
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient DBU accelerated synthesis of 18F-labelled trifluoroacetamides†
Antonio Bermejo
Gómez
 abc,
Miguel A.
Cortés González
bc,
Marvin
Lübcke
bc,
Magnus J.
Johansson
d,
Christer
Halldin
ce,
Kálmán J.
Szabó
*bc and
Magnus
Schou
*ace
abc,
Miguel A.
Cortés González
bc,
Marvin
Lübcke
bc,
Magnus J.
Johansson
d,
Christer
Halldin
ce,
Kálmán J.
Szabó
*bc and
Magnus
Schou
*ace
aAstraZeneca Personalised Healthcare and Biomarkers, PET Centre at Karolinska Institutet, Karolinska Universitetssjukhuset Solna, R5:02, SE-171 76 Stockholm, Sweden. E-mail: Magnus.Schou@astrazeneca.com
bDepartment of Organic Chemistry, Stockholm University, Arrhenius Laboratory, SE-106 91 Stockholm, Sweden
cStockholm Brain Institute, Karolinska Institutet, SE-171 77 Stockholm, Sweden
dCVMD iMed, Medicinal Chemistry AstraZeneca R&D, Mölndal, SE-431 83, Sweden
eDepartment of Clinical Neuroscience, Karolinska Institutet, S-17176 Stockholm, Sweden
First published on 10th November 2016
Abstract
Nucleophilic 18F-fluorination of bromodifluoromethyl derivatives was performed using [18F]Bu4NF in the presence of DBU (1,8-diazabicyclo[5.4.0]undec-7-ene). This novel procedure provided a diverse set of [18F]trifluoroacetamides in good to excellent radiochemical conversions. A mechanism where DBU acts as organomediator in this transformation is proposed.
Due to the favourable properties of fluorine, approximately one fifth of newly registered small drug molecules are organofluorine compounds.1 Moreover, the artificial isotope 18F (t1/2 109.8 min) has suitable nuclear properties for in vivo imaging in human subjects using positron emission tomography (PET).2 Hence, there is a large demand for efficient fluorination methodologies in both medicinal3 and radiosynthetic chemistry.4
Despite the remarkable developments in PET radiochemistry during the past decade,4 lack of efficient radiochemical methodology may still be a bottleneck in drug discovery and development. It is particularly rate-limiting for microdosing studies, in which the distribution of the radiolabeled drug molecule, previously developed without the intent to be radiolabeled, is studied during the PET measurement.5 In view of our long-term objective to facilitate PET studies during drug discovery and development, we turned our attention to the development of a new method for introducing fluorine-18 into trifluoroacetamide (–NCOCF3) groups. Although these groups are important motifs in medicinal chemistry,6 their radiolabelling has so far been remarkably neglected in the literature. On the other hand, more attention has been directed to the development of methods for the installation of [18F]CF3 groups into other type of motifs and two distinct approaches have been taken for this purpose. The first proceeds via direct substitution of a leaving group in the appropriate difluoromethyl analog with 18F-fluoride (Scheme 1).7–12 Traditionally, these transformations have been thermally activated, but more recently metal salts were successfully used as activators.11 The second approach comprises a two-step procedure in which the [18F]CF3 functionality is incorporated into aromatic substrates via [18F]CuCF3 species.8–10,13
