
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ion mobility separation of deprotonated oligosaccharide isomers – evidence for gas-phase charge migration†
W. B.
Struwe‡
*a,
C.
Baldauf
*b,
J.
Hofmann
b,
P. M.
Rudd
a and
K.
Pagel
*bc
aNational Institute of Bioprocessing, Research and Training (NIBRT), Fosters Avenue, Dublin, Ireland. E-mail: weston.struwe@bioch.ox.ac.uk
bFritz Haber Institute of the Max Planck Society, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: baldauf@fhi-berlin.mpg.de
cInstitut für Chemie und Biochemie der Freien Universität Berlin, Takustr. 3, 14195 Berlin, Germany. E-mail: kevin.pagel@fu-berlin.de
First published on 27th September 2016
Abstract
There has been increasing evidence that certain isomeric glycans can be separated efficiently by ion mobility-mass spectrometry when deprotonated ions are analyzed. To better understand the fundamentals behind these separations, we here investigate the impact of ionisation mode and adduct formation using IM-MS, density-functional theory and ab initio molecular dynamics.
Carbohydrates constitute a major class of macromolecules and play a critical role in cellular signalling, biomolecular interactions, protein structure and function.1 These “glycans” are complex biomolecules composed of many different monosaccharides that are arranged in elaborate structures. In contrast to biopolymers with a linear sequence such as DNA and proteins, carbohydrate building blocks can be connected by various linkages resulting in branched structures with a complex stereochemistry. The range of glycan structures is considerable and even subtle differences can modulate protein function. Robust analytical strategies are essential to delineate glycan structure–function relationships and the need for rapid screening methods is particularly important in biotherapeutic drug design.2 Considerable efforts have been made in developing techniques to routinely characterize individual structures in complex glycan mixtures with the majority being based on HPLC and LC-MS methodologies.3–5 HPLC is desirable owing to its sensitivity and high-throughput capabilities, while LC-MS is information-rich providing retention time, precursor mass and fragmentation spectra that together can distinguish structural isomers. There are, however, intrinsic limits to both strategies mostly determined by the resolution and reproducibility of the separation or the lack of diagnostic fragments. The combination of ion mobility (IM) and mass spectrometry (MS) represents a relatively new tool for glycoconjugate analysis, but a fundamental understanding of how gas-phase glycan ions behave remains and is vital to fully appreciate the value of IM-MS for glycomics.
IM separates ions based on the time required to traverse a region of inert neutral gas under the influence of a weak electric field. Separation is based on the physical size, shape, and charge of the analyte and, in conjunction with MS, reports a drift time and m/z value. Drift times can be used to calculate a collision cross section (CCS), which is an intrinsic value reflecting the rotationally averaged structure of the ion and is unique for each molecule. Theoretical CCS values can be determined using computational methods,6–9 but there are limited examples for oligosaccharides. Previous IM-MS reports for glycan analysis were predominantly focused on ions containing metal cation10–15 and/or anion adducts16,17 The CCSs of such glycan adducts have been shown to be diagnostic in many cases, but the differences are not always sufficient to distinguish isomers in complex mixtures. We recently showed that separation of deprotonated synthetic trisaccharide isomers is possible by IM-MS,18 but the basis for the exceptional separation of [M − H]− ions remains unknown. Here we systematically evaluate the impact of ionisation mode and adduct formation on the gas-phase structure of glycans by combining IM-MS and first-principles theory.
Our investigation is focused on the two human milk oligosaccharide (HMO) isomers lacto-N-hexaose (LNH) and lacto-N-neo-hexaose (LNnH). Both structures consist of the same six monosaccharides and differ only in the regiochemistry at the lower antenna, with the last galactose (Gal) residue linked to the penultimate N-acetylglucosamine (GlcNAc) via a β1,3 glycosidic bond in LNH and a β1,4 glycosidic bond in LNnH (Table 1). Such a minor structural difference makes it very difficult to separate and distinguish these isomers using established chromatographic techniques. The HPLC retention times of LNH and LNnH are identical when analysed separately (Fig. S1, ESI†). Similarly, tandem MS yield identical fragmentation patterns in both positive (Fig. S2, ESI†) and negative mode (Fig. S3, ESI†).
