reviewed.121 As expected, interaction energies of anion–π+ complexes are dominated by strong electrostatic effects exhibiting very large binding energies (>80 kcal mol−1).
Orvay el al.122 also exploited anion–π+ interactions in five proton transfer compounds using 4,6-di(1H-imidazol-1-yl)pyrimidine and different counterions. In the crystal structures of these five salts, anion–π interactions involving the aromatic rings play a fundamental role for the generation of three-dimensional supramolecular frameworks in the solid state. Two selected examples are illustrated in Fig. 20A and B.
 | ||
| Fig. 20 (A, B) Fragments of the X-ray crystal structures containing 4,6-di(1H-imidazol-1-yl)pyrimidine moieties. The relevant anion–π interactions are indicated by dashed lines. The CSD reference codes are indicated. Distances in Å. | ||
The first one (YOGJIP) is characterized by the formation of a remarkable supramolecular architecture through two anion–π+ (chloride ions) and one lp–π (water molecule) interactions involving all three aromatic rings of the diprotonated 4,6-di(1H-imidazol-1-yl)pyrimidine moiety. That is, an oxygen atom lone pair of the water molecule is oriented toward the π-cloud of the pyrimidine ring, forming a strong lp–π interaction. In addition, two chloride ions interact with the imidazolium rings, forming two anion–π+ interactions. In the second one (YOGJOV), two [CdCl4]2− anions and two imidazolium moieties participate in the generation of anion–π/π–π/π–anion assemblies (see Fig. 20B).
Several interesting crystal structures of 4′-(4-pyridyl)-2,2′:6′,2′′-terpyridine (PTP) salts have been recently reported by Manna et al.123 They found that the structural architecture depends on the type of anion (mononuclear or trinuclear) and on the pH from which the solid crystallized. As illustrated in Fig. 21 for one of the structures (LODJAR), the [pyridine-H]+ rings concurrently participate in hydrogen-bonding (N–H⋯Br−) and anion–π+ interactions.
 | ||
| Fig. 21 Fragment of the X-ray crystal structure containing 4′-(4-pyridyl)-2,2′:6′,2′′-terpyridine moieties. The relevant anion–π interactions are indicated by dashed lines. The CSD reference code is also indicated. Distances in Å. | ||
In this experimental work, the role of the anion–π+ interaction in the solid state has also been investigated theoretically, providing additional evidence for its role as a decisive supramolecular force responsible for the packing geometry of these salts. The same research group124 has further investigated the role of the anion–π+ interactions in the generation of supramolecular assemblies using a similar ligand, i.e. the triply protonated 4′-(4-pyridyl)-3,2′:6′,3′′-terpyridine. Combining X-ray crystallography with theoretical studies, they showed that π+–π and various anion–π+ interactions are the major driving forces in the stabilization of the assemblies observed in these solid-state structures (see Fig. 22).