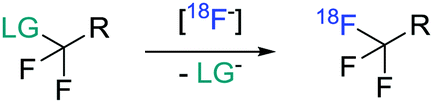 | ||
| Scheme 1 General synthesis of 18F-CF3 moieties by displacement of a leaving group. | ||
We started investigating the transformation of difluorobromoacetamide 1a into the corresponding 18F-trifluoromethyl analog [18F]2a using [18F]Bu4NF in a set of solvents with varying temperatures (Table 1, entries 1–5). The reaction proceeded with low to modest radiochemical conversion (RCC; the fraction of radioactivity incorporated into the desired product) in all examined solvents, including DCE (entry 1) acetonitrile (entry 2) and DMSO (entry 3). The highest RCC (35%) was observed in DMF. There was no obvious difference between kryptofix/K18F and [18F]Bu4NF as the fluorinating species (entries 5 vs. 4). Interestingly, the use of metal salts as additives did not lead to improvements in the RCC either (entries 6–8). In light of these results where reported methods failed, we turned our attention to nitrogen based nucleophilic organic activators for the 18F-fluorination.14 Thus, the transformation of 1a into [18F]2a was investigated in DMF at 100 °C using 1 equiv. of activator. Though DABCO and DBN did not provide an improvement in RCC (entries 9 and 12 vs. 4), gratifyingly, DMAP and pyridine increased the RCC to 59% and 68%, respectively (entries 10 and 11). A further increase in RCC to 71%, twice compared to control conditions without activator (cf. entries 4 and 13), was obtained using DBU. When two further guanidine derivatives were used, TBD and MTBD, an even higher RCC was observed (entries 15 and 16, respectively). However, when the reactions with TBD and MTBD were repeated in the cold lab (using 19F-containing TBAF) extensive hydrodebromination of the starting material 1a was also detected. Because formation of this by-product would complicate the purification and isolation of [18F]2a and since the reaction with DBU proceeded with a high RCC without hydrodebromination (or other side reactions), DBU was selected as the activator in the further studies.
|
|
||||
|---|---|---|---|---|
| Entry | Solventb | Temp. (°C) | Additivec | RCCd (%) |
| a Unless otherwise noted: 1a (0.06 mmol, 19 mg), solvent 0.4 mL, additive 0.06 mmol (1 equiv.). b DMF = N,N-dimethylformamide; DCM = dichloromethane; DCE = 1,2-dichloroethane. c DMAP = 4-dimethylaminopyridine; DABCO = (1,4-diazabicyclo[2.2.2]octane); DBN = 1,5-diazabicyclo[4.3.0]non-5-ene; DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene; TBD = 1,5,7-triazabicyclo[4.4.0]dec-5-ene; MTBD = 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene. d Estimated by radio-HPLC. e Using KF[18F]/K2.2.2. f 2 equiv. | ||||
| 1 | DCE | 85 | — | 3 ± 2 (n = 2) |
