
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium catalyzed synthesis of difluoromethyl cyclopropanes†
Katharina J.
Hock
,
Lucas
Mertens
and
Rene M.
Koenigs
 *
*
RWTH Aachen University, Institute of Organic Chemistry, Landoltweg 1, D-52074 Aachen, Germany. E-mail: rene.koenigs@rwth-aachen.de
First published on 2nd November 2016
Abstract
Difluoromethyl-substituted cyclopropanes still remain one of the most challenging class of substrates. Despite significant progress in modern chemistry, an atom-economic and concise synthesis has not been described to date. Herein, we describe the first method for their catalytic, one-step synthesis using difluoromethyl diazomethane and a rhodium(II) catalyst.
In recent years, novel methods for the introduction of difluoromethyl groups have attracted the interest of organic chemists from all fields.1,2 Although the difluoromethyl group differs only in a single fluorine atom from the well-studied trifluoromethyl group, it possesses significantly altered properties making difluoromethylated compounds attractive synthetic targets. While trifluoromethyl groups are inert from a chemical and biological perspective, the difluoromethyl group possesses lower lipophilicity and increased polarity. Moreover, it is an alcohol or thiol bioisostere and can act as a hydrogen bond donor and is thus highly attractive to drug discovery programs.1b
Over the past decade, trifluoromethyl diazomethane found regular application in organic synthesis and led to a flourishing area of research.3,4 Interestingly, while trifluoromethyl diazomethane is known since 1943,5 the difluoromethyl-substituted analogue was first described only in 20156 and applications of this novel reagent in organic synthesis are still scarce.6,7 Against this background, we became interested in the catalytic synthesis of difluoromethyl-substituted cyclopropanes, which should be accessible via this newly introduced reagent.8
