
The endeavour to advance graphene–semiconductor composite-based photocatalysis
Nan
Zhang
ab and
Yi-Jun
Xu
*ab
aState Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou, 350002, P. R. China
bCollege of Chemistry, New Campus, Fuzhou University, Fuzhou, 350108, P. R. China. E-mail: yjxu@fzu.edu.cn; Fax: +86 591 83779326; Tel: +86 591 83779326
First published on 6th November 2015
Abstract
Graphene (GR)–semiconductor composite-based photocatalytic systems have received ever-increasing attention due to the attractive possibilities they provide to alleviate environmental and energy issues. Extensive endeavours have been made to construct high-performance GR–semiconductor composite photocatalysts for solar energy conversion. In this review, recent advances in developing strategies to assemble efficient GR–semiconductor composite photocatalysts are highlighted. These advances can be mainly classified into three aspects. The first is the optimization of individual components, including maximization of the functions of graphene and optimization of the photoactive semiconductors. The second is interface engineering between graphene and semiconductors. The third is the design and optimization of GR–semiconductor composite photocatalysts from a system-level consideration. Finally, it is proposed that combining these advances together with theoretical investigations will take us further along the road to advancing GR–semiconductor composite-based photocatalysis. Truly smart GR–semiconductor composite photocatalysts with robust structural and functional infrastructure are anticipated to be forthcoming.
 Nan Zhang | Nan Zhang is now pursing her Ph.D. degree under the supervision of Prof. Yi-Jun Xu at State Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, P. R. China. Her main research interests include the fabrication of core–shell and carbon-based nanocomposites for potential target applications in heterogeneous photocatalysis. |
 Yi-Jun Xu | Yi-Jun Xu is a full professor now working at State Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, P. R. China. His current research interests primarily focus on the assembly and applications of semiconductor-based nanostructured materials, such as graphene-based, one-dimensional and core–shell structure-based composite photocatalysts, in the field of heterogeneous photocatalysis. |
1. Introduction
The breakthrough made in 2004 ignited research enthusiasm on the one-atom-thick planar crystal of carbon atoms, graphene (GR), in a plethora of potential fields.1 Due to its unique properties, including the two-dimensional (2D) planar structure, superior electrical conductivity, high transparency, and theoretically large specific surface area, GR has been regarded as an ideal high performance candidate to construct GR-based composites for energy storage and conversion.2–10 Recent years have witnessed an exponential increase in publications on GR–semiconductor composite-based artificial photosynthesis systems for environmental remediation, fuel production and selective organic transformations.4–16 Thus far, the reported GR–semiconductor composite photocatalysts mainly include GR–metal oxide, GR–metal chalcogenide, GR–metal halide, GR–metallate, GR–layered double hydroxide, etc.4 In these GR–semiconductor composites, GR is often utilized as an electron reservoir to accept or transport the photogenerated electrons from the semiconductor, by which the recombination of electron–hole pairs is retarded and the ability of charge carriers to participate in photoredox reactions, and thus the photocatalytic performance, are improved, as illustrated in Fig. 1. However, it should be noted that there is a significant gap between the numerous reports on GR–semiconductor composite photocatalysts and on how to sufficiently deliver the unique, great potential of GR, such as the large 2D flat structure and excellent electrical conductivity.4,5,13,17–19 It seems a little too arbitrary and not objective to simply attribute the photoactivity improvement of GR–semiconductor composites to the excellent electrical conductivity of GR, because GR's forebears, carbon nanotubes (CNTs) and fullerene (C60), are able to do the same thing as GR.13,17–22 Analogous to GR, both CNTs and C60 can also act as electron mediators to accept and shuttle photoexcited electrons from the semiconductor,21,22 thereby boosting the lifetime and transfer efficiency of carbon–semiconductor composite-based photocatalytic systems, and thus improving the photocatalytic efficiency.17–20,23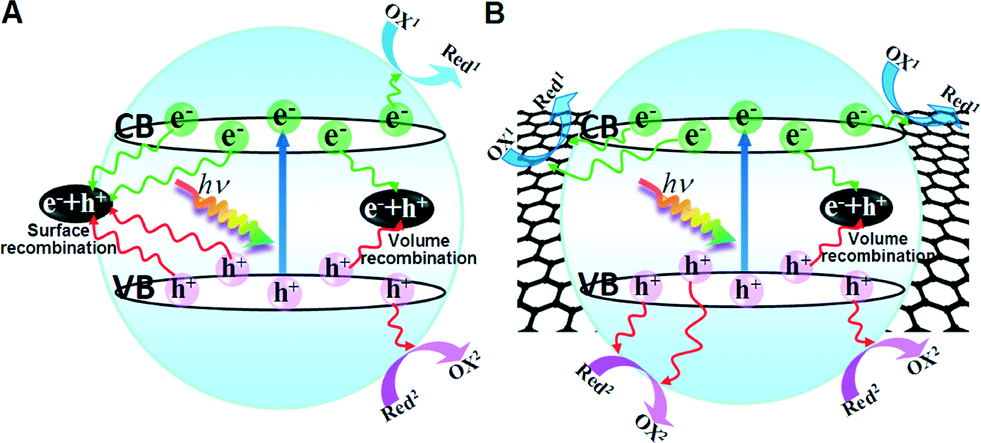 | ||
| Fig. 1 Schematic illustrations of charge carrier transfer for (A) semiconductor and (B) GR–semiconductor composite photocatalysts. | ||
In 2010, our research group first argued that TiO2–GR composite photocatalysts obtained via a random hard integration of solid TiO2 (P25) nanoparticles with graphene oxide (GO) are in essence the same as other TiO2–carbon (CNTs, C60 and activated carbon) counterparts in enhancing the photoactivity of TiO2, although GR by itself has unique structural and electronic properties.17 This work was timely when the GR-driven “gold rush” has been sweeping over the world. At least, it rationally reminded researchers not to impose much hype on the “miracle of GR” in much the same way as its carbon forebears (fullerenes and carbon nanotubes).4,13,17–20,24 Indeed, this key issue was critically discussed by the editorials published in Nature in 2011,24 which was based on a meeting on the subject “Graphene: The Road to Applications”.25 Although the hunt is on for applications that can exploit GR's remarkable properties,4,17–20,23,26–37 the work necessary to find out how it can be better harnessed remains incomplete,24 and this issue becomes more prominent in the field of GR–semiconductor composite-based artificial photocatalysis.4,5
Considering such situations, instead of summarizing the applications of GR–semiconductor composite photocatalysts, in this review, we would like to focus on highlighting the endeavours that have been made to design and construct high-performance GR–semiconductor composite photocatalysts for solar energy conversion, through bringing the advantages of each ingredient and their interfacial domain into better play. The progress achieved in this aspect is of great importance to pave the way for the further development of GR–semiconductor composite photocatalysts with specific applications, but has not yet been exclusively summarized with sufficient attention.4,5,9,12–15,38–44 This highlight will cover effective strategies not only for the maximization of the functions of GR, but also for the tuning of photoactive semiconductors, interface engineering between GR and semiconductors, and optimization from a system-level consideration. It is expected that this review will provide instructive information on the design and construction of next-generation artificial photosynthesis systems based on GR–semiconductor composites with high performance.
2. Optimization of individual components
2.1 To maximize the functions of graphene
The functions of graphene (GR) in the GR–semiconductor composite photocatalysts reported in the literature mainly involve serving as an electron reservoir to accept and transport photogenerated electrons from the semiconductors, enhancing the adsorption capacity for reaction substrates, and tuning the light absorption range and intensity of the composites.4,5,9,12–15,38–44 The former two result from the excellent electrical conductivity and theoretically large specific surface area of GR, respectively, and are often regarded as the advantages of GR over its carbon forebears (e.g., CNTs and C60) to more effectively boost the photocatalytic performances of semiconductors. However, it should be noted that such expected superiority of GR can only be intrinsically associated with single-layer and defect-free GR sheets.4,5,13 The graphene used to construct GR–semiconductor composite photocatalysts is nearly always derived from the reduction of graphene oxide (GO). The as-obtained GR is also called reduced graphene oxide (RGO), which is defective and multi-layered due to the irreversible oxidation and aggregation processes, and exhibits much lower electrical conductivity and specific surface area than the ideal GR sheets.4,5,13 To alleviate these problems, some approaches have been developed to maximize the functions of GR in GR–semiconductor composite photocatalysts, especially its electron acceptor and adsorbent roles.Our group has prepared a series of solvent-exfoliated graphene (SEG)–TiO2 composites with different weight ratios of SEG through in situ and ex situ methods using TiF4 and solid TiO2 (P25) nanoparticles as the precursors of TiO2, respectively.49 For comparison, the RGO–TiO2 counterparts were also synthesized. Notably, unlike GO which has abundant hydrophilic functional groups,51 SEG is hydrophobic (Fig. 2A), which is unfavourable for the nucleation and growth of semiconductors on its surface during the in situ synthesis procedure. This is partially responsible for its relatively limited use in the wet chemistry synthesis of GR–semiconductor composite photocatalysts, as compared to GO.4,5,9,12–15,38–44 In order to resolve this deficiency, a moderate amount of a polymer, i.e., polyvinylpyrrolidone (PVP), was introduced as an interface linker to functionalize the surface of SEG.49,52 This slight modification endows the SEG surface with additional hydrophilic functional groups, which can be regarded as being “roughly” similar to the abundant functional groups of GO, and makes SEG well dispersed and stable in the aqueous phase. Notably, the mild functionalization of SEG with PVP at room temperature does not introduce a significant amount of defects into the SEG surface.
