
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chloromethyl-triazole: a new motif for site-selective pseudo-acylation of proteins†
Richard C.
Brewster
a,
Georgina C.
Gavins
a,
Barbara
Günthardt
a,
Sarah
Farr
a,
Kimberly M.
Webb
b,
Philipp
Voigt
 b and
Alison N.
Hulme
*a
b and
Alison N.
Hulme
*a
aEaSTCHEM School of Chemistry, The University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh EH9 3FJ, UK. E-mail: Alison.Hulme@ed.ac.uk
bThe Wellcome Trust Centre for Cell Biology, The University of Edinburgh, Michael Swann Building, Max Born Crescent, Edinburgh, EH9 3BF, UK
First published on 13th September 2016
Abstract
Rapid, site-selective modification of cysteine residues with chloromethyl-triazole derivatives generates pseudo-acyl sLys motifs, mimicking important post-translational modifications. Near-native biotinylation of peptide and protein substrates is shown to be site-selective and modified histone H4 retains functional activity.
Advances in techniques for the site-selective functionalization of proteins have revolutionised approaches to investigating the effects of post-translational modifications (PTMs) in recent years.1 Cysteine is an attractive target for chemical modification due to its relatively low abundance in proteins and the high reactivity of the sulfhydryl group compared to other amino acid side chain functional groups which ensures good selectivity.2 Indeed, in proteins such as histones where there are almost no native cysteine residues, a popular route to the systematic investigation of lysine modifications has been the selective modification of cysteine mutants to give near-native analogues of the parent PTM.3 This has been achieved either through the formation of a cleavable disulfide,4 or through S-alkylation to give thia-lysine derivatives (sLys),5 in which the only perturbation between the native lysine-containing histone and its cysteine-containing analogue is the switch of a CH2 for S. A range of amide derivatives of the side-chain of lysine are known as PTMs,6 with one example being the ‘native’ or endogenous biotinylation of proteins. The epigenetic role of biotin as a PTM of histones has been investigated recently.7 However, to date these studies have been hindered by the lack of a suitable reagent to chemically modify proteins with a near-native mimic of post-translational biotinylation, causing their conclusions to be questioned.8
sLys derivatives mimicking the different methylation states of lysine have been generated through the reaction of methylated chloro- or bromo-ethylamines.5c Shokat et al. have shown that the alkylation reaction proceeds via an intermediate aziridine/aziridinium, which is subsequently ring-opened by the nucleophilic cysteine.5c In contrast, in investigating the alkylation of cysteine to create acetyl lysine mimics, Cole et al. have shown that alkyl halide 1a is virtually unreactive (Scheme 1).5b Similarly, attempted ring-opening of a pre-formed acyl aziridine (not shown) has been shown to result in acyl transfer to the cysteine rather than formation of the acylated sLys derivative.5b An alternative strategy, employing a thiol-ene reaction of N-vinyl acetamide 2a has been used to generate acetyl lysine mimics on histones.5a However, our attempted synthesis of the vinyl amide of biotin 2b (either directly, or from bromo-ethyl derivative 1b) was unsuccessful. We were thus drawn to an alternative approach in which a triazole mimic of the amide bond would be targeted,9 and the enhanced reactivity of the ‘pseudo-benzylic’ halide α to the triazole would give an efficient, high yielding alkylating reagent (3b, Scheme 1).