To date, there are only two reports claiming 1-difluoromethyl-2-aryl cyclopropanes, yet without describing the synthesis.9 The synthesis of difluoromethyl cyclopropanes in general has been studied only to a very limited extent.10 Leadbeater and co-workers and Hanamoto and co-workers reported the synthesis of this class of substances applying ring closure reactions using tedious multi-step syntheses.10a,b De Meijere and co-workers reported a multi-step synthesis of difluoromethyl cyclopropanes starting from cyclopropyl esters and by application of hazardous and highly reactive fluorinating reagents for the introduction of the difluoromethyl group.10c To the best of our knowledge no catalytic methods for the efficient one-step preparation from readily available starting materials has been described yet (Scheme 1).
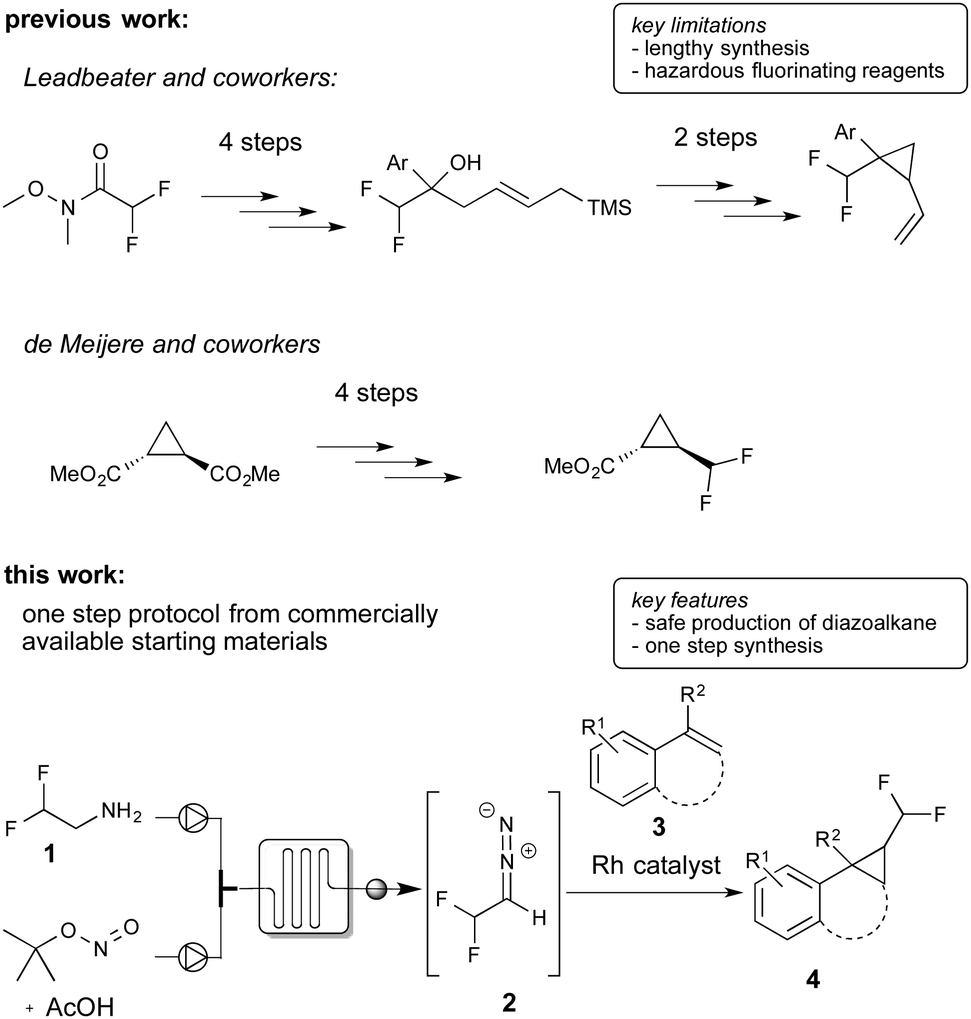 | ||
| Scheme 1 Synthetic approaches towards difluoromethyl cyclopropanes. | ||
Difluoromethyl-substituted cyclopropanes find few applications,9,11 such as in the ROR-gamma modulator (6)9a or as insecticides (7).9b,11b The most prominent compound containing a difluoromethyl cyclopropane is voxilaprevir (5), which is currently in phase II clinical trials for treatment of hepatitis C virus infections (Fig. 1).11c,d
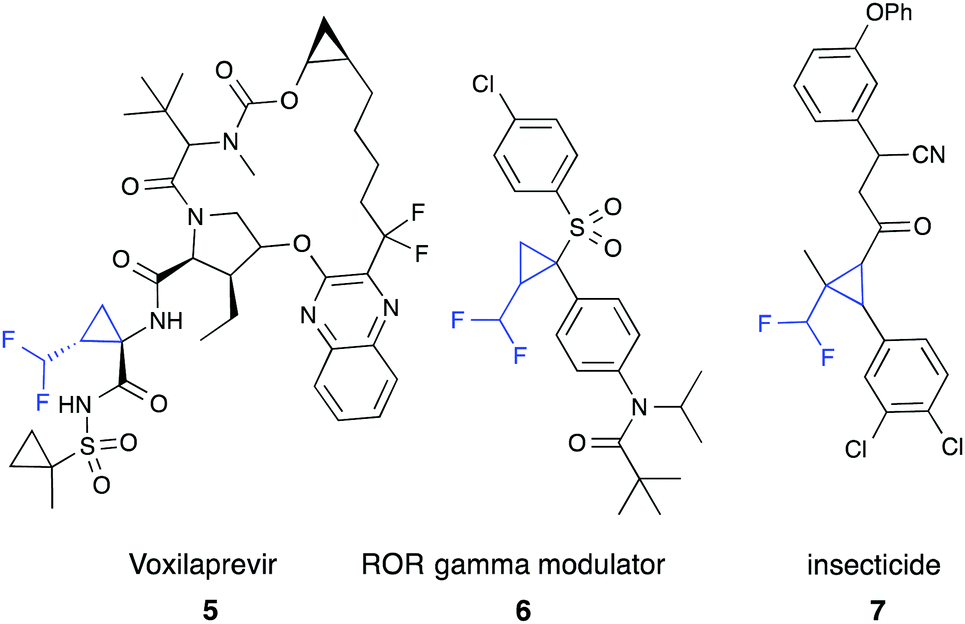 | ||
| Fig. 1 Difluoromethyl cyclopropanes in medicinal and agrochemical research. | ||