| Name/structurea | CCSexp | PACCScalcb | ||||
|---|---|---|---|---|---|---|
| [M + Na]+ | [M − H]− | [M + Na]+ | M | [M − H]− | ||
| a Chemical structures are depicted as cartoons: yellow circles, galactose; blue circles, glucose; blue squares, N-acetylglucoseamine; horizontal lines, β1,4 bonds; upwards diagonals, β1,6 bonds; downwards diagonals, β1,3 bonds; the reducing end is located on the right. b Theoretical CCS values were calculated for the lowest potential-energy structures using the PA method.6,7 | ||||||
| LNH |
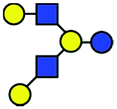
|
232.6 ± 0.2 | 255.2 ± 1.6 | 224 | 250 | 233 |
| LNnH |
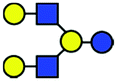
|
224.9 ± 0.2 | 227.6 ± 1.6 | 227 | 222 | 225 |
| LNT |
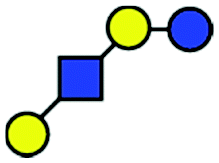
|
170.5 ± 1.0 | 172.2 ± 1.0 | 185 | 177 | 177 |
| LNnT |
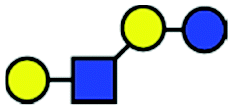
|
169.6 ± 1.0 | 186.3 ± 0.8 | 173 | 189 | 168 |
The gas-phase separation of sodiated, protonated, chlorinated and deprotonated oligosaccharide ions was examined using traveling-wave (TW) IM-MS with nitrogen as drift gas. We first analysed an equimolar mixture of synthetic LNH and LNnH oligosaccharides (Fig. 1a) and recorded their arrival time distributions (ATDs). The ATDs of the [M + Na]+ ions (m/z 1095) of LNH and LNnH are virtually identical (6.11 ms) and no separation can be observed. In contrast the extracted ATDs of the deprotonated LNH and LNnH ions (m/z 1071) showed significantly different drift times and were baseline separated. Compared to LNnH (6.06 ms), the drift time of the LNH [M − H]− ion increases considerably (7.35 ms) indicating a more extended three-dimensional structure. The ATDs of protonated and chlorinated LNH/LNnH ions, on the other hand, were again similar (Fig. S4, ESI†). Drift times were converted to CCS values, which enable a more universal comparison (Table 1).13,17 Sodiated, chlorinated and protonated ions shared similar CCSs for LNH ([M + Na]+ = 233 Å2, [M + Cl]− = 245 Å2 and [M + H]+ = 226 Å2) and LNnH ([M + Na]+ = 225 Å2, [M + Cl]− = 244 Å2 and [M + H]+ = 229 Å2), while significantly different values are obtained for deprotonated [M − H]− ions, with LNH being considerably larger in size (255 Å2) than LNnH (228 Å2). To confirm the observations from synthetic LNH/LNnH and to substantiate IM-MS analysis for complex biological samples, we analysed free oligosaccharides isolated from pig, horse, and human milk. LNnH is a predominant structure in pig and horse milk19 whereas human milk contains sialylated LNH and LNnH.20 Following enzymatic de-sialylation of the HMO sample the extracted arrival time of the [M − H]− ion (m/z 1071) gave two separate peaks that matched the drift times of the LNH and LNnH oligosaccharide standards (Fig. 1a). Expectedly, the arrival times of the [M − H]− ions from the pig and horse samples yielded a single peak corresponding to the LNnH isomer (Fig. S5, ESI†).