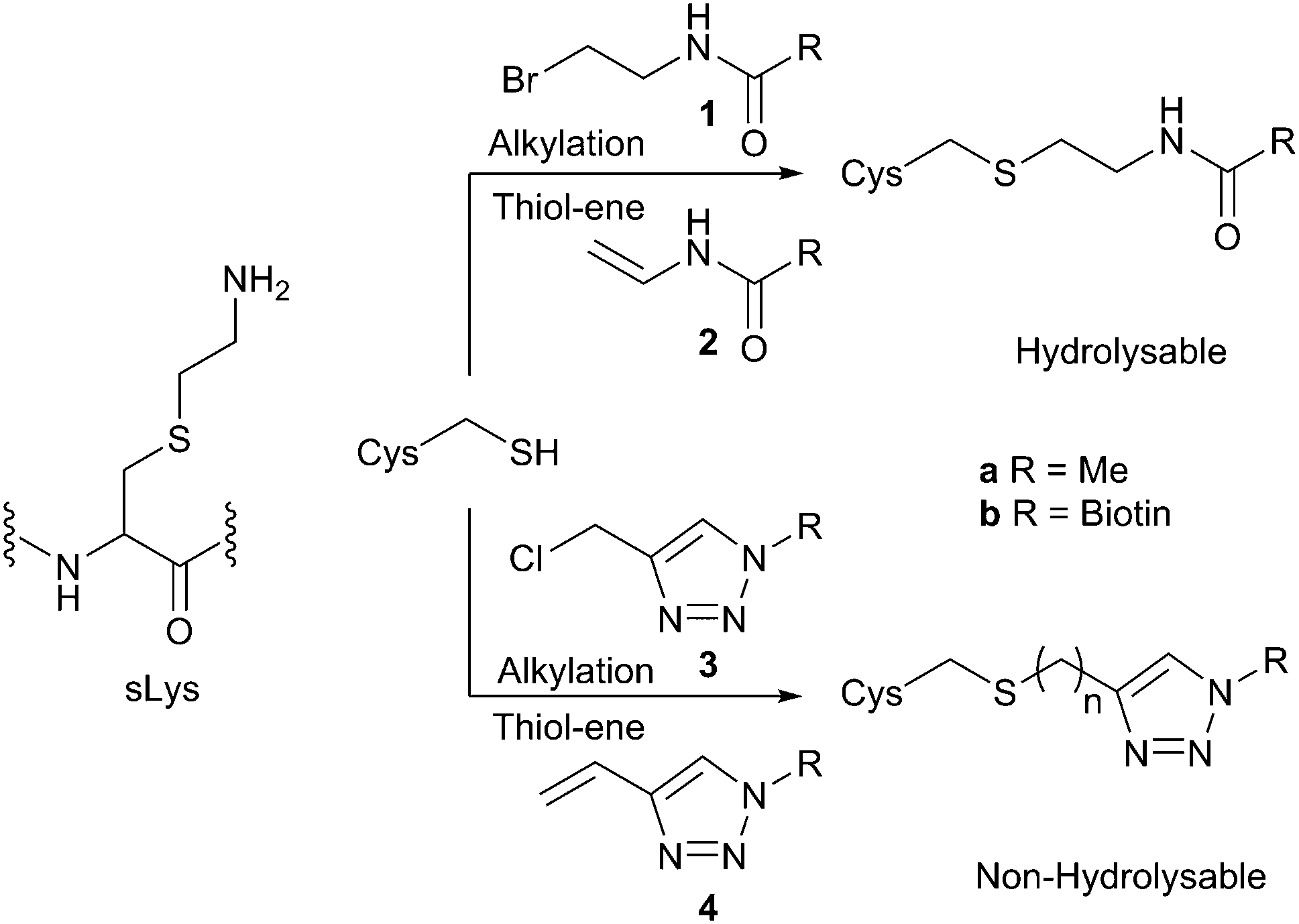 | ||
| Scheme 1 Routes to the formation of acyl, or pseudo-acyl sLys derivatives. | ||
Synthesis of norbiotin azide 5 (Scheme 2) was achieved by a Curtius rearrangement of the carboxylic acid side chain on biotin 6,10 followed by Boc deprotection and a diazo transfer reaction on the amine hydrochloride 7, using imidazole-1-sulfonyl azide hydrochloride 8.11 The CuAAC click reaction of azide 5 with homopropargyl bromide proceeded well under standard coupling conditions (65%).‡ However, in reactions with propargyl chloride and propargyl bromide only the azide starting material 5 was recovered under a range of CuAAC conditions. Gratifyingly, the desired product (which possesses both the optimum length of linkage for a near-native connection and enhanced reactivity) could be obtained by a CuAAC reaction of 5 with propargyl alcohol, and subsequent conversion to the chloromethyl-triazole 3b using SOCl2. An alternative chlorination protocol,12 which employed treatment of the pseudo-benzylic intermediate alcohol with tosyl chloride gave chloromethyl-triazole 3b in reduced yields. Attempts to synthesise the corresponding bromide from the intermediate alcohol using SOBr2 were very poor yielding, and the use of other brominating reagents was hindered by the poor solubility of the intermediate hydroxymethyl-triazole in organic solvents.