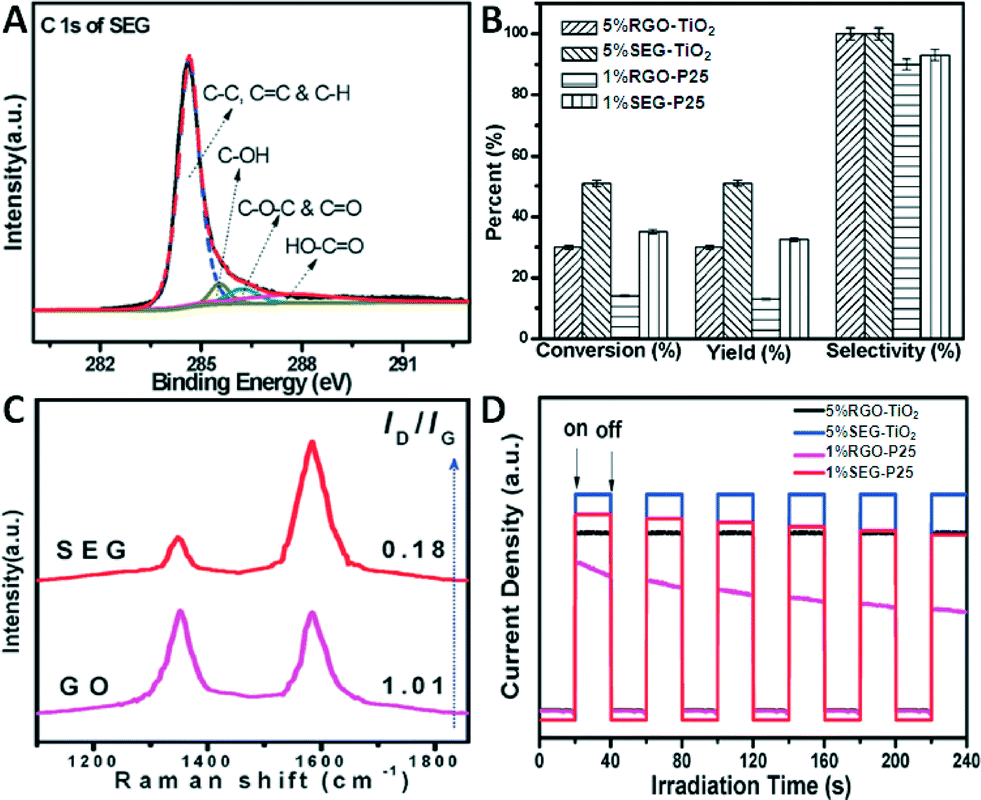 | ||
| Fig. 2 (A) C 1s X-ray photoelectron spectroscopy (XPS) spectrum of SEG; (B) photocatalytic selective oxidation of benzyl alcohol to benzaldehyde over the optimum GR (SEG, RGO)–TiO2 composites under visible light irradiation for 4 h at room temperature; (C) Raman spectra of GO and SEG; (D) photocurrent transient responses of the optimum GR (SEG, RGO)–TiO2 composites in 0.2 M Na2SO4 aqueous solution without bias versus the Ag/AgCl electrode under visible light irradiation. Reproduced with permission from ref. 49. Copyright 2012 PCCP Owner Societies. | ||
The photocatalytic performance of GR (SEG, RGO)–TiO2 composites in the aerobic selective oxidation of alcohols has been evaluated.49 Among the two series of SEG–TiO2 composites, the 5% SEG–TiO2 and 1% SEG–P25 exhibit the best photocatalytic performance under visible light irradiation for the tested reaction. The optimum ratios of GR in the SEG–TiO2 composites are the same as those for RGO–TiO2, indicating that the optimal synergistic interaction between GR and TiO2 can be achieved by controlling the ratio of GR. Excessive addition of GR in the GR (SEG, RGO)–TiO2 composites results in a decrease in the photoactivity, which can be attributed to the decrease in the amount of the primary photoactive ingredient TiO2 and the lowered intensity of light that can penetrate through the depth of the reaction solution, due to the introduction of black GR.
Besides, as shown in Fig. 2B, the optimal 5% SEG–TiO2 and 1% SEG–P25 both display enhanced visible light photoactivities toward selective oxidation of benzyl alcohol as compared to their counterparts, 5% RGO–TiO2 and 1% RGO–P25. The activity enhancement can be ascribed to the fact that SEG has a lower defect density than RGO, as revealed by the intensity ratios of the D and G bands (i.e., ID/IG) in the Raman spectra (Fig. 2C), which measure the relative concentrations of local defects or disorder (particularly sp3 hybridized defects) compared to the sp2 hybridized graphene domains.23 Therefore, SEG is able to make better use of the electrical conductivity of GR to promote the separation and transport of photogenerated charge carriers from TiO2. This can be further verified by the photoelectrochemical analysis. It can be seen from Fig. 2D that the optimum 5% SEG–TiO2 and 1% SEG–P25 show higher photocurrent density under visible light irradiation as compared to their RGO-based counterparts, indicating that SEG is able to more efficiently improve the separation of photogenerated charge carriers. Besides, the electrochemical impedance spectroscopy (EIS) Nyquist plots of the samples reveal that improved interfacial charge transfer is obtained for the SEG–TiO2 composites as compared to RGO–TiO2. This study demonstrates that decreasing the defects density in GR has a significant positive influence on the utilization of the electrical conductivity of GR and thus the photoactivity enhancement of GR–semiconductor composite photocatalysts.
For instance, Mou et al. fabricated N-doped GR (NGR) by a thermal solid-state reaction of graphene oxide (GO) and urea, and used it to synthesize NGR–TiO2 nanoparticle composites through a solvothermal treatment approach.61Fig. 3A shows the photoactivity over time of pure TiO2, NGR–TiO2, and RGO–TiO2 without N doping toward photocatalytic H2 production from a triethanolamine (TEOA) aqueous solution under Xe lamp illumination. As compared to the pure TiO2, the NGR–TiO2 photocatalysts exhibit enhanced photocatalytic performance for H2 evolution. When the weight ratio of NGR in the NGR–TiO2 composites is ca. 2%, the highest photocatalytic activity is obtained. Besides, the optimum 2% NGR–TiO2 shows higher photoactivity than 2% RGO–TiO2, suggesting that NGR is superior to RGO as a cocatalyst for photocatalytic H2 production.
 | ||
| Fig. 3 (A) The time course of hydrogen production from a 50 mL aqueous solution containing 10 vol% TEOA with different photocatalysts (20 mg, 0.4 mg mL−1). pH: 10.8; light source: a 150 W Xe lamp. (B) C 1s XPS spectrum of 2% NGR–TiO2. Reproduced with permission from ref. 61. Copyright 2014 American Chemical Society. | ||
To reveal the corresponding origins of the enhanced photocatalytic activity, a series of characterizations were conducted. The X-ray photoelectron spectroscopy (XPS) results in Fig. 3B show that the peaks in the C 1s XPS spectrum of 2% NGR–TiO2 can be fitted into three peaks centred at 284.6, 285.4, and 287.7 eV, which correspond to the C–C, sp2 C–N, and sp3 C–N species, respectively. Clearly, the proportion of sp2 C–N bonds induced by the N doping is higher than that of sp3 C–N bonds, which is favourable for enhancing the electrical conductivity of GR. This can be confirmed by the results of measurements using the four-point probe method under ambient conditions. The electrical conductivity of NGR is ca. 40 S cm−1, much higher than that of RGO obtained in the solvothermal reduction of GO (3.5 S cm−1).61 In addition, the higher photocurrent density under light irradiation and the smaller impedance arc of 2% NGR–TiO2 than TiO2 and 2% RGO–TiO2 indicate that the introduction of NGR in the composites can more efficiently facilitate electron–hole separation and transfer, thus leading to the enhanced photocatalytic performance of the NGR–TiO2 composites.
A 3D AgX (X = Br, Cl)/GR aerogels (GAs) structure was fabricated through a facile wet-chemistry approach by using the GAs as building blocks.69 As exemplified in Fig. 4A, the AgBr nanoparticles are uniformly distributed throughout the surface of the 3D GAs hierarchical porous structure, whereas the pristine AgBr particles are much larger in size and tend to agglomerate into irregularly shaped blocks. Obviously, the presence of the GAs dramatically inhibits the agglomeration of AgBr during the synthesis process, which should contribute to improving the photocatalytic activity of AgBr/GAs.