| 2 | CH3CN | 60 | — | 29 ± 5 (n = 2) |
| 3 | DMSO | 170 | — | 3 ± 1 (n = 2) |
| 4 | DMF | 100 | — | 35 ± 13 (n = 3) |
| 5 | DMF | 100 | —e | 22 ± 6 (n = 3) |
| 6 | DMF | 100 | (PPh3)3CuOAc | <1 (n = 2) |
| 7 | DCM | RT | AgOTf | 2 ± 0 (n = 2) |
| 8 | DCE | 80 | AgOTff | 4 ± 1 (n = 2) |
| 9 | DMF | 100 | DABCO | 41 ± 7 (n = 3) |
| 10 | DMF | 100 | DMAP | 59 ± 16 (n = 3) |
| 11 | DMF | 100 | Pyridine | 68 ± 9 (n = 3) |
| 12 | DMF | 100 | DBN | 40 ± 7 (n = 3) |
| 13 | DMF | 100 | DBU | 71 ± 8 (n = 3) |
| 14 | DMF | 100 | DBUe | 65 ± 13 (n = 2) |
| 15 | DMF | 100 | TBD | 81 ± 8 (n = 3) |
| 16 | DMF | 100 | MTBD | 76 ± 12 (n = 3) |

With the improved conditions in hand, we explored the scope of the DBU-mediated reaction with different tertiary (1a–1o) and secondary (1p and 1r) bromodifluoroacetamides (Scheme 2). Substrates with cyclic or linear alkyl chains (1a–1b), or including alkene functionalities (1c), gave the labelled amides [18F]2a–2c in good to excellent RCC (71, 68 and 84%, respectively). The benzylic derivatives 1d–1f and the morpholine, oxetane, proline and ketal derivatives 1g–1j could also be conveniently transformed with [18F]TBAF in the presence of DBU to [18F]2d–2j (61–92% RCC). In metal catalyzed/mediated reactions, sulfur has a tendency to coordinate to the metal atom and thus inhibit the reaction. With the current methodology, thioether derivative [18F]2k was obtained in excellent RCC (84%), thus suggesting an apparent insensitivity of the protocol to thiol functionalities. Next, substrate 1l, with an additional amide function, and 1m–1o, with some important nitrogen containing heterocycles commonly used in medicinal chemistry, were converted smoothly into trifluoromethyl derivatives [18F]2l–2o in excellent RCCs (77, 90, 75 and 80%, respectively). Finally, we focused our attention to the more challenging secondary bromodifluoroacetamide substrates 1p and 1r. As expected, [18F]2p and [18F]2r were obtained in substantially lower RCCs than the tertiary amide derivatives using direct DBU-mediated fluorination. Considering that the introduction of a benzyl group into 1r provided a threefold increase in RCC (cf. [18F]2fvs. [18F]2r) we decided to explore a protecting group strategy for the secondary amide substrates. However, much to our disappointment, N-Boc (tert-butoxycarbonyl) protection was not a viable solution for this purpose as it provided only marginal improvement, or even a reduction, in the RCC for [18F]2q and [18F]2s.
 | ||
| Scheme 2 Substrate scope in the 18F-labelling mediated by DBU. 1a–1u: bromodifluoroacetamides, 2a–2u: trifluoroacetamides. RCCs estimated by radio-HPLC. Boc = tert-butoxycarbonyl. | ||