 | ||
| Scheme 2 Synthesis of biotin alkylation reagent 3b. (a) DPPA. TEA, tBuOH, reflux, 18 h, 73%; (b) AcCl, EtOH, CHCl3, 4 h, 93%; (c) 8, K2CO3, CuSO4·5H2O, MeOH, 75%; (d) propargyl alcohol, Cu(MeCN)4BF4, DMF, 20 h, 80%; (e) SOCl2, 0.5 h, 88%. | ||
To probe the selectivity and reactivity of the new chloromethyl-triazole reagent, a series of peptide models was used. Monitoring the reaction by HPLC showed that alkylation of the cysteine in a short peptide sequence (Pep1: naphthalene-mPEG-GACR-OH) with 3b under buffered conditions was complete in under 6 hours [Pep1 (5.0 mM), 3b (100 mM), DTT (20 mM), HEPES (pH 8.0), rt];§ no double alkylation product was detected by LC-MS (Fig. S1, ESI†). Using the same reaction conditions, further peptide sequences incorporating lysine and histidine residues (Pep2: FITC-βAla-GKAACF-NH2, Pep3: FITC-βAla-HGKAACF-NH2) showed that alkylation by 3b was cysteine-selective (Fig. S2, ESI†). The rate of alkylation of Pep1 by chloromethyl-triazole 3b was then compared with that of other methylated chloro- (9a−c), or bromo-ethylamines (10a) typically used to create sLys methyl lysine mimics (Fig. 1).3a As expected, the rate of reaction was observed to increase with increasing methylation of the amine (9c > 9b > 9a),5c and the bromoethylamine 10a was shown to react significantly faster than its chloroethylamine equivalent 9a. However, chloromethyl-triazole 3b reacts significantly faster than any of these reagents, with conversion levels of ∼80% reached in under 3 h. Whilst this rate of reaction is not as fast as that of a maleimide reagent (which is essentially instantaneous under equivalent conditions), it does provide a synthetically viable route for the selective and efficient alkylation of cysteine residues to give pseudo-acyl sLys derivatives.
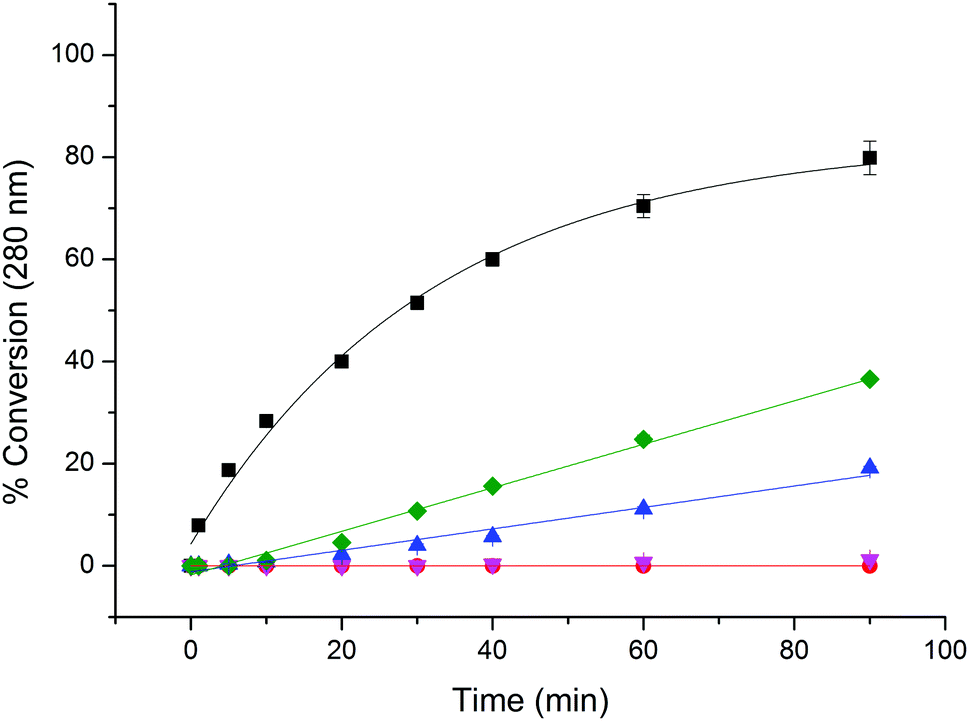 | ||
| Fig. 1 Relative rates of reaction of Pep1 (naphthalene-mPEG-GACR-OH) with 2-chloroethylamine (9a, red), N-methyl 2-chloroethylamine (9b, magenta), N,N-dimethyl 2-chloroethylamine (9c, green), 2-bromoethylamine (10a, blue), and chloromethyl-triazole biotin (3b, black). Reactions conducted in the presence of 20 eq. alkylating reagent, 1 M HEPES pH 8, 5 eq. DTT, rt. Reactions were monitored by RP-HPLC (280 nm), and % conversion data represent the mean from 3 independent experiments. Error bars: ± standard deviation. | ||