We started our investigations towards difluoromethyl-substituted cyclopropanes by evaluating different batch transformations. First, we investigated phase transfer protocols,4c,i that find regular application in reactions using trifluoromethyl diazomethane, yet the desired product could not be observed using different cyclopropanation catalysts. Similarly, preformation of difluoromethyl diazomethane and subsequent dropwise addition of the reagent to styrene and different cyclopropanation catalysts failed completely (Scheme 2). This observation is in sharp contrast to trifluoromethyl diazomethane and the corresponding cyclopropanation reactions.4
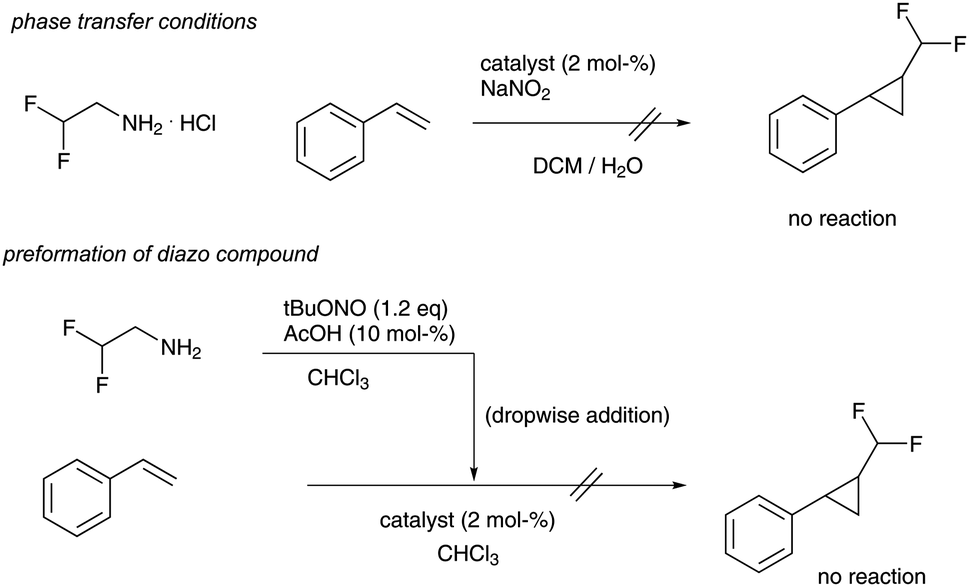 | ||
| Scheme 2 Attempts to difluoromethyl cyclopropane synthesis in batch reactions (catalysts investigated = Rh2OAc4, Ru(TPP)CO, Fe(TPP)Cl, Rh2Piv4). | ||
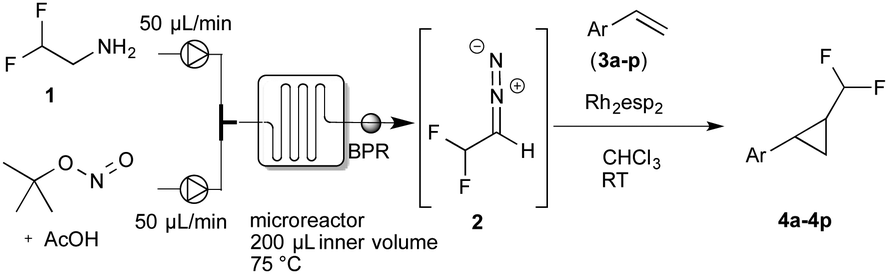
Recently, we reported on the continuous-flow synthesis of fluoroalkyl-substituted diazoalkanes and their application in pyrazole and pyrazoline synthesis.7,12,13 As conventional batch reactions did not provide the desired product, we became interested in evaluating the flow protocol in this cyclopropanation reaction.
For the continuous-flow generation of difluoromethyl diazomethane, we used a micromixer (Little Things Factory, MR-LAB Type MST, mixer volume: 200 μL) to mix a stream of difluoroethylamine with a mixture of tBuONO and AcOH. We then examined a range of different conditions, e.g. temperature, amount of acetic acid and residence time in the microreactor, by monitoring aliquots of the outlet by NMR spectroscopy. 40 mol% of acetic acid and a residence time of 2 minutes in the microreactor at 75 °C proved to yield the desired difluoromethyl diazomethane in about 40% yield. With the optimal conditions for the preparation of difluoromethyl diazomethane in hand, we next assessed the cyclopropanation reaction of styrene with difluoromethyl diazomethane, prepared in continuous-flow, using a broad variety of different cyclopropanation catalysts (Table 1).