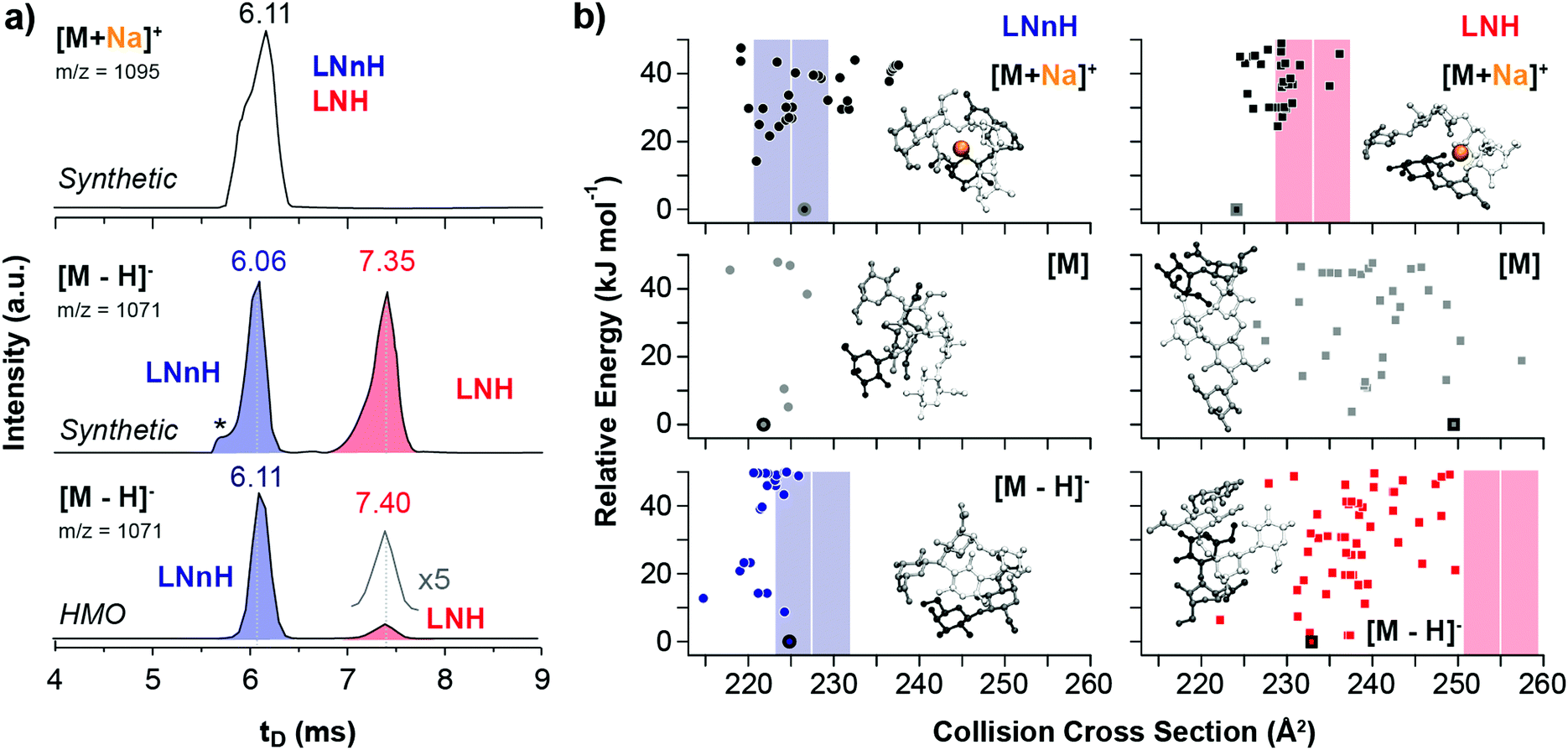 | ||
| Fig. 1 Experimental and theoretical CCSs of LNnH and LNH. (a) Arrival time distributions (ATDs) of an equimolar mixture of LNnH and LNH as sodium adducts (top) and deprotonated ions (middle and bottom). [M + Na]+ ions have similar drift times and cannot be distinguished via IM-MS, while baseline separation is achieved for [M − H]− ions. The asterisk indicates a synthetic impurity, which is present in both synthetic standards, but is not present in samples extracted from human milk (HMO, bottom). (b) Theoretical CCSs and lowest energy structures of LNnH and LNH as sodiated (top), neutral (middle) and deprotonated species (bottom). Theoretical CCSs were computed using the PA method.6,7 Experimental CCSs are shown as coloured bars; the width of each bar corresponds to the typical CCS error of approximately 1.5%. In the model structures, the reducing end lactose moiety is depicted in white; the disaccharides at the 3 and 6 antennae are shown in black and grey, respectively. | ||
Baseline separation of LNH and LNnH can be achieved for deprotonated species [M − H]−, but is this a general phenomenon that can be applied in routine analysis? This question is currently difficult to answer due to the lack of systematic IM-MS data of deprotonated isomers. To test this hypothesis at least in part, we analysed the structurally-related set of milk tetrasaccharide isomers lacto-N-tetraose (LNT) and lacto-N-neo-tetraose (LNnT). Also here, a similar dependence between metal adduction/deprotonation and IM-MS separation was observed. The CCSs of the [M + Na]+ ions of LNT and LNnT are comparable (Table 1) whereas the [M − H]− ions are baseline separated and differ in CCS by over 7% (Fig. S6, ESI†). Changing the drift gas from nitrogen to helium, which for smaller molecules was shown to significantly affect the separation,21 yielded similar trends (Fig. S7, ESI†).