 | ||
| Fig. 22 Fragment of the X-ray crystal structure containing 4′-(4-pyridyl)-3,2′:6′,3′′-terpyridine moieties. The relevant anion–π interactions are indicated by dashed lines. The CSD reference code is also indicated. Distances in Å. | ||
Shown in Fig. 23A–C are illustrations of solid-state structures where anion–π+ interactions (found by our CSD analysis)† are particularly relevant (η6 anion–π+ interactions) but not described by the original authors. For instance Chen et al.125 have reported the X-ray structure of the 5,6-dioxo-1,10-phenanthrolinium bromide (see Fig. 23A). The bromide anions induce the formation of sandwich complexes in the solid state, forming two symmetrically equivalent anion–π+ interactions with the dioxo ring. The shortest Br−⋯ring centroid distance of 3.22 Å is 0.33 Å shorter than the sum of the van der Waals radii of B (1.85) and C (1.70) namely 3.55 Å. The shortest Br−⋯C distance of 3.36 Å is 0.19 Å shorter than this benchmark of overlapping van der Waals shells. 4,4′-Bipyridine has been combined with polyoxometalates POMs to generate several organic–inorganic supramolecular hybrid crystals, prepared from acidic solutions, that are organized through anion–π+ interactions.126 In Fig. 23B we show a selected example where a Wells–Dawson polyoxoanion is involved. This type of POM has 54 surface oxygen atoms including 18 terminal oxygen atoms (only bonded to a W atom) and 36 μ2-O atoms (bonded to two W atoms). The terminal O atoms establish extensive anion–π+ interactions in the crystal structure (see Fig. 23B). These interactions along with hydrogen bonds connect the POMs and the diprotonated bipyridine cations, generating a remarkable 3D supramolecular network. Finally, Albrecht and co-workers127 have synthesised and characterized a novel phthalazine-triazole ligand (1,4-bis(1-methyl-1H-1,2,3-triazol-4-yl)phthalazine) with the purpose of generating new ruthenium(II) complexes. They also studied its reactivity in several methylation reactions. Particularly, when the ligand was reacted with MeOTf (OTf = trifluoromethyl sulfonate) a double methylation was observed and the resulting product was characterized by X-ray analysis (see Fig. 23C). Interestingly, the double methylated ligand exhibits anion–π+ interactions with both the six- and five-membered rings that are positively charged in the solid state.
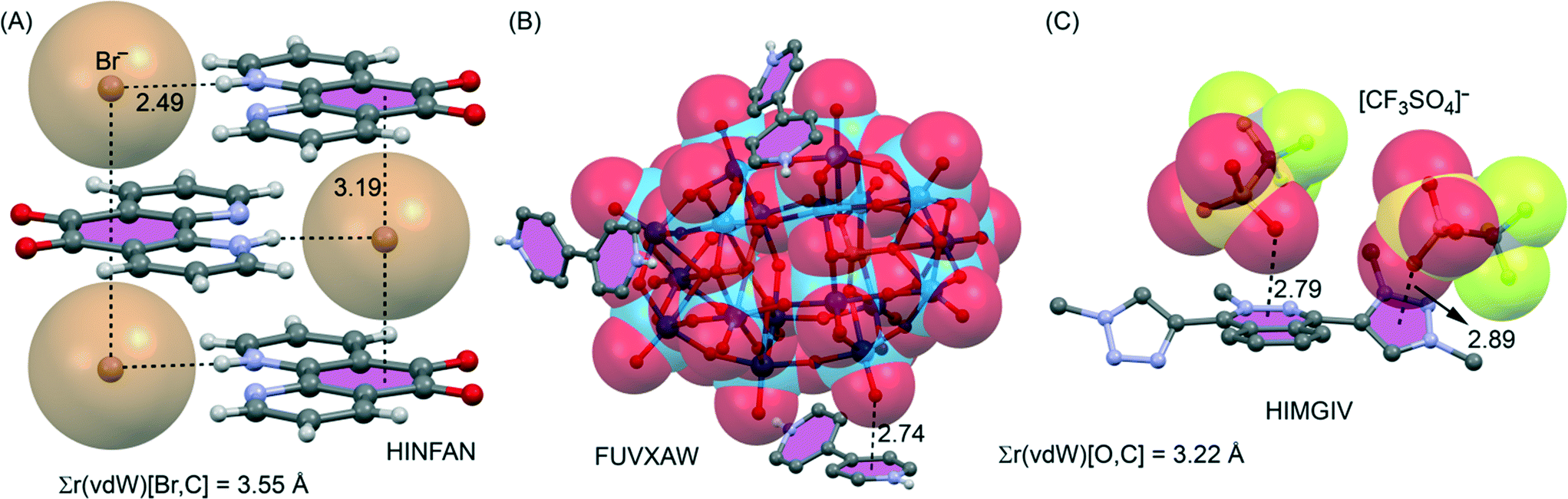 | ||
| Fig. 23 (A–C) Illustrations of examples where an η6 anion–π interaction involving a formally cationic aromatic was found in the CSD,† which has escaped the attention of the authors of the original manuscripts. Distances in Å. The sum of van der Waals radii [Σr(vdW)] values is also given. | ||
Concluding remarks