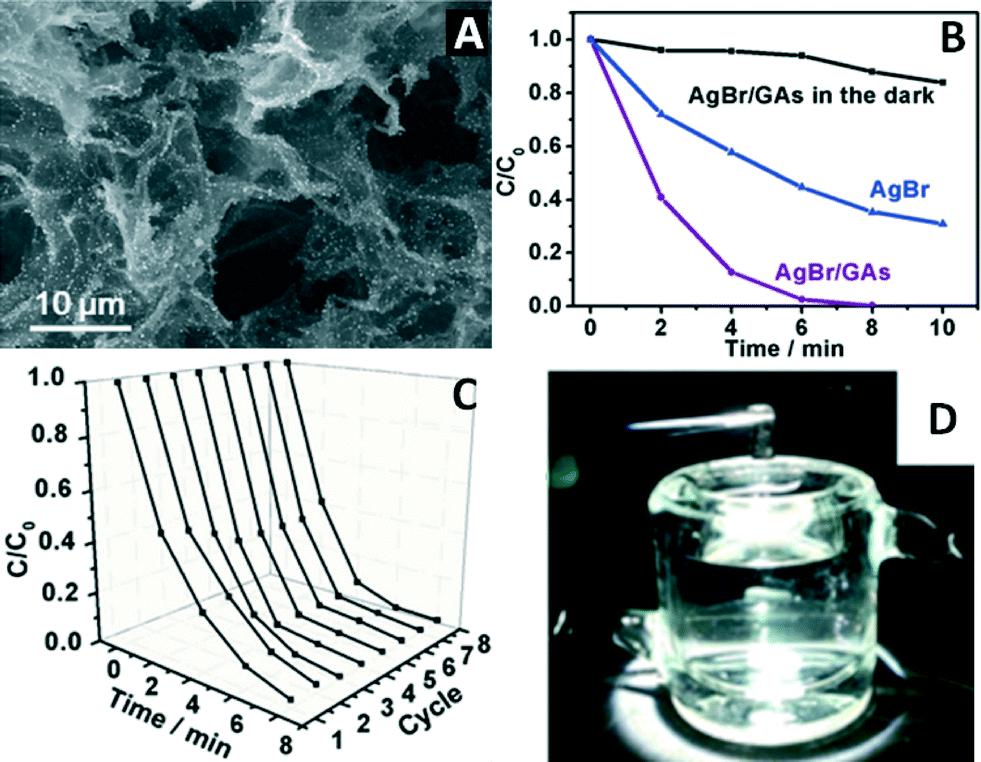 | ||
| Fig. 4 (A) Typical SEM image of AgBr/GAs; (B) photocatalytic oxidation of methyl orange (MO) by AgBr/GAs and AgBr under visible light (λ > 420 nm) and the adsorption curve for AgBr/GAs in the dark; (C) eight successive dynamic curves for the photodegradation of MO over AgBr/GAs; (D) illustration of the recycling of AgBr/GAs from the completed reaction. Reproduced with permission from ref. 69. Copyright 2015 Wiley-VCH. | ||
The photocatalytic performance of AgBr/GAs was investigated for the oxidative degradation of methyl orange (MO) and the reduction of Cr(VI). Taking the former reaction as an example here, it can be seen from Fig. 4B that after visible light irradiation (>420 nm) for 8 min, MO can be completely degraded by AgBr/GAs, while only 65% is removed by pristine AgBr during the same time interval. According to the pseudo-first-order fitting, the degradation rate constant of AgBr/GAs is 0.72 min−1, six times higher than that of bare AgBr. Obviously, the introduction of 3D GAs can significantly enhance the photocatalytic activity of AgBr.
The improved photocatalytic performance of AgBr/GAs in the degradation process can be ascribed to two possible reasons. (i) The adsorption capacity of the 3D GAs plays an important role due to the high specific surface area of AgBr/GAs (ca. 106 m2 g−1). As revealed by the adsorption curve of AgBr/GAs in the dark (Fig. 4B), the AgBr/GAs displays an admirable adsorption capacity, which can even adsorb 16% of MO in 10 min without light irradiation. Notably, this adsorption capacity is much lower than the photodegradation ability of AgBr/GAs over the same time period, indicating that the photocatalytic activity is the main driving force in removing the pollutants and that the favourable adsorption properties of GAs serve as effective assistance.69 (ii) Since AgBr nanoparticles grow tightly on the GAs in situ, the photogenerated electrons could be promptly injected into the GR sheets through a percolation process under light irradiation.
Fig. 4C demonstrates the cycling tests of AgBr/GAs for the photodegradation of MO. The photocatalytic activity of AgBr/GAs distinctly increases along with increasing cycling number during the first four circulation processes. This should be attributed to the localized surface plasmon resonance (SPR) of Ag0, which is produced during the photocatalytic process. The photoactivity of AgBr/GAs remains unchanged during the last four circulation processes. Thus, it is inferred that the amount of metallic Ag reaches a maximum after four cycles, which was confirmed by the X-ray photoelectron spectroscopy (XPS) results. The mechanism of stopping the formation of Ag0 after four cycles is considered as follows: the SPR-excited electrons on the surface of metallic Ag develop polarization fields, which produce many regions with negative and positive charges close to the surface of AgBr. The polarization fields can force the excited electrons further away from the surface of AgBr and close to the GAs, which prevents the surface electrons from combining with Ag+ ions to continuously generate Ag0. The quality and morphology of the AgBr/GAs composite material can be almost completely maintained after the photocatalytic reaction. Besides, the recycling process is very easy and just involves directly taking the 3D photocatalytic composite from the reaction system with tweezers, which excludes centrifugation, sonication, and drying processes (Fig. 4D). Obviously, as a capable substrate for a photocatalyst, GAs possess general applicability and are anticipated to promote the evolution of photocatalysis toward commercial applications.
2.2 Optimization of semiconductors
The coupling of semiconductors with appropriate band gap values with GR to form GR–semiconductor composites has been widely investigated.4,5,9,12–15,38–44 As the semiconductors are the photoactive components in most of the GR–semiconductor composites, their physicochemical properties, such as the crystal phase, crystallinity, crystal facets, size, dimensionality, morphology, etc., also have great effects on the performance of the photocatalysts, because these factors not only play a key role in the generation and transfer of charge carriers on the surface of the semiconductors,73–82 but also influence the integration/interaction between the semiconductors and GR.83–85 Therefore, the optimization of the semiconductors should also be taken into account when constructing efficient GR–semiconductor composite photocatalysts. In this section, we will mainly highlight the efforts to understand the effects of three typical semiconductor properties, i.e., the size, dimensionality and exposed crystal facets, on the performance of GR–semiconductor composite photocatalysts. | ||
| Fig. 5 Photocatalytic reduction of Cr(VI) to Cr(III) over (A) GR/ZnO-S1 composites and (B) GR/ZnO-S2 composites under UV light irradiation; the insets are the apparent rate constant (ka); typical SEM images of (C) 5% GR/ZnO-S1 and (D) 5% GR/ZnO-S2. Reproduced with permission from ref. 83. Copyright 2013 Elsevier. | ||
It is easy to see that, with addition of the appropriate weight ratios of GR, the GR/ZnO-S1 composites with smaller ZnO particle size exhibit enhanced UV light photoactivity as compared to bare ZnO-S1. However, all the GR/ZnO-S2 composites with relatively large ZnO particle size show lower photoactivity than bare ZnO-S2. These results indicate that the size of the ZnO particles plays an important role in the synergetic interaction between ZnO and GR, and thus affects the overall photocatalytic performance of the GR–ZnO composites. The recycling photoactivity testing shows that, for GR/ZnO-S1, the photocorrosion of ZnO-S1 is efficiently inhibited by hybridization with GR, whereas this is not the case for GR/ZnO-S2.