The observed difference in drift time and CCS of the deprotonated species in LNH/LNnH and LNT/LNnT strongly suggests different gas-phase structures of the individual isomers. To investigate this in detail, we performed computer simulations and generated candidate structures of the neutral and sodiated species of both isomeric pairs using Amber99 force field22 sampling. Next, density-functional theory geometry relaxations with FHI-aims23 that utilize the PBE functional24 augmented with a pairwise correction for long-range van der Waals forces25 (PBE + vdW) were employed. For the resulting minima structures, CCSs were computed employing the projection approximation (PA) method; qualitatively similar results were obtained using the more elaborate trajectory method (Fig. S9, ESI†).6,7 Overall the experimental and theoretical CCSs of sodiated species of LNH and LNnH match (Fig. 2b and Table 1). In both cases [M + Na]+ ions adopt similar conformations that results from the attractive positive charge of the sodium cation that ‘captures’ as many of the oxygen atoms as sterically and energetically possible. Similar favourable electrostatics have also been observed for sodiated and lithiated peptides.26 In both cases we also observe a global optimum that is separated by a relatively large energy gap (>10 kJ mol−1) from the next stable conformers. In contrast to the good correlation between experimental and simulated LNH and LNnH [M + Na]+ CCSs, dissimilar results are found for the sodiated species of LNT and LNnT.
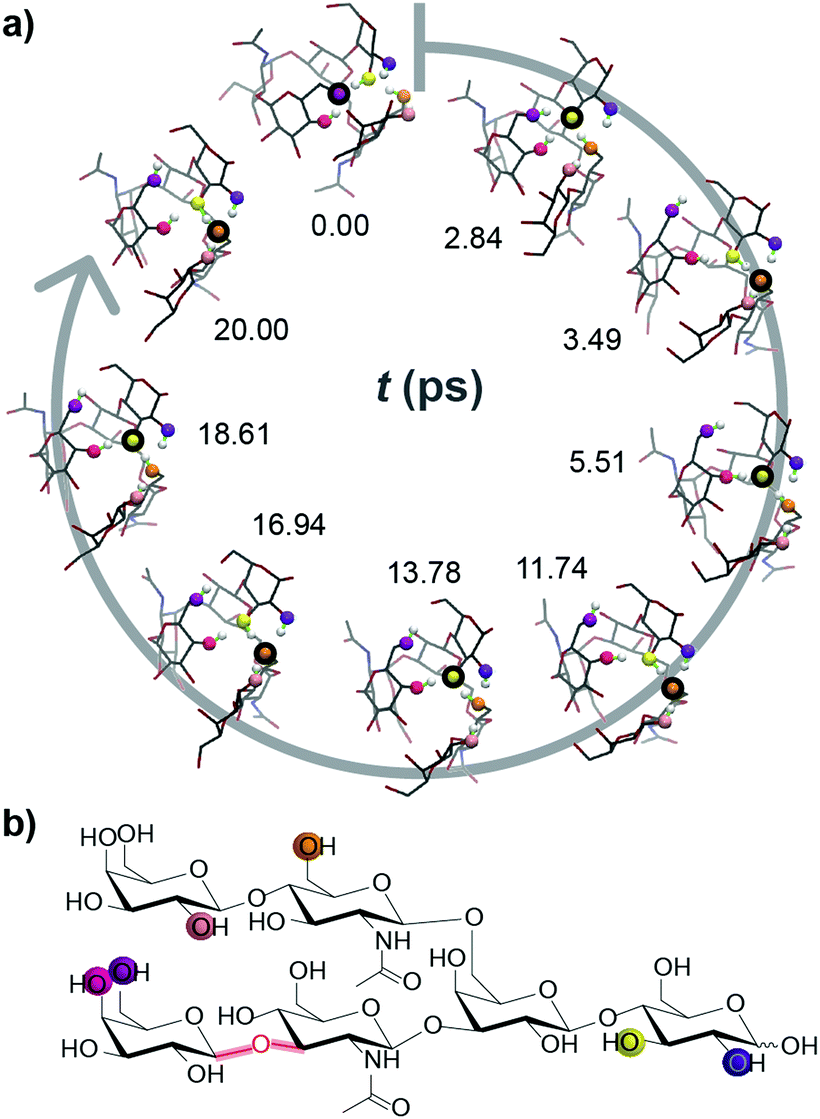 | ||
| Fig. 2 Ab initio MD simulations show rapid charge migration in deprotonated LNH ions. (a) Snapshots at different simulation times. The coloured spheres indicate the OH groups involved in charge migration. The position of the charge within each structure is highlighted by a black circle. (b) Chemical structure of LNH. The coloured spheres correspond the OH groups in (a) which are deprotonated over the 20 ps simulation. | ||
The theoretical CCSs calculated from the neutral species on the other hand, agree with the experimental [M − H]− CCS values. For the four oligosaccharides investigated here, the deviation between experiment and theory is typically around 2%, which is very close to the 1.5% experimental error of the method. To determine if this correlation is purely coincidental or if it indeed reflects the true gas-phase structure and therefore explains the experimental observations, we examined the conformational space of deprotonated species of LNH and LNnH. Individual protons were removed from each of the 18 hydroxy groups of low-energy structures of the respective neutral forms (for details see ESI†). The resulting structures were relaxed (PBE + vdW) and CCS values were computed by the PA method (Fig. 