There clearly remains an element of unpredictability in crystal engineering, as even the coordination geometry of a transition metal complex is not always as predicted. The degree of serendipity logically increases with weaker interactions, which include the anion–π interaction. Nevertheless, anion–π interactions are undoubtedly a force to be reckoned with for a proper post factum rationalization of supramolecular assemblies in crystals. Moreover, while serendipity remains, our recently acquired understanding of anion–π interactions – as highlighted in this work – has led to a degree of predictability that was absent only about two decades ago. It is our hope that the design strategies outlined in this highlight will inspire (supra)molecular designers to advance this predictability even more. We furthermore anticipate that with an increased control over anion–π interactions will follow opportunities for the design of novel materials, catalysts, sensors, and biologically active molecules.Acknowledgements
The financial support from MINECO of Spain (projects CTQ2014-57393-C2-1-P and CONSOLIDER INGENIO 2010 CSD2010-00065, FEDER funds) is gratefully acknowledged.Notes and references
- H. J. Schneider, Angew. Chem., Int. Ed., 2009, 48, 3924 CrossRef CAS PubMed.
- H. J. Schneider and A. Yatsimirski, Principles and methods in supramolecular chemistry, Wiley, Chichester, 2000 Search PubMed.
- J. M. Lehn, Supramolecular chemistry concepts and perspectives, Wiley–VCH, Weinheim, 1995 Search PubMed.
- F. Vögtle, Supramolecular chemistry: an introduction, Wiley, New York, 1993 Search PubMed.
- (a) P. D. Beer, P. A. Gale and D. K. Smith, Supramolecular chemistry, Oxford University Press, Oxford, 1999 Search PubMed; (b) J. W. Steed and J. L. Atwood, Supramolecular chemistry, Wiley, Chichester, 2000 Search PubMed.
- H. Destecroix, C. M. Renney, T. J. Mooibroek, T. S. Carter, P. F. N. Stewart, M. P. Crump and A. P. Davis, Angew. Chem., Int. Ed., 2015, 54, 2057 CrossRef CAS PubMed.
- P. Murray-Rust and W. D. S. Motherwell, J. Am. Chem. Soc., 1979, 101, 4374 CrossRef CAS.
- P. Murray-Rust, W. C. Stallings, C. T. Monti, R. K. Preston and J. P. Glusker, J. Am. Chem. Soc., 1983, 105, 3206 CrossRef CAS.
- N. Ramasubbu, R. Parthasarathy and P. Murray-Rust, J. Am. Chem. Soc., 1986, 108, 4308 CrossRef CAS.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386 CrossRef CAS PubMed.
- P. Politzer and J. S. Murray, ChemPhysChem, 2013, 14, 278 CrossRef CAS PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178 RSC.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748 RSC.
- A. Bundhun, P. Ramasami, J. S. Murray and P. Politzer, J. Mol. Model., 2013, 19, 2739 CrossRef CAS PubMed.
- (a) A. Bauzá, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317 CrossRef PubMed; (b) A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496 CrossRef PubMed; (c) A. Bauzá and A. Frontera, ChemPhysChem, 2015, 16, 3108 CrossRef PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303 CrossRef CAS PubMed.
- N. Zacharias and D. A. Dougherty, Trends Pharmacol. Sci., 2002, 23, 281 CrossRef CAS PubMed.
- N. S. Scrutton and A. R. C. Raine, Biochem. J., 1996, 319, 1 CrossRef CAS PubMed.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459 CrossRef CAS.
- A. Frontera, P. Gamez, M. Mascal, T. J. Mooibroek and J. Reedijk, Angew. Chem., Int. Ed., 2011, 50, 9564 CrossRef CAS PubMed.
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Chem. Soc. Rev., 2008, 37, 68 RSC.
- C. Caltagirone and P. A. Gale, Chem. Soc. Rev., 2009, 38, 520 RSC.