|
|
||||
|---|---|---|---|---|
| Entrya | Catalyst | Loading (mol%) | Solvent | Yieldb (%) |
| a A solution of difluoroethylamine (0.2 M, 4 eq.) in CHCl3 and a mixture of AcOH (1.6 eq.) and tBuONO (0.24 M, 4.8 eq.) in CHCl3 were transferred by syringe pump into the microreactor (Little Things Factory, MR Lab, Type MST, inner volume 200 μL) at a constant flow rate of 50 μL min−1 for each syringe. The microreactor was heated to 75 °C and the outlet of the microreactor was connected to a back pressure regulator (40 psi). The reaction solution from the microreactor was added to a standard reaction flask at room temperature under Ar atmosphere containing styrene (0.4 mmol, 1 eq.) and the appropriate catalyst in 4 mL CHCl3. b Yields determined by 19F-NMR of the crude reaction mixture using α,α,α-trifluorotoluene as internal standard. c Isolated yield after column chromatography. d Reaction temperature of the cyclopropanation reaction 0 °C. | ||||
| 1 | Fe(TPP)Cl | 2 | CHCl3 | Traces |
| 2 | Ru(TPP)CO | 2 | CHCl3 | No rct. |
| 3 | Co(salen) | 2 | CHCl3 | No rct. |
| 4 | CuOTf | 2 | CHCl3 | No rct. |
| 5 | [Cp*IrCl2]2 | 2 | CHCl3 | No rct. |
| 6 | [Cp*RhCl2]2 | 2 | CHCl3 | No rct. |
| 7 | Pd(acac)2 | 2 | CHCl3 | No rct. |
| 8 | Rh2OAc4 | 2 | CHCl3 | 12 |
| 9 | Rh2Piv4 | 2 | CHCl3 | 6 |
| 10 | Rh2TPA4 | 2 | CHCl3 | No rct. |
| 11 | Rh2esp2 | 2 | CHCl3 | 24 |
| 12 | Rh2esp2 | 5 | CHCl3 | 67c |
| 13 | Rh2esp2 | 5 | CHCl3 | 13d |
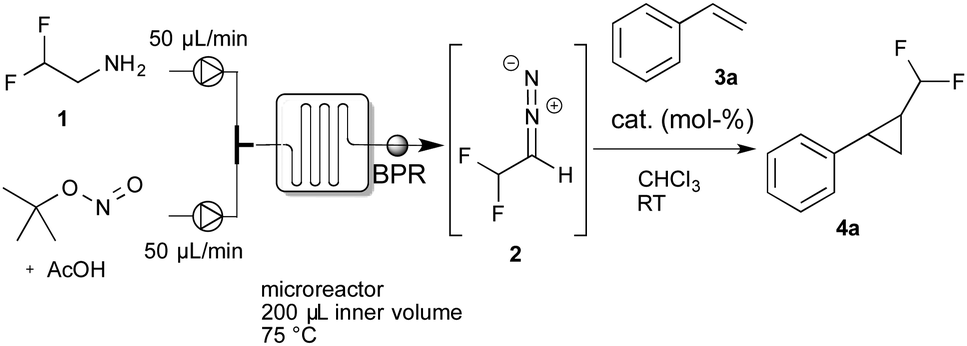
Initially, we examined iron and ruthenium porphyrine complexes, which were already demonstrated to be versatile catalysts for the preparation of trifluoromethyl substituted cyclopropanes. To our surprise, only trace amounts of the desired difluoromethyl-substituted cyclopropane could be observed using Fe[TPP]Cl. Other typical cyclopropanation catalysts, such as Ru[TPP]CO, Co[salen] complexes or Cu(I), or Ru(III) and Ir(III) complexes failed completely and no product was observed.
Interestingly, both Rh2OAc4 and Rh2Piv4 gave minor amounts of the desired cyclopropanation product, while Rh2TPA4 failed to yield the cyclopropane. These results are in sharp contrast to previously reported cyclopropanation reactions using trifluoromethyl diazomethane, which we assume to result from (a) the highly instable nature of difluoromethyl diazomethane and (b) from facile β-hydride elimination of the intermediate metal–carbene complex when using difluoromethyl diazomethane. We thus hypothesized, that sterically more demanding catalysts would reduce the rate of β-hydride elimination and result in higher yield of the desired cyclopropane.
To our delight, Rh2esp214 proved significantly better and we could observe increased yields of the cyclopropanation product. Encouraged by these results we increased the catalyst loading to 5 mol% and we could finally isolate the desired difluoromethylated cyclopropane 4a for the first time in 67% yield.