In order to shed light on the structure–photoactivity relationship, a series of characterizations were performed on the optimum 5% GR/ZnO-S1 composite, which shows higher photoactivity than bare ZnO-S1, and its analogue 5% GR/ZnO-S2, which shows lower photoactivity than bare ZnO-S2. The morphology characterization reveals that there is a relatively good interfacial contact between GR and the ZnO-S1 particles with small size distribution in 5% GR/ZnO-S1, while the interfacial contact between GR and the ZnO-S2 particles with large size distribution is relatively poor in 5% GR/ZnO-S2 (Fig. 5C and D). Such a particle size effect leads to enhanced separation and transfer of charge carriers for GR/ZnO-S1, as evidenced by the photoelectrochemical and photoluminescence (PL) analysis, which thus results in the improved photoactivity and efficient anti-photocorrosion observed for GR/ZnO-S1. Our results suggest that the semiconductor particle size has an important effect on the photocatalytic performance of GR–semiconductor composite photocatalysts, which should be considered in the design and optimization of efficient GR–semiconductor composite photocatalysts.
 | ||
| Fig. 6 TEM images of (A) GR–TiO2 nanowire (NW) and (B) GR–TiO2 nanoparticle (NP) hybrid nanostructures; (C) photodegradation of methylene blue (MB) under solar light over TiO2 NPs, TiO2 NWs, GR–TiO2 NPs and GR–TiO2 NWs; (D) EIS Nyquist plots and (E) cyclic voltammograms of as-made thin films of TiO2 NPs, TiO2 NWs, GR–TiO2 NPs and GR–TiO2 NWs on FTO glass. Reproduced with permission from ref. 84. Copyright 2012 American Chemical Society. | ||
When being applied in the photocatalytic degradation of methylene blue (MB) under solar light illumination, both GR–TiO2 composites display enhanced performance as compared to their respective blank TiO2 counterparts. Furthermore, GR–TiO2 NWs exhibit higher photoactivity than GR–TiO2 NPs toward the photodegradation reaction (Fig. 6C). The electrochemical impedance spectroscopy (EIS) Nyquist plots of TiO2 NPs, TiO2 NWs and the corresponding GR–TiO2 composites (Fig. 6D) reveal that, besides the depressed semicircles at high frequencies for the GR–TiO2 composites compared with their pure TiO2 counterparts, GR–TiO2 NWs exhibit a smaller semicircle than GR–TiO2 NPs. The smaller semicircle for GR–TiO2 NWs indicates diminished resistance in the solid-state interface layer and decreased charge transfer resistance across the solid–liquid junction on the surface on forming hybrid structures of TiO2 NWs with GR. Fig. 6E shows the cyclic voltammograms of the samples. GR–TiO2 NWs and TiO2 NWs exhibit the smallest peak-to-peak separations (ΔEp), indicating highly improved reaction reversibility. Furthermore, the hybrid structure of GR–TiO2 NWs shows a larger current density as compared to GR–TiO2 NPs, demonstrating an enhanced rate of electron transfer for GR–TiO2 NWs.
These observations can be ascribed to the fact that (i) TiO2 NWs have a uniform distribution on GR, resulting in direct contact between the semiconductor 1D TiO2 NWs and the GR, which provides an efficient path for the transfer of excited electrons from the TiO2 NWs to the GR sheets; and (ii) in contrast with the zigzag path in agglomerated NPs, the NW structure can provide a straight transfer path for excited charge carriers and suppress carrier scattering, which is a common phenomenon in nanoparticle systems. Therefore, electron–hole pair recombination is more efficiently retarded in GR–TiO2 NWs than in GR–TiO2 NPs. Besides, the strong adsorption of MB by GR–TiO2 NWs means that more MB molecules are likely to be close to the holes for oxidation. These synergetic effects lead to significantly higher photocatalytic capability for GR–TiO2 NWs compared with GR–TiO2 NPs. This work demonstrates that the optimization of semiconductor dimensionality provides another approach for improving the performance of GR–semiconductor composite photocatalysts.
 | ||
| Fig. 7 TEM images of (A) GR–TiO2-101, (B) GR–TiO2-001, and (C) GR–TiO2-100; the insets are the corresponding HRTEM images; (D) XPS spectra of the C 1s regions of GO and GR–TiO2 composites; (E) rates of H2 evolution from methanol solution catalyzed by TiO2 nanocrystals and GR–TiO2 composites under UV light irradiation. Reproduced with permission from ref. 85. Copyright 2014 Wiley-VCH. | ||
The UV-vis diffuse reflectance spectra show that the band structures of TiO2 nanocrystals and GR–TiO2 composites are dependent on the crystal facets. The absorption edge of GR–TiO2-001 is almost the same as that of the corresponding TiO2 nanocrystals, indicating that carbon atoms are not incorporated into the lattice of TiO2-001 to change its band gap. For GR–TiO2-101 and GR–TiO2-100, the absorption edges are red shifted, indicating the narrowing of the band gaps of TiO2-101 and TiO2-100. The chemical states of carbon in the GR–TiO2 composites were measured by X-ray photoelectron spectroscopy (XPS). It can be seen from Fig. 7D that, for GR–TiO2-101 and GR–TiO2-001, there are only three peaks in the C 1s spectra corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–O and OC–O, suggesting that TiO2 nanocrystals with exposed {101} and {001} facets are connected with GR through C–O–Ti bonds, which are formed from Ti(OH)4 and GO during the hydrothermal synthesis. Notably, besides these three peaks, a new peak located at 283.4 eV is found in the XPS C 1s spectrum of GR–TiO2-100, which corresponds to Ti–C bonds. Since the atomic structures of the {100}, {101}, and {001} facets are quite different, the formation of Ti–C bonds between GR and TiO2 {100} facets may be facilitated by the stepped surface structure, in which both Ti and O atoms are exposed. The different contacts between different exposed TiO2 crystal facets and GR may have an influence on their photocatalytic performance.
C, C–O and OC–O, suggesting that TiO2 nanocrystals with exposed {101} and {001} facets are connected with GR through C–O–Ti bonds, which are formed from Ti(OH)4 and GO during the hydrothermal synthesis. Notably, besides these three peaks, a new peak located at 283.4 eV is found in the XPS C 1s spectrum of GR–TiO2-100, which corresponds to Ti–C bonds. Since the atomic structures of the {100}, {101}, and {001} facets are quite different, the formation of Ti–C bonds between GR and TiO2 {100} facets may be facilitated by the stepped surface structure, in which both Ti and O atoms are exposed. The different contacts between different exposed TiO2 crystal facets and GR may have an influence on their photocatalytic performance.
The photocatalytic performance of the obtained GR–TiO2 composites was measured through H2 production from methanol solution under UV light irradiation. As presented in Fig. 7E, pure TiO2 nanocrystals show low H2 evolution rates due to the fast recombination of photogenerated electrons and holes of TiO2 in the absence of cocatalysts (such as Pt). The introduction of GR dramatically improves the H2 evolution rates of the GR–TiO2 composites as compared to pure TiO2. Notably, the photocatalytic activities of the GR–TiO2 composites for H2 production are dissimilar, and follow the order: GR–TiO2-100 > GR–TiO2-101 > GR–TiO2-001.
The interfacial charge transfer rates were estimated according to the time-resolved photoluminescence spectra of the samples. The charge transfer rates of GR–TiO2-100, GR–TiO2-101, and GR–TiO2-001 were calculated to be 2.67 × 109, 2.34 × 109, and 1.93 × 109 s−1, respectively, which is the same as the order of their photocatalytic performance. This implies that the interfacial charge transfer rate is the key factor affecting the photocatalytic performance. Clearly, the charge transfer rates are greatly dependent on the crystal facets of the TiO2 nanocrystals. Photoelectrochemical measurements also confirmed the dependence relationship between the charge separation efficiency in the GR–TiO2 composites and the crystal facets. This can be attributed to the fact that the different TiO2 crystal facets possess dissimilar atomic structures, leading to differences in the electronic structures and interfacial connections with GR, as demonstrated by the XPS spectra (Fig. 7D), and thus resulting in the dissimilarity in the photoactivities of GR–TiO2 with different exposed crystal facets.
3. Interface engineering between graphene and semiconductors
In order to effectively harness the electrical conductivity of GR for enhancing the photoactivity of GR–semiconductor composites, sufficient interfacial contact between the GR and semiconductor ingredients is crucial because the photogenerated charge carrier transfer predominantly occurs across the interface domain.4–10,12–20,23,26–32 Therefore, interface engineering between GR and semiconductors can be regarded as an important way to improve the performance of GR–semiconductor composite photocatalysts. Thus far, some strategies have been developed to achieve intimate interfacial contacts between GR and semiconductors.18,28,87–92 Among the various methods, soft integration and layer-by-layer self-assembly are two representative and effective approaches, and are the focus of this section.3.1 Soft integration
At present, wet chemistry methods for the synthesis of GR–semiconductor composites are widely adopted for which GO, featuring abundant oxygenated functional groups, is often used as the precursor of GR.4–10,12–16,18–20,27–30,93,94 Yet, simply integrating GO with semiconductor ingredients does not take full advantage of the attractive surface properties of GO, and thus it also does not sufficiently utilize the electrical conductivity of GR.4,13,17,18,31In view of this, in 2011, our group optimized the synthesis method to obtain efficient GR–semiconductor TiO2 composites by interface engineering of the unique 2D mat of GR with TiO2.18 In this approach, soluble TiF4 is used as the precursor of TiO2 instead of hard solid nanoparticles. During the soft integration procedure, GO can play a key role in structurally directing the nucleation and growth of the soluble precursor of semiconductor TiO2, which preferentially results in an intimate interfacial contact; after subsequent reduction of GO to GR, the GR–TiO2 composites are obtained.