Methylation of lysine residues in histones is a well-documented epigenetic modification,13 and detailed protocols for the synthesis of near-native sLys analogues from the corresponding KxxC mutant histones have been published.5 Biotinylation of histone H4 (H4K12(bio)14 and H4K16(bio)15) has also been proposed to play an epigenetic role; with H4K16bio shown to affect chromatin condensation levels in studies conducted using non-native maleimide-PEG2-biotin reagents. Gratifyingly, treatment of the histone 4 mutant H4K12C with chloromethyl triazole 3b, under standard histone alkylation conditions [protein (0.9 mM), 3b (90 mM), DTT (20 mM), HEPES (pH 7.8), guanidine (4 M), rt]16 proceeded to completion in only 4 h. Purification using a size exclusion spin cartridge to desalt and remove excess alkylating reagent, and mass spectrometric analysis showed only the sLys biotin alkylated histone accompanied by very low levels of a double alkylation product (Fig. 2a). We have previously demonstrated that a range of non-hydrolysable triazole derivatives of biotin show very strong binding to avidin, with Kd values in the pM range.17 In this instance, functional activity of the H4K12C triazole biotin adduct was demonstrated by Western blot using an anti-biotin antibody (Fig. 2b), and the assembly of histone complexes incorporating site-selective biotinylation is currently under investigation.
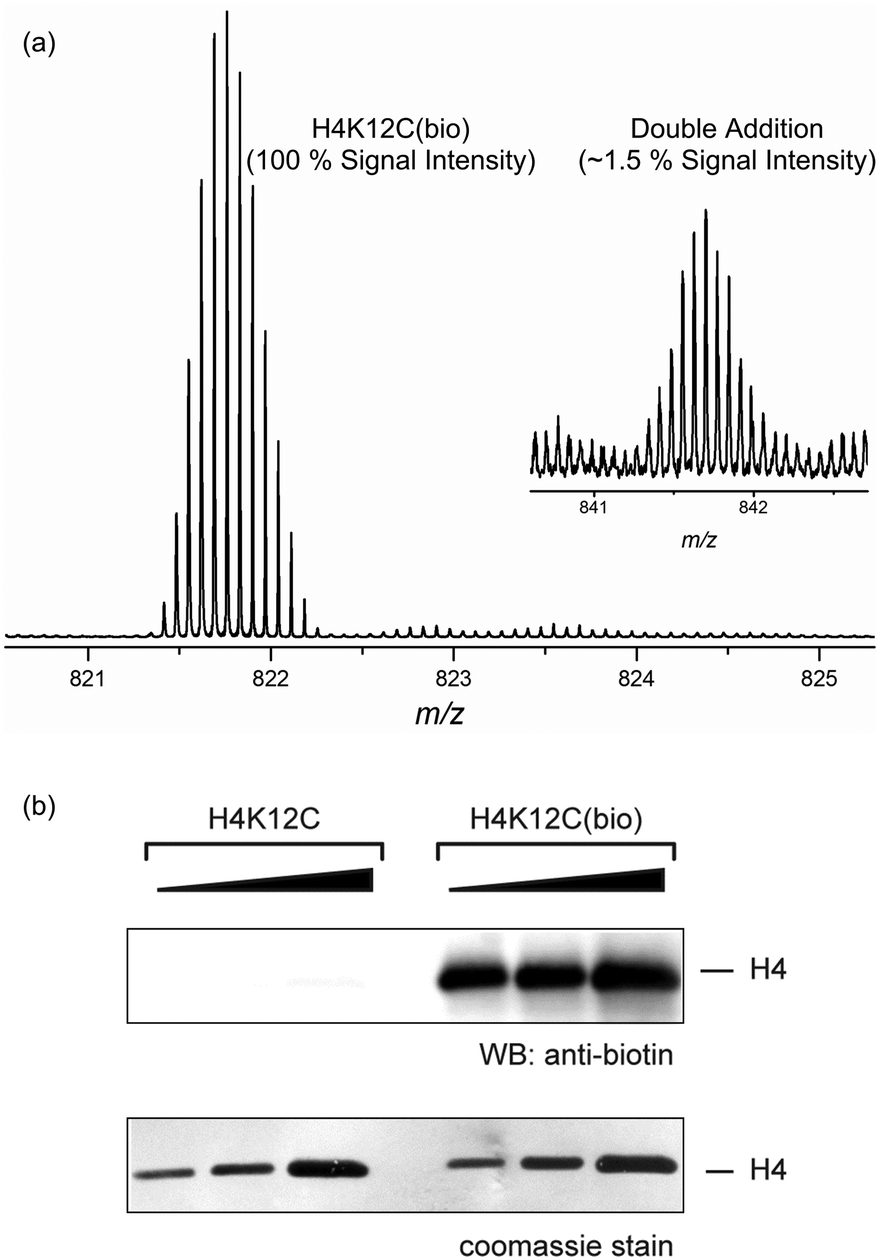 | ||
| Fig. 2 (a) Mass spectrometric analysis of the product of reaction of H4K12C with chloromethyl-triazole biotin reagent 3b. (b) Western blot of the sLys biotin derivative of H4K12C using anti-biotin (Bethyl). | ||