In further experiments we investigated the substrate scope of this transformation (Table 2). Using our continuous-flow protocol for the preparation of difluoromethyl diazomethane, we could show that a range of different styrenes can be converted to the corresponding difluoromethyl-substituted cyclopropanes in moderate to good yield. This transformation is particularly useful, as our newly developed protocol allows for the first time an efficient synthesis of difluoromethyl-substituted cyclopropanes in an operationally simple one-step protocol from cheap, commercially available olefins and amines.
|
|
|||
|---|---|---|---|
| Cyclopropanea | Yieldb (%) | Cyclopropanea | Yieldb (%) |
| a A solution of difluoroethylamine (0.2 M, 4 eq.) in CHCl3 and a mixture of AcOH (1.6 eq.) and tBuONO (0.24 M, 4.8 eq.) in CHCl3 were transferred by syringe pump into the microreactor (Little Things Factory, MR Lab, Type MST, inner volume 200 μL) at a constant flow rate of 50 μL min−1 for each syringe. The microreactor was heated to 75 °C and the outlet of the microreactor was connected to a back pressure regulator (40 psi). The reaction solution the microreactor was added to a standard reaction flask under Ar atmosphere containing olefine (0.4 mmol, 1 eq.) and Rh2esp2 (5 mol%) in 4 mL CHCl3. b Isolated yield after column chromatography. c Determined by NMR. | |||
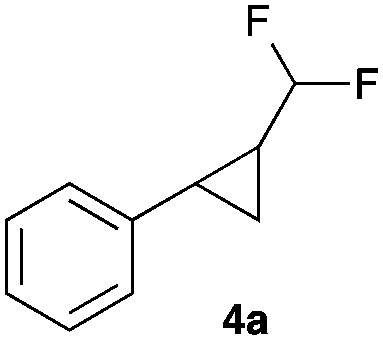
|
67 |
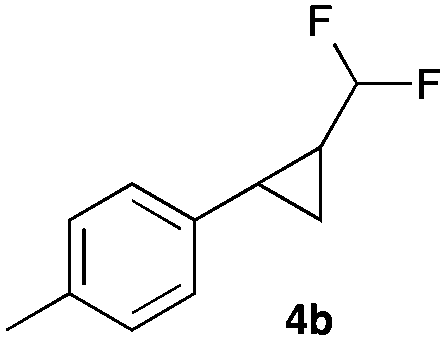
|
52 |
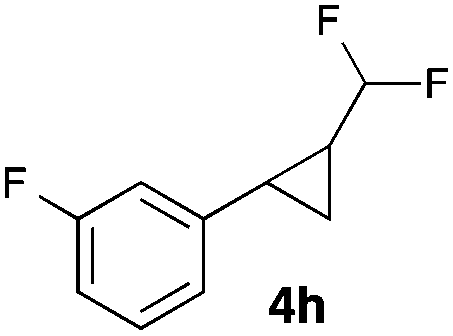
|
68 |
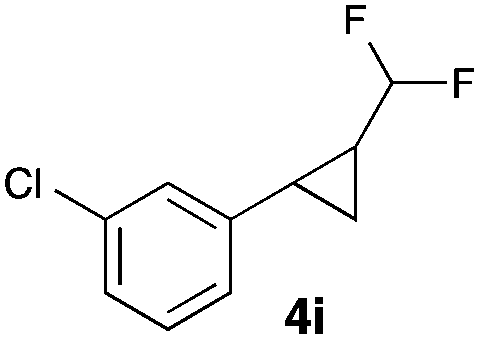
|
34 |
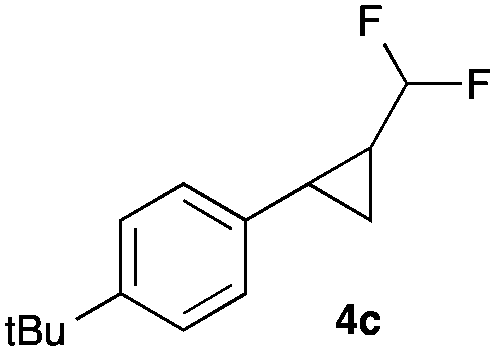