It can be seen from Fig. 8A that the GR sheet and semiconductor TiO2 ingredients are integrated by way of an intimate interfacial contact. The stacking GR layers can also be identified in the edge area of TiO2–5% GR. However, with the current synthesis approach, the interfacial integration of CNTs and TiO2 is ineffective, as proved by the TEM image in Fig. 8B. This demonstrates the superior and easily accessible structure-directing ability of GO (the precursor of GR) as a “solution processable surfactant”, as compared with CNTs. Notably, such an intimate complexation of TiO2 and GR cannot be obtained through the simple random integration of solid TiO2 (P25) nanoparticles and GR sheets (Fig. 8C) because the complexation of solid TiO2 nanoparticles with GO cannot effectively utilize the “structure-directing” role of GO, which results from its unique 2D structure with abundant oxygen-containing functional groups on the basal plane and edge that provide reactive sites to interact with organic/inorganic systems. When the water-soluble inorganic salt TiF4 is used as the precursor of TiO2, it can sufficiently interact with the functional groups of well-dispersed GO in the aqueous phase, thereby leading to the intimate integration of the GR sheet and TiO2 ingredients. In such a situation, the excellent electron conductivity of GR can be utilized efficiently, thus boosting the separation of photogenerated electron–hole pairs more effectively.
 | ||
| Fig. 8 TEM images of (A) TiO2–5% GR, (B) TiO2–5% CNT and (C) P25–0.5% GR; (D) contrast experiments on the selective oxidation of benzyl alcohol to benzaldehyde under visible light irradiation for 4 h over TiO2–5% GR, TiO2–5% CNT, P25–1% GR and P25–1% CNT. Reproduced with permission from ref. 17 and 18. Copyright 2010 and 2011 American Chemical Society. | ||
The photocatalytic performance of GR–TiO2 and CNT–TiO2 in the selective oxidation of alcohols in benzotrifluoride (BTF) solvent under ambient conditions was evaluated. As shown in Fig. 8D, the optimum TiO2–5% GR composite obtained through the soft integration method exhibits remarkably enhanced visible light photoactivity as compared to its TiO2–5% CNT counterpart and the optimum P25–1% GR prepared by the random hard integration procedure. This can be ascribed to the more intimate interfacial contact between GR and TiO2 formed during the soft integration process, which leads to better separation of the photogenerated carriers over GR–TiO2, as verified by the photoelectrochemical measurements.
Besides, the photostability of GR during the photocatalytic selective oxidation process has also been investigated. The C 1s X-ray photoelectron spectroscopy (XPS) spectra of fresh TiO2–5% GR and TiO2–5% GR after visible light irradiation for 4 h or 10 h show that the normalized intensity of the main peak exhibits almost no change, indicating that the GR sheets in the TiO2–5% GR composite are chemically stable in this photocatalytic reaction system. This can be ascribed to two reasons. One is the fact that the soft integration method leads to strong interactions between the GR and the TiO2 that is formed in situ on the surface of the GR sheets, which contributes to stabilizing the GR sheets.4 On the other hand, hydroxyl radicals (˙OH), which have been proven to be the main species that induces photodegradation of GR,95–97 cannot be formed in the BTF solvent.29,93,98,99 Therefore, under such photocatalytic reaction condition, no obvious degradation of GR has been observed.
This work conceptually demonstrates how to synthesize a more efficient GR–semiconductor photocatalyst by interface engineering, and highlights the key importance of optimizing the synthesis method to achieve efficient GR–semiconductor photocatalysts.18 Moreover, the comparison between the GR–TiO2 and CNT–TiO2 counterparts could promote in-depth fundamental understanding of the similarities and differences between GR and CNTs in controlling the morphology of GR (or CNT)–semiconductor composites and enhancing the photocatalytic performance of semiconductors.
3.2 Layer-by-layer self-assembly
When engineering the interface between GR and semiconductors, the stacking of GR sheets should be minimized in order to make better use of the 2D surface and high surface area of GR to enlarge the interface domain in the GR–semiconductor composites, which will favour charge carrier transfer and thus enhancement of the photoactivity of the composites. Layer-by-layer (LbL) self-assembly, as a versatile bottom-up nanofabrication technique, exhibits prominent advantages over conventional approaches for molecular-level control over the structure and composition of composites with simple benchmark operations.100–104 This method offers a promising approach to engineering the interface between GR and the semiconductor on the molecular scale. Besides, the 2D planar morphology and negatively charged surface of GO provide new options for LbL assembly of high-quality GR–semiconductor hybrids.105Recently, Liu's research group reported a significant advance in constructing LbL assembled GR–semiconductor hybrid film photocatalysts on a fluorine-doped tin oxide (FTO) substrate with controllable film thickness and architecture.105 Through the LbL self-assembly of tailor-made negatively charged CdS quantum dots (QDs) and positively charged GR nanosheets (GNs), they successfully realized the judicious integration of CdS QDs with GNs in an alternating manner, as illustrated in Fig. 9A. Such an integration manner allows the monodispersed CdS QDs to cover the GR sheets densely and homogeneously (Fig. 9B), and results in the formation of an interconnected structure between CdS QDs and GR in the GNs–CdS QDs composite film.
 | ||
| Fig. 9 (A) Schematic illustration for LbL self-assembly of GNs–CdS QDs multi-layered films; (B) TEM image of GNs–CdS QDs composite film obtained after five deposition cycles and peeled off from the FTO substrate; (C) transient photocurrent responses of CdS QDs and GNs films obtained after five deposition cycles, and (GNs–CdS QDs)n (n = 1, 5, 10, 15, 20 cycles) multi-layered films; (D) transient photocurrent responses of GNs–CdS QDs composite films with different stacking fashions in 0.1 M Na2S aqueous solution under visible light irradiation (λ > 420 nm) at zero bias versus a Pt counter electrode (left panel) and the corresponding schematic illustrations (right panels). Reproduced with permission from ref. 105. Copyright 2014 American Chemical Society. | ||
Furthermore, the photoelectrochemical and photocatalytic performances of the GNs–CdS QDs composite films can be appropriately tuned by simple control of the deposition cycles. It can be seen from Fig. 9C that the photocurrent of the GNs–CdS QDs composite film is larger than those of CdS QDs and GNs films obtained after the same number of deposition cycles. The enhanced photoelectrochemical performance of the GNs–CdS QDs composite films can be attributed to the alternating deposition of CdS QDs and GNs, which takes full advantage of the 2D planar structure of GNs, leading to the formation of an intimate interfacial contact. In this way, photoexcited electrons in the CdS QDs can readily and efficiently transfer from the conduction band of CdS to the neighbouring GNs scaffold. The GNs serve as an efficient electron collector and transporter, thus suppressing the recombination of photogenerated electron–hole pairs.
Besides, for comparison, some other structures of GNs–CdS QDs were also constructed. It can be seen from Fig. 9D that (GNs–CdS QDs)15 obtained by alternate LbL self-assembly of GNs and CdS QDs exhibits the optimal photocurrent response as compared to (CdS QDs-5 + GNs-1)3 and CdS QDs-15 + GNs-15 (the numbers indicate the deposition cycles). This can be ascribed to the fact that the single layer deposition of GNs and CdS QDs endows the alternating composite film with the best intimate interfacial contact and thus good photon absorption. However, the (CdS QDs-5 + GNs-1)3 model shows less efficient interfacial contact between the GNs and CdS QDs. In the CdS QDs-15 + GNs-15 hybrid film, the photon energy is mostly absorbed by the top GNs layer and the interfacial contact between the components is poor, thus leading to the most unfavourable photocurrent.
This finding not only indicates the importance and effectiveness of well-designed interface engineering for enhancement of the photoactivity of GR–semiconductor composites, but also represents a big step forward in meeting the application requirements of GR–semiconductor composite photocatalysts, considering the easy separation and recovery of the GR–semiconductor composite photocatalysts as well as their tunable architecture and photocatalytic performance.
4. Design and optimization from a system-level consideration
Photocatalysis, as an artificial technique, is focused on the simulation of the natural photosynthesis observed in green plants for the conversion of solar energy into chemical energy.106–109 Biological systems, which have the highest degree of complexity and exquisite functions in the natural world, show the great significance of system-level considerations in the smooth working of the whole system.110 Enlightened by this, to utilize the superior structural and electronic properties of GR in the optimization of GR–semiconductor composite photoactivity, a system-level consideration, including interface optimization, should be of great importance in any composite function.4,5 A system-level consideration accentuates the study of the interactions between the individual components of a system and how these interactions affect the performance of the final system, as highlighted by Yang and Tarascon.111In this sense, it is therefore of crucial importance to optimize the interfacial domain between GR and the semiconductor because it is intimately correlated with the separation and transfer of photogenerated charge carriers across the interface.4,5,13,93 However, this significant issue in improving the photoactivity of GR–semiconductor composites is not just a simple issue of tighter interfacial connection between GR and semiconductors, but it also involves the optimization and manipulation of the interfacial atomic charge carrier transfer pathways that result from the rational synergic interactions between the respective individual components.4,5,13,93 This fundamental key issue was first addressed in a recent report by our group,93 which put forward a simple, general and conceptually new strategy to improve the photocatalytic performance of GR–semiconductor CdS composites via the addition of tiny amounts of metal ions (M = Ca2+, Cr3+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+, and Zn2+) into the interfacial layer while maintaining the intimate interfacial contact between GR and CdS.