In an effort to improve the yields for this important class of compounds, the protocol was modified to include an 18F-labelled ester as intermediate (Scheme 3).15 Because of the basic radiofluorination reaction media, extensive hydrolysis was observed during the synthesis of ester [18F]4a and the resulting RCC was only 2%. Gratifyingly, however, the more stable menthol ester [18F]4b could be obtained in a 64% RCC and further converted, without intermediate purification, into the desired secondary amides [18F]2p and [18F]2r with high RCCs (76% and 72% respectively). To evaluate the utility of the current methodology for the labelling of druglike molecules it was applied to the 18F-labelling of [18F]2t (AZD5423), a nonsteroidal glucocorticoid agonist developed for the treatment of respiratory diseases.12,16 Because [18F]2t is a secondary amide, direct labelling in the presence of DBU proceeded with only a modest RCC (7%, Scheme 2) and starting from the N-Boc protected derivative 1u failed. However, when using the above sequence (Scheme 3), with [18F]4b as a key intermediate, we obtained the target molecule in a 12% RCC under non-optimized conditions.
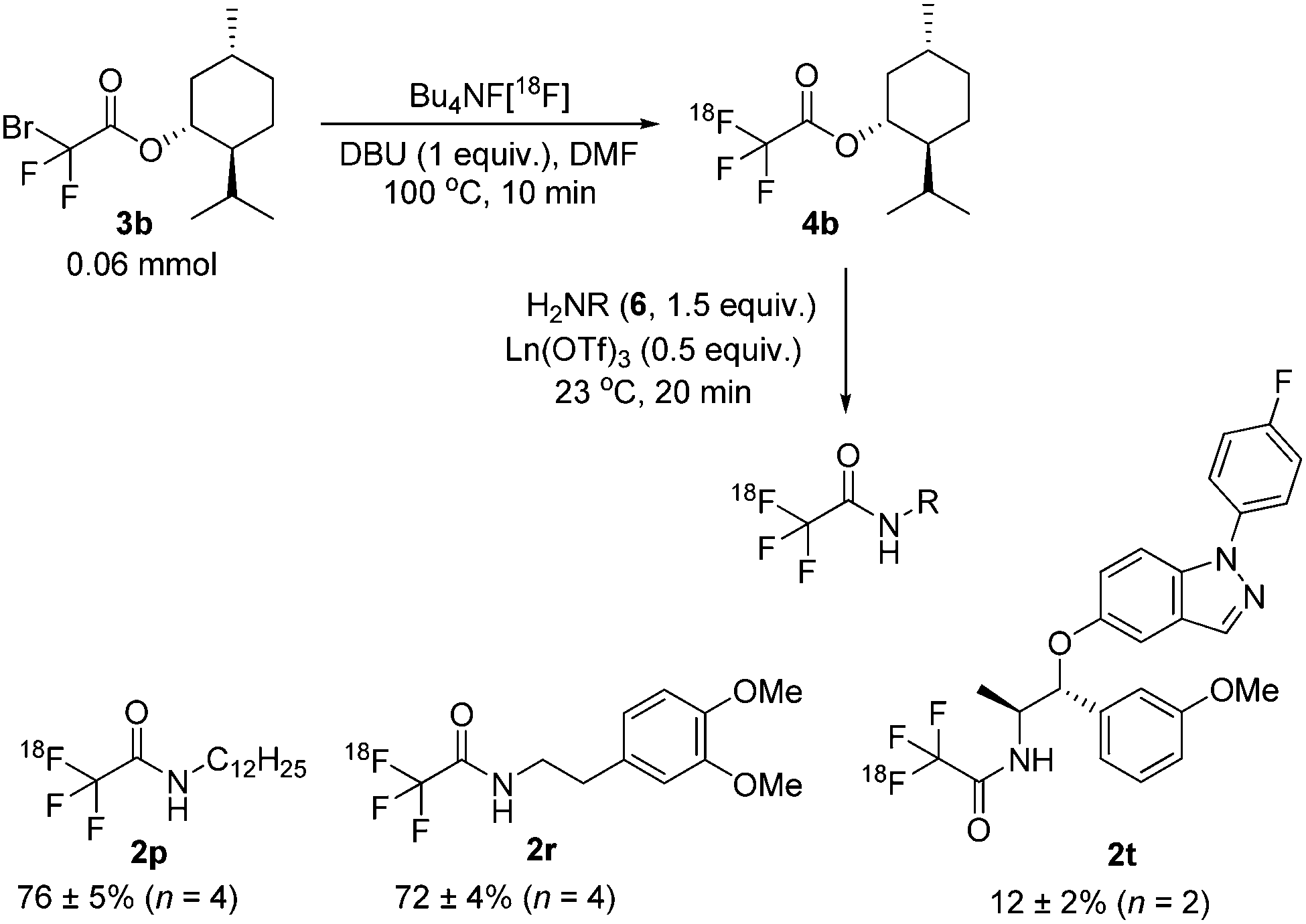 | ||
| Scheme 3 Radiolabelling of [18F]4b and derivatization to secondary amides. RCCs were estimated by radio-HPLC. | ||
In a preparative run, starting from 762 MBq of [18F]Bu4NF, [18F]2a was isolated by semi-preparative HPLC in a 44% radiochemical yield (RCY, not corrected for decay) with a specific activity (S.A. = radioactivity per molar amount of product) of 0.10 GBq μmol−1. Although an S.A. of 0.1 GBq μmol−1 is useful for PET microdosing studies, it is insufficient for examining drug-target engagement.17 Accordingly, a more general application of the methodology for PET studies require an increase of the S.A of the product. In control experiments when [18F]Bu4NF was omitted from the reaction mixture, it was found that the formation of [19F]2a was largely dependent on the amount of 1a used in the reaction.9 This indicates that 19F from the CF2Br group of 1a assists in the formation of [19F]2a, which lowers the S.A. of the product. This problem can be circumvented by lowering the concentration of 1a in the reaction mixture. Indeed, by a 100-fold reduction in the amount of 1a (from 60 to 0.6 μmol, see ESI†) and starting from 7.16 GBq of [18F]Bu4NF, [18F]2a (284 MBq, 4% RCY) was obtained with an 84-fold improved specific radioactivity (8.4 GBq μmol−1). Compared to literature examples,7,11 this is a decent level of S.A. for a [18F]CF3 species, which inherently suffers from low S.A. Furthermore, since we observed that the formation of cold 2a was unaffected by the radioactivity used in the reaction, the S.A. of [18F]2a is expected to be proportional also to the starting radioactivity. Thus, we envisage that the S.A. could be easily increased further under automated conditions when a full cyclotron production may be used for the radiofluorination.18 This would provide a product with a S.A. useful also for drug-target engagement studies. According to our studies neither the RCY nor the S.A. can be increased by increasing the amount of DBU (1 equiv.) in the reaction mixture.