|
57 |
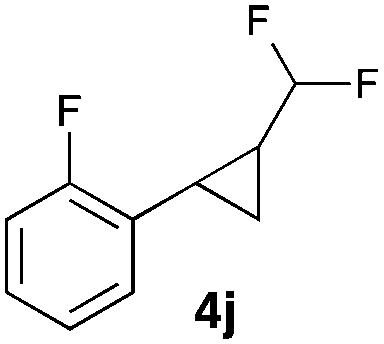
|
49 |
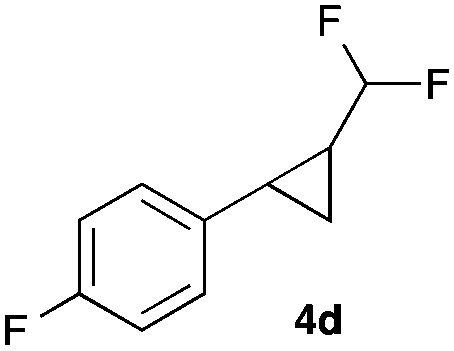
|
42 |
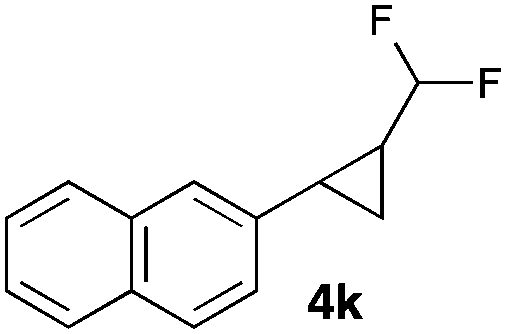
|
32 |
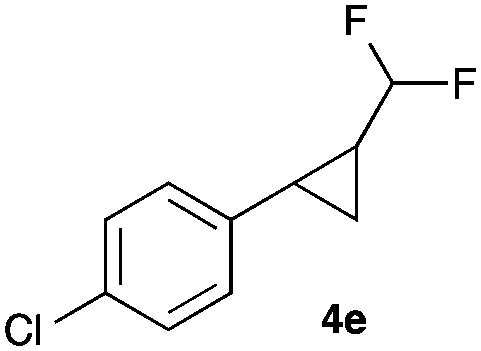
|
56 |
|
63 |
| 45 | |||
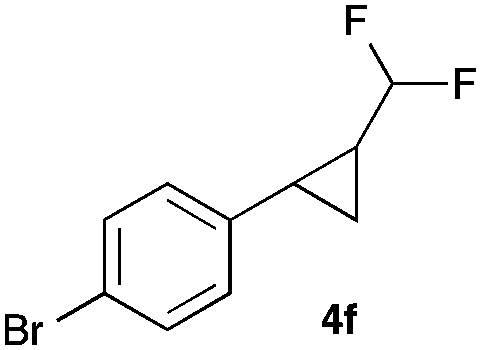
|
59 |
|
50 |
| 49 | |||
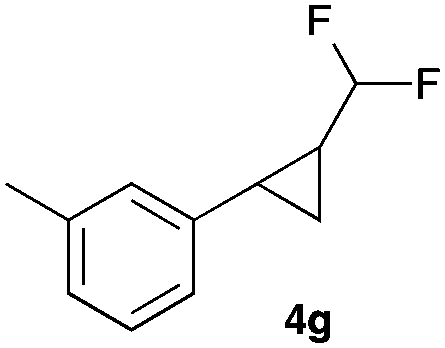
|
60 |
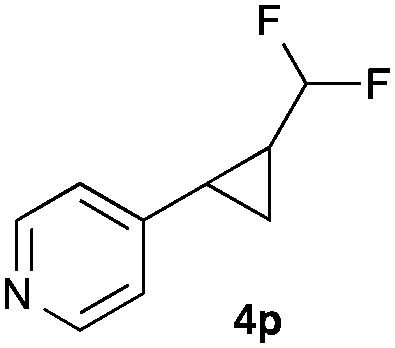
|
<10c |
In general electron donating and electron-withdrawing substituents are well tolerated and the corresponding cyclopropanes can be isolated in moderate to good yield. Moreover, different substitution patterns, such as meta-substituents are compatible under these reaction conditions and the desired reaction products can be obtained in good yield.
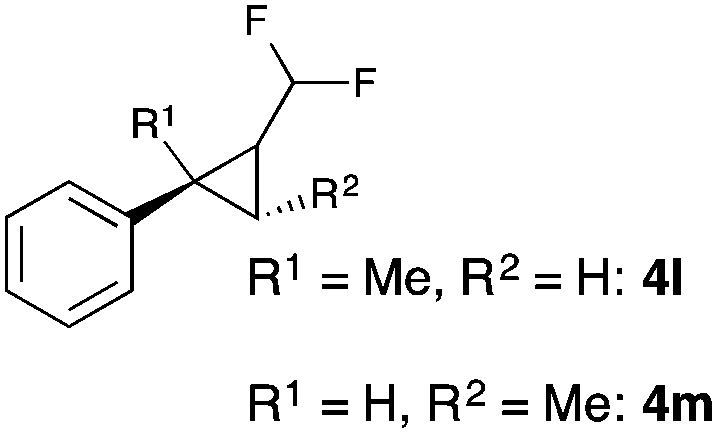
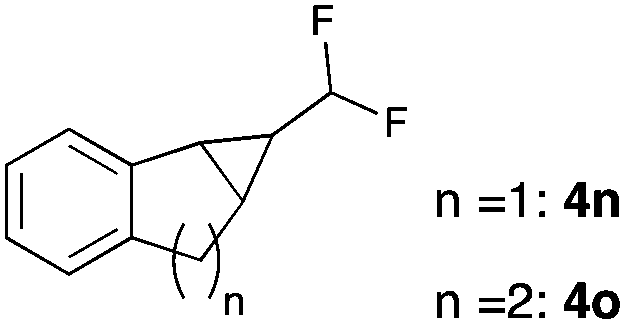
Further investigations concentrated on substituted styrenes. We probed α-methyl styrene and trans-β methyl styrene under these reaction conditions and were delighted to observe that the corresponding cyclopropanes (4l and 4m) can be isolated in good yield. Similarly, indene and dihydronaphthalene gave the desired cyclopropanation products (4n and 4o) in moderate yield. Heterocyclic compounds (4p) proved to give only unsatisfactory yield of the desired cyclopropane. Allylbenzene, as a non-aromatic olefinic substrate, did not provide the desired product. The diastereomeric ratio of all cyclopropanes obtained was in the range of 1.2–1.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Only in the case of 4o we observed a slightly higher diastereomeric ratio of 3:1.