1b). The theoretical CCS values for the rather compact LNnH conformers show little deviation and agree with the experimentally derived CCSs; for deprotonated LNH on the other hand, the CCSs of the predicted conformational ensemble replicate the general trend of LNH being larger in size, but are still notably smaller than the experimental value of 255 Å2 (Fig. 1b, lower right). In addition, the covered CCS range of 230 to 240 Å2 is much broader than in the case of LNnH.
In order to challenge this static picture of individual potential energy surface minima, we performed ab initio molecular dynamics (MD) simulations on exemplary conformations of deprotonated LNH. The derived CCSs vary substantially (Fig. S8, ESI†) and get, with values as high as 250 Å2 and the peak of the CCS distribution around 240 Å2, closer to the experimental value. The most interesting and surprising observation from the ab initio MD simulations, however, is that the position of the deprotonation site is not static. Instead, it migrates rapidly by protons hopping towards the negative charge, which leads to multiple new deprotonation sites. In Fig. 2 and a movie in the ESI,† the traversing of a deprotonation during a 20 ps ab initio MD simulation is shown. Already within such short time, six different OH groups from four distinct residues are deprotonated; the site traverses over the anionic molecule and the negative charge is delocalized. This is a very intriguing result and helps to explain the good agreement between theoretical CCSs of LNH neutral data to experimental [M − H]− CCS values: the rapid movement of the deprotonated site blurs the charge over the molecule within the ms time scale of the IM-MS separation. As a result, the experimental CCS represents an average of a multitude of different molecular structures with distinct protonation sites. Therefore, we postulate that the IM-MS measured result for the deprotonated LNH oligosaccharide is in fact closer to the conformers of the uncharged, neutral structures than to the static structures with a localized negative charge.
The simulation of gas-phase glycans poses challenges that have to be overcome to reliably predict CCS values: (i) ab initio MD timescales (ps) do not reflect the life-span of glycan ions during the experiment (ms). (ii) The conformational space of neutral glycans is already vast, but predicting deprotonation seems almost elusive given the findings from the MD simulations. (iii) There is an uncertainty regarding the energy functions to use, e.g. the generalized gradient approximation functional that we employed slightly underestimates the proton-hopping barriers.27 (iv) The force fields that are commonly applied in glycan simulations, however, would not even be able to describe the proton movement discussed here.