A very interesting feature of the above study is the acceleration effect of DBU in the bromide-18F exchange process (cf.Table 1 entries 4 and 13). In order to rationalize this effect, we monitored the reaction of 1a with DBU using 13C NMR spectroscopy (Scheme 4). The 13C NMR spectra of DBU and isolated 1a are given in Scheme 4b and c. When 1a and DBU was mixed in DMF-d7 and heated at 100 °C (simulating the reaction conditions according to Table 1, entry 4), the 13C NMR signals of DBU underwent systematic changes (Scheme 4a). The signal at 46.5 ppm (assigned to C11) was shifted to 40.5 ppm and substantially broadened. The 13C signal at 38.1 ppm (assigned to C6) also underwent a similar change shifting to 33.6 ppm. The shift of the 13C NMR signals clearly shows that chemical environment of the above carbon atoms in DBU is significantly changed on addition of 1a. Most probably DBU displaces the bromide of 1a to form intermediate 7 (Scheme 4). The broadening of the shifts at 40.0 and 33.6 ppm may indicate C–F coupling and/or hindered rotation along the C(F2)–N bond in 7. The 13C NMR spectrum of the mixture of DBU + 1a (Scheme 4a) remained unchanged in the temperature range of 85–100 °C. When the reaction mixture of DBU + 1a was cooled to room temperature intermediate 7 decomposed to DBU and 1a. This indicates that formation of 7 is reversible14 and its formation requires heating to at least to 85 °C. Based on the above, we suggest that DBU and CF2Br precursors 1 form intermediates such as 7 prior to the reaction with [18F]TBAF. In 7 two quaternary centers forms a strained C(F2)–N bond, which is probably easier to cleave than the C(F2)–Br bond. The behaviour of DBU as a nucleophilic catalyst is known in the literature.14 However, this is the first study, when DBU is applied for mediating the displacement of bromide from CF2Br group and application for 18F labelling.
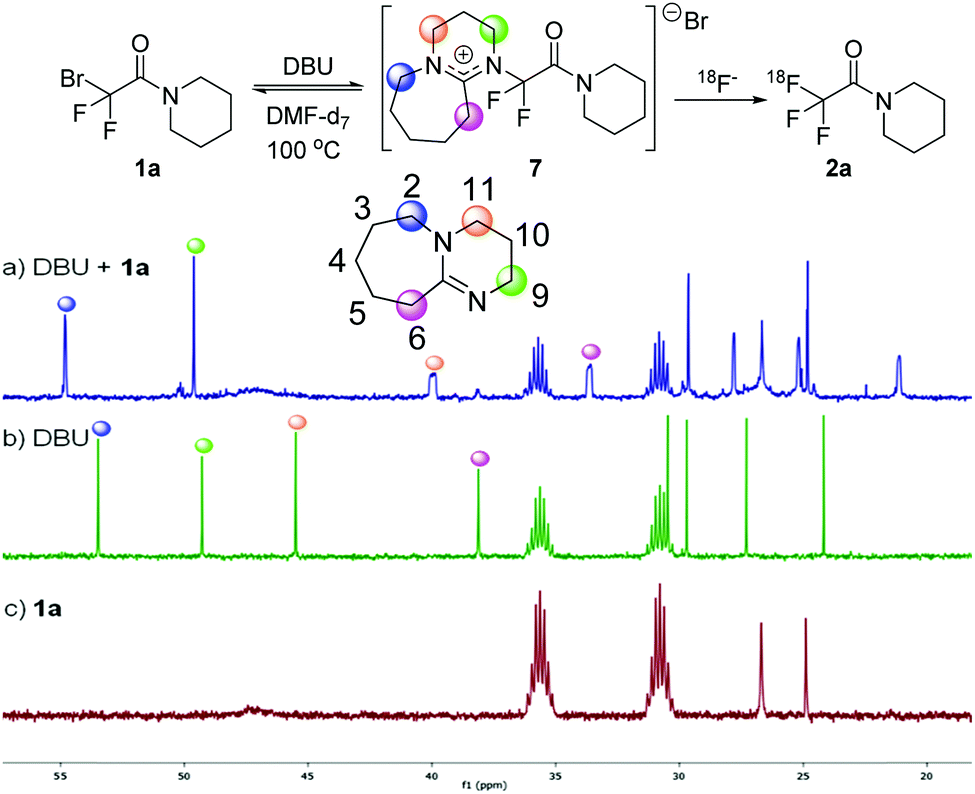 | ||
| Scheme 4 13C NMR studies for monitoring the formation of intermediate 7. 13C NMR spectra (125 MHz) at 100 °C in DMF-d7. (a) DBU + 1a. (b) DBU. (c) 1a. | ||
In summary, we have shown for the first time that 18F-labelled trifluoroacetamides can be efficiently synthesized from easily accessible CF2Br derivatives and [18F]TBAF in the presence of DBU (or analogues). The 18F-labelled tertiary trifluoroacetamide products are typically formed with high RCC. For 18F-labelling of secondary trifluoroacetamides, we propose a modified work-flow via synthesis of an 18F-labelled