- P. Gamez, T. J. Mooibroek, S. J. Teat and J. Reedijk, Acc. Chem. Res., 2007, 40, 435 CrossRef CAS PubMed.
- B. P. Hay and V. S. Bryantsev, Chem. Commun., 2008, 2417 RSC.
- A. Bauzá, D. Quiñonero, P. M. Deyà and A. Frontera, New J. Chem., 2013, 37, 2636 RSC.
- J. R. Galán–Mascarós, A. M. Z. Slawin, J. D. Woollins and D. J. Williams, Polyhedron, 1996, 15, 4603 CrossRef.
- H. J. Schneider, F. Werner and T. Blatter, J. Phys. Org. Chem., 1993, 6, 590 CrossRef CAS.
- H. J. Schneider, Angew. Chem., Int. Ed. Engl., 1991, 30, 1417 CrossRef.
- K. Hiraoka, S. Mizuse and S. Yamabe, J. Phys. Chem., 1987, 91, 5294 CrossRef CAS.
- K. Hiraoka, S. Mizuse and S. Yamabe, J. Chem. Phys., 1987, 86, 4102 CrossRef CAS.
- J. S. Murray, P. Lane, T. Clark, K. E. Riley and P. Politzer, J. Mol. Model., 2012, 18, 541 CrossRef CAS PubMed.
- D. Quiñonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Angew. Chem., Int. Ed., 2002, 41, 3389 CrossRef.
- M. Mascal, A. Armstrong and M. D. Bartberger, J. Am. Chem. Soc., 2002, 124, 6274 CrossRef CAS PubMed.
- I. Alkorta, I. Rozas and J. Elguero, J. Am. Chem. Soc., 2002, 124, 8593 CrossRef CAS PubMed.
- D. Y. Kim, N. J. Singh and K. S. Kim, J. Chem. Theory Comput., 2008, 4, 1401 CrossRef CAS.
- B. L. Schottel, J. Bacsa and K. R. Dunbar, Chem. Commun., 2005, 46 RSC.
- B. Han, J. J. Lu and J. K. Kochi, Cryst. Growth Des., 2008, 8, 1327 CAS.
- M. Mascal, I. Yakovlev, E. B. Nikitin and J. C. Fettinger, Angew. Chem., Int. Ed., 2007, 46, 8782 CrossRef CAS PubMed.
- R. J. Götz, A. Robertazzi, I. Mutikainen, U. Turpeinen, P. Gamez and J. Reedijk, Chem. Commun., 2008, 3384 RSC.
- M. Albrecht, M. Müller, O. Mergel, K. Rissanen and A. Valkonen, Chem. – Eur. J., 2010, 16, 5062 CrossRef CAS PubMed.
- C. Estarellas, A. Frontera, D. Quiñonero and P. M. Deyà, Angew. Chem., Int. Ed., 2011, 50, 415 CrossRef CAS PubMed.
- S. Chakravarty, Z. Z. Sheng, B. Iverson and B. Moore, FEBS Lett., 2012, 586, 4180 CrossRef CAS PubMed.
- D. D. Jenkins, J. B. Harris, E. F. Howell, R. J. Hinde and J. Baudry, J. Comput. Chem., 2013, 34, 518 CrossRef CAS PubMed.
- (a) R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor, J. Mareda, C. A. Schalley and S. Matile, Nat. Chem., 2010, 2, 533 CrossRef CAS PubMed; (b) N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225 RSC.
- K. E. Ranaghan, J. E. Hung, G. J. Bartlett, T. J. Mooibroek, J. N. Harvey, D. N. Woolfson, W. A. van der Donk and A. J. Mulholland, Chem. Sci., 2014, 5, 2191 RSC.
- P. Gamez, T. J. Mooibroek, S. J. Teat and J. Reedijk, Acc. Chem. Res., 2007, 40, 435 CrossRef CAS PubMed.
- P. Ballester, Recognition of Anions, Springer Berlin, Heidelberg, 2008 Search PubMed.