The preparation of such CdS–(GR–M) composites is based on a two-step wet chemistry process, as shown in Fig. 10A.93 The first step is to anchor a tiny amount of metal ions onto the GO surface through the attractive electrostatic interactions between the positively charged metal ions and the negatively charged oxygenated functional groups on the GO surface in water, resulting in GO–M. Then, after an in situ solvothermal treatment process, the CdS nanoparticles uniformly carpet the 2D surface of GO along with the reduction of GO to GR, thereby giving rise to CdS–(GR–M) composites with intimate interfacial contact (Fig. 10B). This strategy wisely introduces a tiny amount of metal ions into the interlayer matrix between the GR and the semiconductor CdS, which hardly alters the intimate interfacial contact, the crystallite size and phase of the CdS particles, or the light absorption properties of the composites, as compared to their CdS–GR counterparts without metal ions. This consequently offers a reasonable framework to comparatively study the effect of metal ions as interfacial mediators on the separation and transfer of photogenerated charge carriers and the photoactivity of CdS–(GR–M) and CdS–GR composites under visible light irradiation.
 | ||
| Fig. 10 (A) Illustration of the fabrication of CdS–(GR–M) (M = Ca2+, Cr3+, Mn2+, Fe2+, Co2+, Ni2+, Cu2+ and Zn2+) composites; (B) the corresponding TEM images for the optimal CdS–(GR–M) samples; photocatalytic performances of blank CdS, CdS–GR, and CdS–(GR–M) composites with different weight ratios of GR for (C) selective oxidation of benzyl alcohol under visible light irradiation (λ > 420 nm) for 2 h and (D) selective reduction of 4-nitroaniline under visible light irradiation (λ > 420 nm) for 80 min. Reproduced with permission from ref. 93. Copyright 2014 American Chemical Society. | ||
Benchmarked by two typical probe reactions (i.e., aerobic selective oxidation of an alcohol and anaerobic selective reduction of a nitro compound), we find that the CdS–(GR–M) composites exhibit significantly enhanced photoactivity compared with blank CdS and the optimal CdS–5% GR (Fig. 10C and D).93 The optimal weight ratios of GR in the CdS–(GR–M) photocatalysts are the same for the two tested reactions, indicating that the same optimum synergistic interaction between GR–M and the CdS semiconductor can be achieved for these two different selective photocatalytic redox reactions. In addition, it is worth noting that the optimal weight ratio of GR in the CdS–(GR–M) composites is remarkably increased to 10%, and even 30%, in comparison with the optimal CdS–5% GR. In conjunction with the photoelectrochemical and photoluminescence characterizations, a two-fold role of the metal ions in the interfacial layer of CdS–GR has been demonstrated.93 The first role of the metal ions is to act as “generic interfacial mediators” to positively boost the lifetime of the photogenerated charge carriers and the transfer efficiency between CdS and the GR sheet, which contributes to the improvement in photoactivity. The second interesting role of the metal ions is to partially counterbalance the negative “shielding” effect induced by the higher weight ratio of GR.4–10,12–20,23,26–32 Therefore, the strategy of using metal ions as interfacial mediators is capable of balancing the double-edged sword of GR, which thus leads to enhanced net efficiency of GR–semiconductor composite photoactivity.93
Such an interfacial mediator strategy has also proven to be effective in the case of using noble metals as mediators to optimize the charge carrier transfer pathway between GR and semiconductors from a system-level consideration.112–116 For instance, when a tiny amount of Pd nanoparticles are introduced into the interfacial layer between GR and semiconductors (e.g., CdS nanoparticles,112 BiVO4 nanosheets,113 and In2S3 petals114), the resultant ternary composites show remarkably enhanced visible light photoactivity as compared to their optimal binary counterparts toward photoredox processes, including aerobic oxidation of alcohols, anaerobic reduction of nitro compounds and degradation of dyes.112–114 This is ascribed to the optimized spatial transfer pathway of the photogenerated charge carriers across the interface, resulting from the introduction of Pd nanoparticles as mediators into the interfacial layer between GR and semiconductors. The Pd nanoparticles play two roles.112–114 As illustrated in Fig. 11, one role of the Pd is to serve as an electron reservoir to directly trap photogenerated electrons from the semiconductor, and the other role is as an interfacial mediator to promote electron relay in the ternary semiconductor–(GR–Pd) photocatalysts, in which Pd and conductive GR act as dual cocatalysts. Moreover, the negative light “shielding effect” of GR at high weight ratios can be partially counterbalanced in the semiconductor–(GR–Pd) composites.
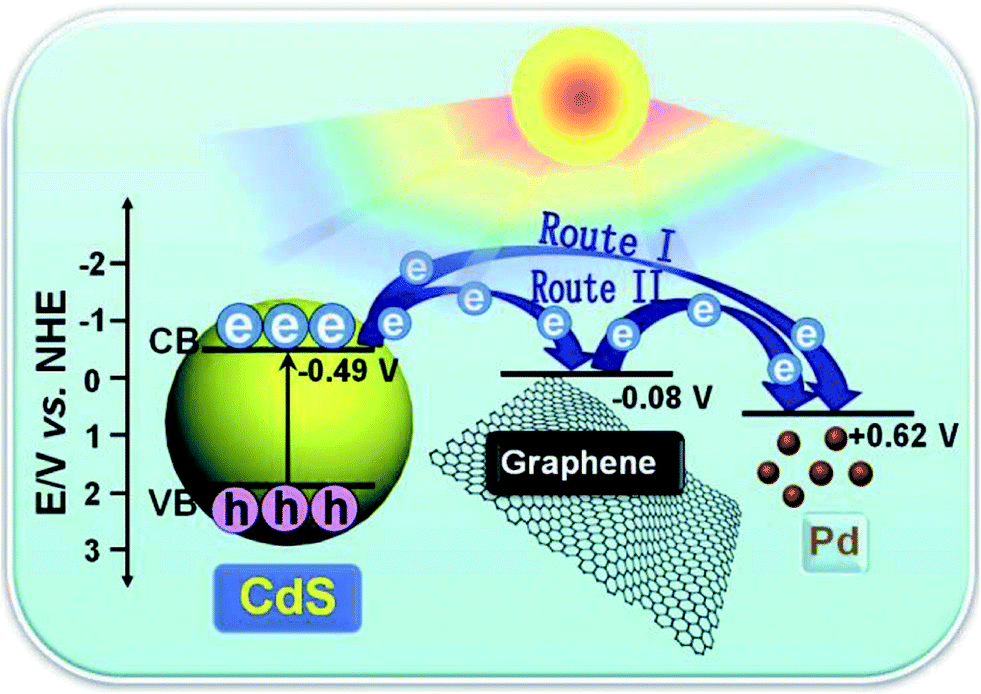 | ||
| Fig. 11 Schematic diagram of the charge carrier transfer in the ternary CdS–(GR–Pd) composites under visible light irradiation. Reproduced with permission from ref. 112. Copyright 2014 The Royal Society of Chemistry. | ||
These results are significant because they highlight that the critical key factor in improving the photocatalytic performance of GR–semiconductor composites relies on the optimization of the atomic charge carrier pathway across the interface between the GR and the semiconductor.93,112–116 It is believed that extension and development of this interfacial mediator strategy would promote theoretical research (e.g., using band bending calculations) and electron transfer kinetics studies (e.g., using femtosecond transient absorption spectroscopy, X-ray spectroscopy, time-resolved spectroscopy techniques, etc.) to precisely depict an elegant model of how metal ions and noble metal nanoparticles as interfacial mediators affect and optimize the charge carrier transfer pathway and dynamics at an atomic level. A clear theoretical scenario would in turn aid the further rational design and fabrication of smarter GR–semiconductor composite photocatalysts. It is reasonable to believe that the system-level planning of theoretical and experimental efforts, which is increasingly important for the development of modern materials science, would steer the photocatalysis community toward sufficient utilization of the superlative potential of GR for achieving truly smart, highly efficient GR–semiconductor composite photocatalysts for specific applications.