In summary, we have demonstrated that a chloromethyl-triazole motif can be used to introduce site selective, near-native mimics of amide-based PTMs into peptide and protein substrates. Using biotin chloromethyl-triazole 3b as an example, the rate of cysteine alkylation was shown to be faster than that of commonly used N-methylated 2-haloethylamines; and functional activity of the resultant pseudo-acyl derivatives was confirmed by Western blot with anti-biotin. Due to the wide range of easily accessible functional azides and simple conversion to the corresponding chloromethyl-triazole, this motif could serve as a robust method for the rapid installation of PTM acetylation mimics into proteins. This chemical derivatisation approach complements acetylation techniques which rely on the genetic introduction of unnatural amino acids,18 which can be hampered by low protein expression levels. Ongoing efforts in our laboratories seek to expand this strategy to sugars, phosphates and fatty acids, and to exploit the high reactivity observed for chloromethyl-triazole based reagents in dual labelling studies.
This work was supported by the BBSRC (Grant Ref. BB/J01446X/1, EASTBIO studentship to RCB), the Wellcome Trust and the Royal Society (joint Grant Ref. 104175/Z/14/Z, Sir Henry Dale Fellowship to PV) and the Wellcome Trust through core funding to The Wellcome Trust Centre for Cell Biology (Grant Ref. 092076).
Notes and references
- (a) T. H. Wright, M. R. J. Vallée and B. G. Davis, Angew. Chem., Int. Ed., 2016, 55, 5896 CrossRef CAS PubMed; (b) A. Maruani, D. A. Richards and V. Chudasama, Org. Biomol. Chem., 2016, 14, 6165 RSC; (c) O. Boutureira and G. L. J. Bernardes, Chem. Rev., 2015, 115, 217 CrossRef PubMed; (d) K. Lang and J. W. Chin, ACS Chem. Biol., 2014, 9, 16 CrossRef CAS PubMed.
- (a) S. Gunnoo and A. Madder, ChemBioChem, 2016, 17, 529 CrossRef CAS PubMed; (b) J. Willwacher, R. Raj, S. Mohammed and B. Davis, J. Am. Chem. Soc., 2016, 138, 867 CrossRef PubMed; (c) O. Konievab and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495 RSC; (d) J. M. Chalker, G. J. L. Bernades, Y. A. Lin and B. G. Davis, Chem. – Asian J., 2009, 4, 630 CrossRef CAS PubMed.
- (a) M. M. Müller and T. W. Muir, Chem. Rev., 2015, 115, 2296 CrossRef PubMed; (b) P. Voigt and D. Reinberg, ChemBioChem, 2011, 12, 236 CrossRef CAS PubMed.
- C. Chatterjee, R. K. McGinty, B. Fierz and T. W. Muir, Nat. Chem. Biol., 2010, 6, 267 CrossRef CAS PubMed.