- A. Frontera, Coord. Chem. Rev., 2013, 257, 1716 CrossRef CAS.
- M. Giese, M. Albrecht and Kari Rissanen, Chem. Rev., 2015, 115, 8867 CAS.
- D. Kim, P. Tarakeshwar and K. S. Kim, J. Phys. Chem. A, 2004, 108, 1250 CrossRef CAS.
- D. Y. Kim, N. J. Singh, J. W. Lee and K. S. Kim, J. Chem. Theory Comput., 2008, 4, 1162 CrossRef CAS.
- C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, ChemPhysChem, 2003, 4, 1344 CrossRef CAS PubMed.
- D. Quiñonero, C. Garau, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Chem. Phys. Lett., 2002, 359, 486 CrossRef.
- A. Bauzá, P. M. Deyà, A. Frontera and D. Quiñonero, Phys. Chem. Chem. Phys., 2014, 16, 1322 RSC.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, New J. Chem., 2003, 27, 211 RSC.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Org. Lett., 2003, 5, 2227 CrossRef CAS PubMed.
- C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, J. Phys. Chem. A, 2004, 108, 9423 CrossRef CAS.
- C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, J. Phys. Chem. A, 2004, 108, 9423 CrossRef CAS.
- C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, Chem. Phys. Lett., 2004, 399, 220 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- T. J. Mooibroek and P. Gamez, Inorg. Chim. Acta, 2007, 360, 381 CrossRef CAS.
- T. J. Mooibroek, C. A. Black, P. Gamez and J. Reedijk, Cryst. Growth Des., 2008, 8(4), 1082 CAS.
- (a) C. Murcia-García, A. Bauzá, A. Frontera and R. Streubel, CrystEngComm, 2015, 17, 6736 RSC; (b) C. Murcia-García, A. Bauzá, G. Schnakenburg, A. Frontera and R. Streubel, CrystEngComm, 2015, 17, 1769 RSC.
- (a) T. J. Mooibroek and P. Gamez, CrystEngComm, 2012, 14, 1027 RSC; (b) T. J. Mooibroek and P. Gamez, CrystEngComm, 2013, 15, 1802 RSC.
- J. Kroon and J. A. Kanters, Nature, 1975, 248, 667 CrossRef.
- I. Alkorta and J. Elguero, J. Phys. Chem. A, 2003, 107, 9428 CrossRef CAS.
- I. Alkorta, D. Quiñonero, C. Garau, A. Frontera, J. Elguero and P. M. Deyà, J. Phys. Chem. A, 2007, 111, 3137 CrossRef CAS PubMed.
- D. Quiñonero, A. Frontera, P. M. Deyà, I. Alkorta and J. Elguero, Chem. Phys. Lett., 2008, 460, 406 CrossRef.
- J. W. Steed, R. K. Juneja and J. L. Atwood, Angew. Chem., Int. Ed. Engl., 1995, 33, 2456 CrossRef.
- M. Staffilani, K. S. B. Hancock, J. W. Steed, K. T. Holman, J. L. Atwood, R. K. Juneja and R. S. Burkhalter, J. Am. Chem. Soc., 1997, 119, 6324 CrossRef CAS.
- K. T. Holman, M. M. Halihan, S. S. Jurisson, J. L. Atwood, R. S. Burkhalter, A. R. Mitchell and J. W. Steed, J. Am. Chem. Soc., 1996, 118, 9567 CrossRef CAS.
- M. Staffilani, G. Bonvicini, J. W. Steed, K. T. Holman, J. L. Atwood and M. R. J. Elsegood, Organometallics, 1998, 17, 1732 CrossRef CAS.
- D. Quiñonero, A. Frontera and P. M. Deyà, ChemPhysChem, 2008, 9, 397 CrossRef PubMed.
- I. A. Gural'skiy, D. Escudero, A. Frontera, P. V. Solntsev, E. B. Rusanov, A. N. Chernega, H. Krautscheid and K. V. Domasevitch, Dalton Trans., 2009, 2856 RSC.