5. Summary and outlook
In summary, there have been extensive efforts made to improve the performance of GR–semiconductor composite photocatalysts in solar energy conversion by developing efficient strategies to design and construct efficient GR–semiconductor composites. The advances achieved in this regard, especially the strategies of generic interfacial mediators to boost GR–semiconductor composite-based photocatalysis93,112–114 and layer-by-layer self-assembly of hybrid films of GR–semiconductor composites,105 highlight the promising scope and potential of fabricating GR–semiconductor composites with optimized photocatalytic performance for practical applications. As one promising prospect for the development of GR–semiconductor composite photocatalysts, if the generic interfacial mediator strategy could be rationally combined with controllable film architectures, highly efficient GR–semiconductor film photocatalysts with tunable structural and electronic properties would be wisely integrated in optimal systems. Such a GR–semiconductor composite-based photocatalytic system is expected to meet two fundamental requirements of practical applications, i.e., high performance and easy separation/recovery.To advance the further development of GR–semiconductor composite-based photocatalysis, more endeavour is needed not only to improve the efficiency of GR–semiconductor composite photocatalysts, but also to understand the microscopic dynamic processes of the generation, recombination, separation, and transfer of charge carriers in the photocatalysts, which would in turn guide us to design and synthesize more efficient GR–semiconductor composite photocatalysts.
Although the construction of the next generation of photocatalytic systems based on GR–semiconductor composites is a long story, considering the usual trajectory of any material from discovery to industry which often takes over 20 years,4,5,24,117 we have been on the road to advancing GR–semiconductor composite-based photocatalysis for practical applications. Hopefully, joint experimental and theoretical efforts will take us further along this promising road, and then truly unique, smart GR-based composite photocatalysts with robust structural and functional infrastructures are anticipated to be forthcoming.
Acknowledgements
The support from the Key Project of National Natural Science Foundation of China (U1463204), the National Natural Science Foundation of China (20903023 and 21173045), the Award Program for Minjiang Scholar Professorship, the Natural Science Foundation of Fujian Province for Distinguished Young Investigator Grant (2012 J06003), the Independent Research Project of State Key Laboratory of Photocatalysis on Energy and Environment (No. 2014A05), the 1st Program of Fujian Province for Top Creative Young Talents, and the Program for Returned High-Level Overseas Chinese Scholars of Fujian Province is kindly acknowledged.References
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- J. D. Roy-Mayhew and I. A. Aksay, Chem. Rev., 2014, 114, 6323–6348 CrossRef CAS PubMed.
- L. Dai, Acc. Chem. Res., 2013, 46, 31–42 CrossRef CAS PubMed.
- N. Zhang, M.-Q. Yang, S. Liu, Y. Sun and Y.-J. Xu, Chem. Rev., 2015, 115, 10307–10377 CrossRef CAS PubMed.
- M.-Q. Yang, N. Zhang, M. Pagliaro and Y.-J. Xu, Chem. Soc. Rev., 2014, 43, 8240–8254 RSC.
- C. Huang, C. Li and G. Shi, Energy Environ. Sci., 2012, 5, 8848–8868 CAS.
- D. R. Dreyer, R. S. Ruoff and C. W. Bielawski, Angew. Chem., Int. Ed., 2010, 49, 9336–9344 CrossRef CAS PubMed.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752–7777 CrossRef CAS PubMed.
- M.-Q. Yang and Y.-J. Xu, Phys. Chem. Chem. Phys., 2013, 15, 19102–19118 RSC.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- C. Han, M.-Q. Yang, B. Weng and Y.-J. Xu, Phys. Chem. Chem. Phys., 2014, 16, 16891–16903 RSC.
- W. Tu, Y. Zhou and Z. Zou, Adv. Funct. Mater., 2013, 23, 4996–5008 CrossRef CAS.
- N. Zhang, Y. Zhang and Y.-J. Xu, Nanoscale, 2012, 4, 5792–5813 RSC.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC.
- P. V. Kamat, J. Phys. Chem. Lett., 2011, 2, 242–251 CrossRef CAS.
- F. Li and Z. Chen, in Graphene Chemistry: Theoretical Perspectives, ed. D.-E. Jiang and Z. Chen, John Wiley & Sons, 2014 Search PubMed.
- Y. Zhang, Z.-R. Tang, X. Fu and Y.-J. Xu, ACS Nano, 2010, 4, 7303–7314 CrossRef CAS PubMed.
- Y. Zhang, Z.-R. Tang, X. Fu and Y.-J. Xu, ACS Nano, 2011, 5, 7426–7435 CrossRef CAS PubMed.
- N. Zhang, Y. Zhang, M.-Q. Yang, Z.-R. Tang and Y.-J. Xu, J. Catal., 2013, 299, 210–221 CrossRef CAS.
- M.-Q. Yang, N. Zhang and Y.-J. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 1156–1164 CAS.
- D. Eder, Chem. Rev., 2010, 110, 1348–1385 CrossRef CAS PubMed.
- X. Lu and Z. Chen, Chem. Rev., 2005, 105, 3643–3696 CrossRef CAS PubMed.
- Y. T. Liang, B. K. Vijayan, K. A. Gray and M. C. Hersam, Nano Lett., 2011, 11, 2865–2870 CrossRef CAS PubMed.
- Editorials, Nature, 2011, 473, 419 Search PubMed.
- Graphene: The Road to Applications, Nature, Cambridge, Massachusetts, USA, 2011, May 11–13 Search PubMed.
- Z. Chen, N. Zhang and Y.-J. Xu, CrystEngComm, 2013, 15, 3022–3030 RSC.
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed.
- J. S. Lee, K. H. You and C. B. Park, Adv. Mater., 2012, 24, 1084–1088 CrossRef CAS PubMed.
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, ACS Nano, 2012, 6, 9777–9789 CrossRef CAS PubMed.
- Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H.-J. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS PubMed.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS PubMed.
- J. Liu, H. Bai, Y. Wang, Z. Liu, X. Zhang and D. D. Sun, Adv. Funct. Mater., 2010, 20, 4175–4181 CrossRef CAS.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS PubMed.
- J. S. Bunch, A. M. van der Zande, S. S. Verbridge, I. W. Frank, D. M. Tanenbaum, J. M. Parpia, H. G. Craighead and P. L. McEuen, Science, 2007, 315, 490–493 CrossRef CAS PubMed.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179–6212 CrossRef CAS PubMed.
- D. Chen, L. Tang and J. Li, Chem. Soc. Rev., 2010, 39, 3157–3180 RSC.
- S. Patil, A. Harle, S. Sathaye and K. Patil, CrystEngComm, 2014, 16, 10845–10855 RSC.
- N. Zhang, Y. Zhang, M.-Q. Yang and Y.-J. Xu, Curr. Org. Chem., 2013, 17, 2503–2515 CrossRef CAS.
- L. Han, P. Wang and S. Dong, Nanoscale, 2012, 4, 5814–5825 RSC.
- I. V. Lightcap and P. V. Kamat, Acc. Chem. Res., 2013, 46, 2235–2243 CrossRef CAS PubMed.
- G. Xie, K. Zhang, B. Guo, Q. Liu, L. Fang and J. R. Gong, Adv. Mater., 2013, 25, 3820–3839 CrossRef CAS PubMed.
- Q. Xiang and J. Yu, J. Phys. Chem. Lett., 2013, 4, 753–759 CrossRef CAS PubMed.
- X. An and J. C. Yu, RSC Adv., 2011, 1, 1426–1434 RSC.
- X. Xie, K. Kretschmer and G. Wang, Nanoscale, 2015, 7, 13278–13292 RSC.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26–41 CrossRef CAS PubMed.
- J. N. Coleman, Acc. Chem. Res., 2013, 46, 14–22 CrossRef CAS PubMed.
- M. Lotya, Y. Hernandez, P. J. King, R. J. Smith, V. Nicolosi, L. S. Karlsson, F. M. Blighe, S. De, Z. Wang, I. T. McGovern, G. S. Duesberg and J. N. Coleman, J. Am. Chem. Soc., 2009, 131, 3611–3620 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, Phys. Chem. Chem. Phys., 2012, 14, 9167–9175 RSC.
- T. Wu, L. Zou, D. Han, F. Li, Q. Zhang and L. Niu, Green Chem., 2014, 16, 2142–2146 RSC.
- Y. Zhang, N. Zhang, Z.-R. Tang and Y.-J. Xu, J. Phys. Chem. C, 2014, 118, 5299–5308 CAS.
- S. Guo, S. Dong and E. Wang, ACS Nano, 2010, 4, 547–555 CrossRef CAS PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- D. Wei and Y. Liu, Adv. Mater., 2010, 22, 3225–3241 CrossRef CAS PubMed.
- H. Liu, Y. Liu and D. Zhu, J. Mater. Chem., 2011, 21, 3335–3345 RSC.
- Z.-R. Tang, Y. Zhang, N. Zhang and Y.-J. Xu, Nanoscale, 2015, 7, 7030–7034 RSC.
- B. Han, S. Liu, Z.-R. Tang and Y.-J. Xu, J. Energy Chem., 2015, 24, 145–156 CrossRef.
- B. Guo, Q. Liu, E. Chen, H. Zhu, L. Fang and J. R. Gong, Nano Lett., 2010, 10, 4975–4980 CrossRef CAS PubMed.
- P. Wang, Z. Wang, L. Jia and Z. Xiao, Phys. Chem. Chem. Phys., 2009, 11, 2730–2740 RSC.