ester reagent, which undergo transesterification with the corresponding amine. The radiosynthetic utility of the method was demonstrated by 18F-labelling of the druglike substance [18F]2t (AZD5423). The preparative scale experiments show that this method is suitable to produce 18F-labelled trifluoroacetamides with sufficiently high specific activity for PET microdosing studies and with slight modification even for drug-target engagement studies (8.4 GBq μmol−1 for [18F]2a). Mechanistic studies suggest that DBU has an activating effect on the displacement of bromide from the CF2Br group of the precursor 1. This study broadens the radiosynthetic scope of [18F]CF3 derivatives and assists in the development of new bromide to 18F exchange methods where thermal activation or metal mediated transformations are unsuccessful.
The authors like to thank the members of the PET group at Karolinska Institutet. Support from AstraZeneca, Stockholm Brain Institute, the Swedish Research Council, the Knut and Alice Wallenberg Foundation are gratefully acknowledged. A. B. G. is grateful to the AstraZeneca Postdoctoral Programme for funding a postdoctoral contract.
Notes and references
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (b) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (c) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed; (d) H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637 CrossRef PubMed; (e) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (f) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed; (g) H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637 CrossRef PubMed.
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998 CrossRef CAS PubMed.
- (a) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (b) C. Alonso, E. Martínez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS PubMed; (c) W. Kong, E. Merino and C. Nevado, Chimia, 2014, 68, 430 CrossRef CAS PubMed; (d) J. R. Wolstenhulme and V. Gouverneur, Acc. Chem. Res., 2014, 47, 3560 CrossRef CAS PubMed; (e) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed.
- (a) K. Chansaenpak, B. Vabre and F. P. Gabbai, Chem. Soc. Rev., 2016, 45, 954 RSC; (b) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719 CrossRef CAS PubMed; (c) C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2015, 54, 3216 CrossRef CAS PubMed.
- M. Schou, K. Varnäs, S. Lundquist, R. Nakao, N. Amini, A. Takano, S. J. Finnema, C. Halldin and L. Farde, Int. J. Neuropsychopharmacol., 2015, 18, 10 Search PubMed.