- A. Bauzá, D. Quiñonero, P. M. Deyà and A. Frontera, Theor. Chem. Acc., 2012, 131, 1219 CrossRef.
- D. Escudero, A. Frontera, D. Quiñonero and P. M. Deyà, J. Comput. Chem., 2009, 30, 75 CrossRef CAS PubMed.
- X. Lucas, C. Estarellas, D. Escudero, A. Frontera, D. Quiñonero and P. M. Deyà, ChemPhysChem, 2009, 10, 2256 CrossRef CAS PubMed.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, J. Phys. Chem. A, 2005, 109, 9341 CrossRef CAS PubMed.
- R. Podgajny, D. Pinkowicz, B. Czarnecki, M. Kozieł, S. Chorąży, M. Wis, W. Nitek, M. Rams and B. Sieklucka, Cryst. Growth Des., 2014, 14, 4030 CAS.
- M. Giese, M. Albrecht, A. Valkonen and K. Rissanen, Eur. J. Org. Chem., 2013, 3247 CrossRef CAS.
- M. Giese, M. Albrecht, T. Repenko, J. Sackmann, A. Valkonen and K. Rissanen, Eur. J. Org. Chem., 2014, 2435 CrossRef CAS.
- M. Giese, M. Albrecht, A. Valkonen and K. Rissanen, Chem. Sci., 2015, 6, 354 RSC.
- J.-J. Liu, Y.-F. Guan, C. Jiao, M.-J. Lin, C.-C. Huang and W.-X. Dai, Dalton Trans., 2015, 44, 5957–5960 RSC.
- J. Fujisawa, N. Tajima, K. Tamaki, M. Shimomura and T. Ishihara, J. Phys. Chem. C, 2007, 111, 1146 CAS.
- T. Fukumoto, M. Hirasawa and T. Ishihara, J. Lumin., 2000, 497, 87 Search PubMed.
- S. Kumar, M. R. Ajayakumar, G. Hundal and P. Mukhopadhyay, J. Am. Chem. Soc., 2014, 136, 12004 CrossRef CAS PubMed.
- J.-Z. Liao, X.-J. Dui, H.-L. Zhang, X.-Y. Wu and C.-Z. Lu, CrystEngComm, 2014, 16, 10530 RSC.
- J.-Z. Liao, H.-L. Zhang, S.-S. Wang, J.-P. Yong, X.-Y. Wu, R. Yu and C.-Z. Lu, Inorg. Chem., 2015, 54, 4345–4350 CrossRef CAS PubMed.
- S. T. Schneebeli, M. Frasconi, Z. Liu, Y. Wu, D. M. Gardner, N. L. Strutt, C. Cheng, R. Carmieli, M. R. Wasielewski and J. R. Stoddart, Angew. Chem., Int. Ed., 2013, 52, 13100 CrossRef CAS PubMed.
- D.-X. Wang and M.-X. Wang, J. Am. Chem. Soc., 2013, 135, 892 CrossRef CAS PubMed.
- D.-X. Wang, Q.-Y. Zheng, Q.-Q. Wang and M.-X. Wang, Angew. Chem., Int. Ed., 2008, 47, 7485 CrossRef CAS PubMed.
- W. Liu, Q.-Q. Wang, Y. Wang, Z.-T. Huang and D.-X. Wang, RSC Adv., 2014, 4, 9339 RSC.
- H. T. Chifotides, B. L. Schottel and K. R. Dunbar, Angew. Chem., Int. Ed., 2010, 49, 7202 CrossRef CAS PubMed.
- G. Aragay, A. Frontera, V. Lloveras, J. Vidal-Gancedo and P. Ballester, J. Am. Chem. Soc., 2013, 135, 2620 CrossRef CAS PubMed.
- C. S. Campos-Fernandez, B. L. Schottel, H. T. Chifotides, J. K. Bera, J. Bacsa, J. M. Koomen, D. H. Russell and K. R. Dunbar, J. Am. Chem. Soc., 2005, 127, 12909 CrossRef CAS PubMed.