:1. Only in the case of 4o we observed a slightly higher diastereomeric ratio of 3:1.
In summary, we have developed a new protocol that allows for the first time a catalytic one-step synthesis of difluoromethyl cyclopropanes. A key element for the development of this protocol is the efficient preparation of difluoromethyl diazomethane in continuous-flow. Subsequent application in rhodium(II) catalyzed cyclopropanation reactions provide for the first time difluoromethyl substituted cyclopropanes in a one-step protocol and good isolated yields.
Notes and references
- Selected references: (a) G. K. S. Prakash and J. Hu, Acc. Chem. Res., 2007, 40, 921 CrossRef CAS PubMed; (b) J. Hu, W. Zhang and F. Wang, Chem. Commun., 2009, 7465–7478 RSC; (c) J. Hu and C. Ni, Sci. Synth., C-1 Build. Blocks Org. Synth., 2014, 2, 409 CAS; (d) C. Ni, J. Liu, L. Zhang and J. Hu, Angew. Chem., Int. Ed., 2007, 46, 786 CrossRef CAS PubMed; (e) C. Ni and J. Ju, Chem. Soc. Rev., 2016, 45, 5441–5454 RSC.
- (a) D. Barnes-Seeman, J. Beck and C. Springer, Curr. Top. Med. Chem., 2014, 14, 855–864 CrossRef CAS PubMed; (b) Fluorine in Pharmaceutical and Medicinal Chemistry: From Biophysical Aspects to Clinical Applications, ed. V. Gouverneur and K. Müller, Imperial College Press, London, 2012 Search PubMed.
- Review on fluoroalkyl-substituted diazoalkanes: L. Mertens and R. M. Koenigs, Org. Biomol. Chem., 2016 10.1039/C6OB01618A.
- Selected references: (a) P. Le Maux, S. Juillard and G. Simonneaux, Synthesis, 2006, 1701 CAS; (b) P. K. Mykhailiuk, S. Afonin, G. V. Palamarchuk, O. V. Shishkin, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2008, 47, 5765 CrossRef CAS PubMed; (c) B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2010, 49, 938 CrossRef CAS PubMed; (d) B. Morandi, B. Mariampillai and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 1101 CrossRef CAS PubMed; (e) T.-R. Li, S.-W. Duan, W. Ding, Y.-Y. Liu, J. R. Chen, L.-Q. Lu and W.-J. Xiao, J. Org. Chem., 2014, 79, 2296–2302 CrossRef CAS PubMed; (f) B. Morandi, J. Cheang and E. M. Carreira, Org. Lett., 2011, 13, 3080 CrossRef CAS PubMed; (g) O. S. Artamonov, E. Y. Slobodyanyuk, D. M. Volochnyuk, I. V. Komarov, A. A. Tolmachev and P. K. Mykhailiuk, Eur. J. Org. Chem., 2014, 3592–3598 CrossRef CAS; (h) C.-L. Zhu, L.-J. Yang, S. Li, Y. Zheng and J.-A. Ma, Org. Lett., 2015, 17, 3442 CrossRef CAS PubMed; (i) E. Y. Slobodyanyuk, O. S. Artamonov, O. V. Shishkin and P. K. Mykhailiuk, Eur. J. Org. Chem., 2014, 2487 CrossRef CAS.
- H. Gilman and R. G. Jones, J. Am. Chem. Soc., 1943, 65, 1458–1460 CrossRef CAS.