- (a) F. Li, A. Allahverdi, R. Yang, G. B. J. Lua, X. Zhang, Y. Cao, N. Korolev, L. Nardenskiöld and C. Liu, Angew. Chem., Int. Ed., 2011, 50, 9611 CrossRef CAS PubMed; (b) R. Huang, M. A. Holbert, M. K. Tarrant, S. Curtet, D. R. Colquhoun, B. M. Dancy, B. C. Dancy, Y. Hwang, Y. Tang, K. Meeth, R. Marmorstein, R. N. Cole, S. Kochbin and P. A. Cole, J. Am. Chem. Soc., 2010, 132, 9986 CrossRef CAS PubMed; (c) M. D. Simon, F. Chu, L. R. Racki, C. C. de la Cruz, A. L. Burlingame, B. Panning, G. J. Narlikar and K. M. Shokat, Cell, 2007, 128, 1003 CrossRef CAS PubMed.
- H. Lin, X. Su and B. He, ACS Chem. Biol., 2012, 7, 947 CrossRef CAS PubMed.
- T. Kuroishi, L. Rios-Avila, V. Pestinger, S. S. K. Wijeratne and J. Zempleni, Mol. Genet. Metab., 2011, 104, 537 CrossRef CAS PubMed.
- S. Healy, B. Perez-Cadahia, D. Jia, M. K. McDonald, J. R. Davie and R. A. Gravel, Biochim. Biophys. Acta, Gene Regul. Mech., 2009, 1789, 719 CrossRef CAS PubMed.
- H. C. Kolb and B. K. Sharpless, Drug Discovery Today, 2003, 8, 1128 CrossRef CAS PubMed.
- (a) T. P. Soares da Costa, W. Tieu, M. Y. Yap, O. Zvarec, J. M. Bell, J. D. Turnidge, J. C. Wallace, G. W. Booker, M. C. J. Wilce, A. D. Abell and S. W. Polyak, ACS Med. Chem. Lett., 2012, 3, 509 CrossRef CAS PubMed; (b) W. Szalecki, Bioconjugate Chem., 1996, 7, 271 CrossRef CAS PubMed.
- E. D. Goddard-Borger and R. V. Stick, Org. Lett., 2007, 9, 3797 CrossRef CAS PubMed.
- R. Ding, Y. He, X. Wang, J. Xu, Y. Chen, M. Feng and C. Qi, Molecules, 2011, 16, 5665 CrossRef CAS PubMed.
- (a) A. J. Bannister and T. Kouzarides, Cell Res., 2011, 21, 381 CrossRef CAS PubMed; (b) E. I. Campos and D. Reinberg, Annu. Rev. Genet., 2009, 43, 559 CrossRef CAS PubMed; (c) C. Martin and Y. Zhang, Nat. Rev. Mol. Cell Biol., 2005, 6, 838 CrossRef CAS PubMed.
- Y. I. Hassan and J. Zempleni, J. Nutr., 2006, 136, 1763 CAS.
- M. P. Singh, S. S. K. Wijeratne and J. Zempleni, Arch. Biochem. Biophys., 2012, 529, 105 CrossRef PubMed.
- M. D. Simon, in Current Protocols in Molecular Biology, ed. F. M. Ausubel, et al., John Wiley & Sons, Hoboken NJ, 2010, Unit 21.18.1–21.18.10 Search PubMed.
- A. I. Germeroth, J. R. Hanna, R. Karim, F. Kundel, J. Lowther, P. G. N. Neate, E. A. Blackburn, M. A. Wear, D. J. Campopiano and A. N. Hulme, Org. Biomol. Chem., 2013, 11, 7700 CAS.
- E. Arbely, E. Natan, T. Brandt, M. D. Allen, D. B. Veprintsev, C. V. Robinson, J. W. Chin, A. C. Joerger and A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8251 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterisation of compounds 3b, 4b, and peptides Pep1–3. Peptide and protein alkylation procedures. See DOI: 10.1039/c6cc06801d. Primary data files can be found at http://dx.doi.org/10.7488/ds/1484 |
| ‡ CuAAC coupling conditions as in Scheme 2(d). The triazole adduct resulting from CuAAC click reaction of azide 5 with homopropargyl bromide was readily converted by elimination to the vinyltriazole biotin reagent 4b (NaOH, EtOH, rt, 3 h, 79%). However, subsequent thiol-ene reaction with Pep1 resulted in only moderate to poor conversion (<30%) under a range of conditions. |
| § Minor modification of standard histone conditions, see: ref. 5c. |
| This journal is © The Royal Society of Chemistry 2016 |