- B. L. Schottel, H. T. Chifotides, M. Shatruk, A. Chouai, J. Bacsa, L. M. Perez and K. R. Dunbar, J. Am. Chem. Soc., 2006, 128, 5895 CrossRef CAS PubMed.
- P. S. Szalay, J. R. Galan-Mascaros, B. L. Schottel, J. Basca, L. M. Perez, I. S. Ichimura, A. Chouai and K. R. Dunbar, J. Cluster Sci., 2004, 15, 503 CrossRef CAS.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed.
- M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 371 CrossRef PubMed.
- M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509 RSC.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650 CrossRef CAS PubMed.
- R. A. Bilbeisi, J. K. Clegg, N. Elgrishi, X. de Hatten, M. Devillard, B. Breiner, P. Mal and J. R. Nitschke, J. Am. Chem. Soc., 2012, 134, 5110 CrossRef CAS PubMed.
- J. Rebek Jr., Acc. Chem. Res., 2009, 42, 1660 CrossRef PubMed.
- H. T. Chifotides, I. D. Giles and K. R. Dunbar, J. Am. Chem. Soc., 2013, 135, 3039 CrossRef CAS PubMed.
- D. A. Safin, A. Pialat, A. A. Leitch, N. A. Tumanov, I. Korobkov, Y. Filinchuk, J. L. Brusso and M. Murugesu, Chem. Commun., 2015, 51, 9547–9550 RSC.
- L. Qin, L.-Y. Yao and S.-Y. Yu, Inorg. Chem., 2012, 51, 2443–2453 CrossRef CAS PubMed.
- A. Bhattacharyya, P. K. Bhaumik, A. Bauzá, P. P. Jana, A. Frontera, M. G. B. Drew and S. Chattopadhyay, RSC Adv., 2014, 4, 58643 RSC.
- A. Garcia-Raso, F. M. Albertí, J. J. Fiol, A. Tasada, M. Barceló-Oliver, E. Molins, D. Escudero, A. Frontera, D. Quiñonero and P. M. Deyà, Inorg. Chem., 2007, 46, 10724 CrossRef CAS PubMed.
- A. Frontera, D. Quiñonero, C. Garau, A. Costa, P. Ballester and P. M. Deyà, J. Phys. Chem. A, 2006, 110, 9307 CrossRef CAS PubMed.
- D. Quiñonero, A. Frontera, C. Garau, P. Ballester, A. Costa and P. M. Deyà, ChemPhysChem, 2006, 7, 2487 CrossRef PubMed.
- A. Frontera, D. Quiñonero, A. Costa, P. Ballester and P. M. Deyà, New J. Chem., 2007, 31, 556 RSC.
- M. J. Monreal, R. J. Wright, D. E. Morris, B. L. Scott, J. T. Golden, P. P. Power and J. L. Kiplinger, Organometallics, 2013, 32, 1423 CrossRef CAS.
- X.-L. Li, J.-L. Kang, X.-L. Zhang, H.-P. Xiao, A.-L. Wang, L. Zhou, S.-M. Fanga and C.-M. Liu, Dalton Trans., 2014, 43, 17226 RSC.
- (a) S. Brooker, N. G. White, A. Bauza, P. M. Deyà and A. Frontera, Inorg. Chem., 2012, 51, 10334 CrossRef CAS PubMed; (b) C. Biswas, M. G. B. Drew, D. Escudero, A. Frontera and A. Ghosh, Eur. J. Inorg. Chem., 2009, 2009, 2238–2246 CrossRef; (c) A. Das, S. M. Choudhury, C. Estarellas, B. Dey, A. Frontera, J. Hemming, M. Helliwell, P. Gamez and S. Mukhopadhyay, CrystEngComm, 2011, 13, 4519 RSC; (d) P. Kar, R. Biswas, M. G. R. Drew, A. Frontera and A. Ghosh, Inorg. Chem., 2012, 51, 1837 CrossRef CAS PubMed.