- P. K. Mykhailiuk, Angew. Chem., Int. Ed., 2015, 54, 6558–6561 CrossRef CAS PubMed.
- L. Mertens, K. J. Hock and R. M. Koenigs, Chem. – Eur. J., 2016, 22, 9542 CrossRef CAS PubMed.
- Selected reviews: (a) H. Lebel, J. F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS PubMed; (b) J. Hansen and H. M. L. Davies, Coord. Chem. Rev., 2008, 252, 545 CrossRef CAS PubMed; (c) H. M. L. Davies and J. R. Denton, Chem. Soc. Rev., 2009, 38, 3061 RSC; (d) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS PubMed.
- Reports claiming 1-difluoromethyl-2-aryl cyclopropanes: (a) R. H. Hutchings, J. H. Jones, J. Chao, I. J. Enyedy and D. Marcotte, WO 2014028669, Biogen Idec Ma., 2014; (b) K. Yoshio, JP 57040440, 1982.
- Reports on the synthesis of difluoromethyl cyclopropanes: (a) C. B. Kelly, M. A. Mercadante, E. R. Carnaghan, M. J. Doherty, D. C. Fager, J. J. Hauck, A. E. MacInnis, L. J. Tilley and N. E. Leadbeater, Eur. J. Org. Chem., 2015, 4071 CrossRef CAS; (b) T. Ishikawa, N. Kasai, Y. Yamada and T. Hanamoto, Tetrahedron, 2015, 71, 1254 CrossRef CAS; (c) A. de Meijere, S. I. Kuzhushkov, D. S. Yufit, C. Grosse, M. Kaiser and V. A. Raev, Beilstein J. Org. Chem., 2014, 10, 2844 CrossRef PubMed.
- Selected references: (a) J. R. Allen, A. Amegadzie, M. P. Bourbeau, J. A. Brown, J. J. Chen, Y. Cheng, M. J. Frohn, A. Guzman-Perez, P. E. Harrington, L. Liu, Q. Liu, J. D. Low, V. V. Ma, J. Manning, A. E. Minatti, T. T. Nguyen, N. Nishmura, M. H. Norman, L. H. Pettus, A. J. Pickrell, W. Qian, S. Rumfelt, R. M. Rzasa, A. C. Siegmund, M. M. Stec, R. White and Q. Xue, WO 2016022724, Amgen, 2016; (b) T. Mori, K. Ujihara, O. Matsumoto, K. Yanagi and N. Matsuo, J. Fluorine Chem., 2007, 128, 1174 CrossRef CAS; (c) A. Cagulada, J. Chan, L. Chan, D. A. Colby, K. K. Karki, D. Kato, K. A. Keaton, S. Kondapally, C. Levins, A. Littke, R. Martinez, D. Pcion, T. Reynolds, B. Ross, M. Sangi, A. J. Schrier, P. Seng, D. Siegel, N. Shapiro, D. Tang, J. G. Taylor, J. Tripp, A. W. Waltman and L. Yu, US 20150175626, Gilead, 2015; (d) M. Rodriguez-Torres, S. Glass, J. Hill, B. Freilich, D. Hassman, A. M. Di Bisceglie, J. G. Taylor, B. J. Kirby, H. Dvory-Sobol, J. C. Yang, D. An, L. M. Stamm, D. M. Brainard, S. Kim, D. Krefetz, W. Smith, T. Marbury and E. Lawitz, J. Viral Hepatitis, 2016, 23, 614 CrossRef CAS PubMed.
- Selected review articles: (a) S. T. R. Mueller and T. Wirth, ChemSusChem, 2015, 8, 245 CrossRef CAS PubMed; (b) R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502 CrossRef CAS PubMed; (c) J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583–4592 RSC; (d) S. V. Ley, Chem. Rev., 2012, 12, 378 CAS; (e) C. Wiles and P. Watts, Green Chem., 2012, 14, 38 RSC; (f) T. H. Rehm, Chem. Eng. Technol., 2016, 39(1), 66 CrossRef; (g) B. J. Deadman, S. G. Collins and A. R. Maguire, Chem. – Eur. J., 2015, 21, 2298 CrossRef CAS PubMed; (h) M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892 RSC.
- Fluoroalkyl substituted diazoalkanes in flow chemistry: B. Pieber and C. O. Kappe, Org. Lett., 2016, 18, 1076 CrossRef CAS PubMed.
- C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc07745e |
| This journal is © The Royal Society of Chemistry 2016 |