- L. Jia, D.-H. Wang, Y.-X. Huang, A.-W. Xu and H.-Q. Yu, J. Phys. Chem. C, 2011, 115, 11466–11473 CAS.
- Z. Mou, Y. Wu, J. Sun, P. Yang, Y. Du and C. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 13798–13806 CAS.
- C. Li and G. Shi, Nanoscale, 2012, 4, 5549–5563 RSC.
- S. Nardecchia, D. Carriazo, M. L. Ferrer, M. C. Gutierrez and F. del Monte, Chem. Soc. Rev., 2013, 42, 794–830 RSC.
- Z. Chen, W. Ren, L. Gao, B. Liu, S. Pei and H.-M. Cheng, Nat. Mater., 2011, 10, 424–428 CrossRef CAS PubMed.
- R. Menzel, S. Barg, M. Miranda, D. B. Anthony, S. M. Bawaked, M. Mokhtar, S. A. Al-Thabaiti, S. N. Basahel, E. Saiz and M. S. P. Shaffer, Adv. Funct. Mater., 2015, 25, 28–35 CrossRef CAS.
- W. Han, C. Zang, Z. Huang, H. Zhang, L. Ren, X. Qi and J. Zhong, Int. J. Hydrogen Energy, 2014, 39, 19502–19512 CrossRef CAS.
- M. Gao, C. K. N. Peh, W. L. Ong and G. W. Ho, RSC Adv., 2013, 3, 13169–13177 RSC.
- Z. Zhang, F. Xiao, Y. Guo, S. Wang and Y. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 2227–2233 CAS.
- Y. Fan, W. Ma, D. Han, S. Gan, X. Dong and L. Niu, Adv. Mater., 2015, 27, 3767–3773 CrossRef CAS PubMed.
- B. Qiu, M. Xing and J. Zhang, J. Am. Chem. Soc., 2014, 136, 5852–5855 CrossRef CAS PubMed.
- D. A. Reddy, J. Choi, S. Lee, R. Ma and T. K. Kim, RSC Adv., 2015, 5, 18342–18351 RSC.
- L. Estevez, A. Kelarakis, Q. Gong, E. H. Da'as and E. P. Giannelis, J. Am. Chem. Soc., 2011, 133, 6122–6125 CrossRef CAS PubMed.
- S. Liu, Z.-R. Tang, Y. Sun, J. C. Colmenares and Y.-J. Xu, Chem. Soc. Rev., 2015, 44, 5053–5075 RSC.
- S. Yurdakal, G. Palmisano, V. Loddo, V. Augugliaro and L. Palmisano, J. Am. Chem. Soc., 2008, 130, 1568–1569 CrossRef CAS PubMed.
- S. Yang, N. Huang, Y. M. Jin, H. Q. Zhang, Y. H. Su and H. G. Yang, CrystEngComm, 2015, 17, 6617–6631 RSC.
- N. Roy, Y. Sohn and D. Pradhan, ACS Nano, 2013, 7, 2532–2540 CrossRef CAS PubMed.
- R. Li, F. Zhang, D. Wang, J. Yang, M. Li, J. Zhu, X. Zhou, H. Han and C. Li, Nat. Commun., 2013, 4, 1432 CrossRef PubMed.
- X. Pan, N. Zhang, X. Fu and Y.-J. Xu, Appl. Catal., A, 2013, 453, 181–187 CrossRef CAS.
- L. Shen, N. Bao, Y. Zheng, A. Gupta, T. An and K. Yanagisawa, J. Phys. Chem. C, 2008, 112, 8809–8818 CAS.
- G. Liu, L. Wang, H. G. Yang, H.-M. Cheng and G. Q. Lu, J. Mater. Chem., 2010, 20, 831–843 RSC.
- A. McLaren, T. Valdes-Solis, G. Li and S. C. Tsang, J. Am. Chem. Soc., 2009, 131, 12540–12541 CrossRef CAS PubMed.
- N. Zhang, R. Ciriminna, M. Pagliaro and Y.-J. Xu, Chem. Soc. Rev., 2014, 43, 5276–5287 RSC.
- Y. Zhang, Z. Chen, S. Liu and Y.-J. Xu, Appl. Catal., B, 2013, 140–141, 598–607 CrossRef CAS.
- X. Pan, Y. Zhao, S. Liu, C. L. Korzeniewski, S. Wang and Z. Fan, ACS Appl. Mater. Interfaces, 2012, 4, 3944–3950 CAS.
- L. Liu, Z. Liu, A. Liu, X. Gu, C. Ge, F. Gao and L. Dong, ChemSusChem, 2014, 7, 618–626 CrossRef CAS PubMed.
- F. Sordello, G. Zeb, K. Hu, P. Calza, C. Minero, T. Szkopek and M. Cerruti, Nanoscale, 2014, 6, 6710–6719 RSC.
- M.-Q. Yang, B. Weng and Y.-J. Xu, Langmuir, 2013, 29, 10549–10558 CrossRef CAS PubMed.
- B. Weng, J. Wu, N. Zhang and Y.-J. Xu, Langmuir, 2014, 30, 5574–5584 CrossRef CAS PubMed.
- T. Lee, S. H. Min, M. Gu, Y. K. Jung, W. Lee, J. U. Lee, D. G. Seong and B.-S. Kim, Chem. Mater., 2015, 27, 3785–3796 CrossRef CAS.
- L. Yuan, M.-Q. Yang and Y.-J. Xu, Nanoscale, 2014, 6, 6335–6345 RSC.
- Y. Wang, W. Wang, H. Mao, Y. Lu, J. Lu, J. Huang, Z. Ye and B. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 12698–12706 CAS.
- C. Han, Z. Chen, N. Zhang, J. C. Colmenares and Y.-J. Xu, Adv. Funct. Mater., 2015, 25, 221–229 CrossRef CAS.
- N. Zhang, M.-Q. Yang, Z.-R. Tang and Y.-J. Xu, ACS Nano, 2014, 8, 623–633 CrossRef CAS PubMed.
- X. Pan, M.-Q. Yang and Y.-J. Xu, Phys. Chem. Chem. Phys., 2014, 16, 5589–5599 RSC.
- J. G. Radich, A. Krenselewski, J. Zhu and P. V. Kamat, Chem. Mater., 2014, 26, 4662–4668 CrossRef CAS.
- O. Akhavan, ACS Nano, 2010, 4, 4174–4180 CrossRef CAS PubMed.
- L. Zhang, S. Diao, Y. Nie, K. Yan, N. Liu, B. Dai, Q. Xie, A. Reina, J. Kong and Z. Liu, J. Am. Chem. Soc., 2011, 133, 2706–2713 CrossRef CAS PubMed.
- M. Zhang, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2008, 47, 9730–9733 CrossRef CAS PubMed.
- M. Zhang, Q. Wang, C. Chen, L. Zang, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2009, 48, 6081–6084 CrossRef CAS PubMed.
- F.-X. Xiao, Z. Zeng and B. Liu, J. Am. Chem. Soc., 2015, 137, 10735–10744 CAS.
- F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111–1114 CrossRef CAS PubMed.
- F.-X. Xiao, Chem. Commun., 2012, 48, 6538–6540 RSC.
- F. Xiao, J. Phys. Chem. C, 2012, 116, 16487–16498 CAS.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- F.-X. Xiao, J. Miao and B. Liu, J. Am. Chem. Soc., 2014, 136, 1559–1569 CrossRef CAS PubMed.
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 RSC.
- L. Yuan and Y.-J. Xu, Appl. Surf. Sci., 2015, 342, 154–167 CrossRef CAS.
- H. Dau and I. Zaharieva, Acc. Chem. Res., 2009, 42, 1861–1870 CrossRef CAS PubMed.
- S. Liu, B. Weng, Z.-R. Tang and Y.-J. Xu, Nanoscale, 2015, 7, 861–866 RSC.
- H. Kitano, Science, 2002, 295, 1662–1664 CrossRef CAS PubMed.
- P. Yang and J.-M. Tarascon, Nat. Mater., 2012, 11, 560–563 CrossRef CAS PubMed.
- C. Han, M.-Q. Yang, N. Zhang and Y.-J. Xu, J. Mater. Chem. A, 2014, 2, 19156–19166 CAS.
- Z.-R. Tang, Q. Yu and Y.-J. Xu, RSC Adv., 2014, 4, 58448–58452 RSC.
- X. Li, N. Zhang and Y.-J. Xu, ChemCatChem, 2015, 7, 2047–2054 CrossRef CAS.
- P. Roy, A. P. Periasamy, C.-T. Liang and H.-T. Chang, Environ. Sci. Technol., 2013, 47, 6688–6695 CrossRef CAS PubMed.
- P. Gao, J. Liu, S. Lee, T. Zhang and D. D. Sun, J. Mater. Chem., 2012, 22, 2292–2298 RSC.
- R. V. Noorden, Nature, 2011, 469, 14–16 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2016 |