- C. Estarellas, A. Frontera, D. Quiñonero and P. M. Deyà, J. Chem. Theory Comput., 2008, 4, 1981 CrossRef CAS.
- D. Quiñonero, A. Frontera, D. Escudero, P. Ballester, A. Costa and P. M. Deyà, ChemPhysChem, 2007, 8, 1182 CrossRef PubMed.
- A. Das, S. Ray Choudhury, B. Dey, S. K. Yalamanchili, M. Helliwell, P. Gamez, S. Mukhopadhyay, C. Estarellas and A. Frontera, J. Phys. Chem. B, 2010, 114, 4998 CrossRef CAS PubMed.
- A. Das, S. Ray Choudhury, C. Estarellas, B. Dey, A. Frontera, J. Hemming, M. Helliwell, P. Gamez and S. Mukhopadhyay, CrystEngComm, 2011, 13, 4519 RSC.
- M. Fourmigue, K. Boubekeur, P. Batail and K. Bechgaard, Angew. Chem., Int. Ed. Engl., 1989, 28, 588 CrossRef.
- R. F. Childs, R. Faggiani, C. J. L. Lock and C. V. Rogerson, J. Org. Chem., 1983, 48, 3043 CrossRef CAS.
- J. J. Fiol, M. Barceló-Oliver, A. Tasada, A. Frontera, À. Terrón and Á. García-Raso, Coord. Chem. Rev., 2013, 257, 2705 CrossRef CAS.
- F. Orvay, A. Bauzá, M. Barceló-Oliver, A. García-Raso, J. J. Fiol, A. Costa, E. Molins, I. Mata and A. Frontera, CrystEngComm, 2014, 16, 9043 RSC.
- P. Manna, S. K. Seth, A. Bauzá, M. Mitra, S. R. Choudhury, A. Frontera and S. Mukhopadhyay, Cryst. Growth Des., 2014, 14, 747 CAS.
- P. Manna, S. K. Seth, M. Mitra, S. R. Choudhury, A. Bauzá, A. Frontera and S. Mukhopadhyay, Cryst. Growth Des., 2014, 14, 5812 CAS.
- M.-H. Chen, S.-F. Xue and Z. Tao, Hecheng Huaxue, 2009, 17, 722 CAS.
- (a) J.-W. Zhao, L.-J. Chen and J.-P. Wang, Huaxue Yanjiu, 2008, 19, 14 CAS; (b) H. Yang, X. Lin, B. Xu, Y. You, M. Cao, S. Gao and R. Cao, J. Mol. Struct., 2010, 966, 33 CrossRef CAS.
- J. Aguiló, A. Naeimi, R. Bofill, H. Mueller-Bunz, A. Llobet, L. Escriche, X. Sala and M. Albrecht, New J. Chem., 2014, 38, 1980 RSC.
Footnote |
| † The CSD version 5.36 (November 2014, including three updates) was consulted using ConQuest version 1.17, while excluding entries before November 2010 (CSD version 5.32). A subset was first created where an aromatic six-membered ring consisting of C and/or N was present. This subset was then subjected to a search for structures where the intermolecular distance between an electron-rich atom (N, P, As, O, S, Se, TE, F, Cl, Br, I, At) and all six of the ring atoms have overlapping van der Waals shells. Manual inspection of the resulting 68 structures revealed several clear examples of anion–π interactions that were not mentioned by the authors of these structures, as highlighted in this contribution. Several of the structures were too disordered for reliable interpretation, some contained clear evidence of lone-pair–π interactions, and some structures contained anion–π features that the authors of the structure intended and/or mentioned. Relaxing the search criteria to 6 × overlapping van der Waals shells + 0.1 Å gave 170 structures and relaxing further to 0.25 Å gave 1057 structures that might well contain numerous additional examples of anion–π interactions. |
| This journal is © The Royal Society of Chemistry 2016 |