- (a) K. Edman, R. Ahlgren, M. Bengtsson, H. Bladh, S. Bäckström, J. Dahmén, K. Henriksson, P. Hillertz, V. Hulikal, A. Jerre, L. Kinchin, C. Kåse, M. Lepistö, I. Mile, S. Nilsson, A. Smailagic, J. Taylor, A. Tjörnebo, L. Wissler and T. Hansson, Bioorg. Med. Chem. Lett., 2014, 24, 2571 CrossRef CAS PubMed; (b) G. M. Gauvreau, L.-P. Boulet, R. Leigh, D. W. Cockcroft, K. J. Killian, B. E. Davis, F. Deschesnes, R. M. Watson, V. Swystun, C. K. Mårdh, P. Wessman, C. Jorup, M. Aurivillius and P. M. O’Byrne, Am. J. Respir. Crit. Care Med., 2015, 191, 161 CrossRef PubMed.
- V. T. Lien and P. J. Riss, BioMed Res. Int., 2014, 2014, 10 Search PubMed.
- T. Rühl, W. Rafique, V. T. Lien and P. J. Riss, Chem. Commun., 2014, 50, 6056 RSC.
- M. Huiban, M. Tredwell, S. Mizuta, Z. Wan, X. Zhang, T. L. Collier, V. Gouverneur and J. Passchier, Nat. Chem., 2013, 5, 941 CrossRef CAS PubMed.
- D. van der Born, C. Sewing, J. D. M. Herscheid, A. D. Windhorst, R. V. A. Orru and D. J. Vugts, Angew. Chem., Int. Ed., 2014, 53, 11046 CrossRef CAS PubMed.
- (a) T. Khotavivattana, S. Verhoog, M. Tredwell, L. Pfeifer, S. Calderwood, K. Wheelhouse, T. Lee Collier and V. Gouverneur, Angew. Chem., Int. Ed., 2015, 54, 9991 CrossRef CAS PubMed; (b) S. Verhoog, L. Pfeifer, T. Khotavivattana, S. Calderwood, T. L. Collier, K. Wheelhouse, M. Tredwell and V. Gouverneur, Synlett, 2016, 25 CAS.
- P. Johnstrom and S. Stone-Elanderl, J. Labelled Compd. Radiopharm., 1995, 36, 537 CrossRef.
- P. Ivashkin, G. Lemonnier, J. Cousin, V. Grégoire, D. Labar, P. Jubault and X. Pannecoucke, Chem. – Eur. J., 2014, 20, 9514 CrossRef CAS PubMed.
- J. E. Taylor, S. D. Bull and J. M. J. Williams, Chem. Soc. Rev., 2012, 41, 2109 RSC.
- (a) H. Morimoto, R. Fujiwara, Y. Shimizu, K. Morisaki and T. Ohshima, Org. Lett., 2014, 16, 2018 CrossRef CAS PubMed; (b) L. Wang, X.-J. Wei, W.-L. Jia, J.-J. Zhong, L.-Z. Wu and Q. Liu, Org. Lett., 2014, 16, 5842 CrossRef CAS PubMed.
- (a) M. Berger, D. Jan, A. Eriksson, B. Gabos, T. Hanson, M. Hemmerling, K. Henriksson, S. Ivanova, M. Lepisto, D. McKerrecher, M. Munck af Rosenschold, S. Nilsson, H. Rehwinkel and C. Taflin, WO2008/076048, 2008; (b) T. Eriksson and T. Hansson, WO2010/008341A1, 2010.
- S. E. Lapi and M. J. Welch, Nucl. Med. Biol., 2013, 40, 314 CrossRef CAS PubMed.
- H. Shi, A. Braun, L. Wang, S. H. Liang, N. Vasdev and T. Ritter, Angew. Chem., Int. Ed., 2016, 55, 10786 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization, radiochemistry and spectral data. See DOI: 10.1039/c6cc08535k |
| This journal is © The Royal Society of Chemistry 2016 |