Albeit not directly related to IM, the ab initio MD findings help to explain the mechanisms of glycan collision-induced dissociation (CID), which produces the characteristic and information-rich cross-ring cleavages unique to deprotonated ions. Analysis of [M − H]− ions results in extensive in-source fragmentation28 conceivably from instability caused by proton rearrangement that triggers spontaneous fragmentation. This is prevented by analysing samples in the presence of anionic adducts that stabilize ions until CID is applied, causing not only adduct loss, but also abstraction of a proton explaining the presence of [M − H]− fragments from adduct precursors. This differs from positive ions, where fragments retain the adduct and occur predominantly between monosaccharide residues, which make them less informative.29
In conclusion, we show here that certain glycan structural isomers can be separated by IM-MS as deprotonated ions, but not as protonated species or sodium/chloride adducts. The majority of published data investigating gas-phase separation of glycan ions have focused on sodiated species or other metal ion adducts, which can provoke glycan species to adopt a more compact structure with the cation fully solvated by carbohydrate OH groups. This adduct-induced folding is also very well reflected in our simulations. As shown here for the first time, the structural space of deprotonated ions can be considerably richer due to the highly mobile deprotonation site. This effect represents a challenge to theory, but appears to be the basis for the more diagnostic CID fragmentation behaviour and the considerably improved IM-MS separation of certain deprotonated glycan ions.
Notes and references
- R. A. Dwek, Chem. Rev., 1996, 96, 683–720 CrossRef CAS PubMed.
- R. J. Sola and K. Griebenow, BioDrugs, 2010, 24, 9–21 CrossRef CAS PubMed.
- K. Marino, J. A. Lane, J. L. Abrahams, W. B. Struwe, D. J. Harvey, M. Marotta, R. M. Hickey and P. M. Rudd, Glycobiology, 2011, 21, 1317–1330 CrossRef CAS PubMed.
- S. Wu, N. Tao, J. B. German, R. Grimm and C. B. Lebrilla, J. Proteome Res., 2010, 9, 4138–4151 CrossRef CAS PubMed.
- S. Wu, R. Grimm, J. B. German and C. B. Lebrilla, J. Proteome Res., 2011, 10, 856–868 CrossRef CAS PubMed.
- G. von Helden, M. T. Hsu, N. Gotts and M. T. Bowers, J. Phys. Chem., 1993, 97, 8182–8192 CrossRef CAS.
- M. F. Mesleh, J. M. Hunter, A. A. Shvartsburg, G. C. Schatz and M. F. Jarrold, J. Phys. Chem., 1996, 100, 16082–16086 CrossRef CAS.
- A. A. Shvartsburg, R. R. Hudgins, P. Dugourd and M. F. Jarrold, J. Phys. Chem. A, 1997, 101, 1684–1688 CrossRef CAS.
- C. Baldauf and M. Rossi, J. Phys.: Condens. Matter, 2015, 27, 493002 CrossRef PubMed.
- M. D. Plasencia, D. Isailovic, S. I. Merenbloom, Y. Mechref, M. V. Novotny and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2008, 19, 1706–1715 CrossRef CAS PubMed.
- L. S. Fenn and J. A. McLean, Phys. Chem. Chem. Phys., 2011, 13, 2196–2205 RSC.
- J. P. Williams, M. Grabenauer, R. J. Holland, C. J. Carpenter, M. R. Wormald, K. Giles, D. J. Harvey, R. H. Bateman, J. H. Scrivens and M. T. Bowers, Int. J. Mass Spectrom., 2010, 298, 119–127 CrossRef CAS.
- K. Pagel and D. J. Harvey, Anal. Chem., 2013, 85, 5138–5145 CrossRef CAS PubMed.
- Y. Huang and E. D. Dodds, Anal. Chem., 2013, 85, 9728–9735 CrossRef CAS PubMed.
- P. Both, A. P. Green, C. J. Gray, R. Sardzik, J. Voglmeir, C. Fontana, M. Austeri, M. Rejzek, D. Richardson, R. A. Field, G. Widmalm, S. L. Flitsch and C. E. Eyers, Nat. Chem., 2014, 6, 65–74 CrossRef CAS PubMed.
- D. J. Harvey, C. A. Scarff, M. Edgeworth, M. Crispin, C. N. Scanlan, F. Sobott, S. Allman, K. Barauh, L. Pritchard and J. H. Scrivens, Electrophoresis, 2013, 34, 2368–2378 CrossRef CAS PubMed.
- J. Hofmann, W. B. Struwe, C. A. Scarff, J. H. Scrivens, D. J. Harvey and K. Pagel, Anal. Chem., 2014, 86, 10789–10795 CrossRef CAS PubMed.
- J. Hofmann, H. S. Hahm, P. H. Seeberger and K. Pagel, Nature, 2015, 526, 241–244 CrossRef CAS PubMed.
- S. Albrecht, J. A. Lane, K. Marino, K. A. Al Busadah, S. D. Carrington, R. M. Hickey and P. M. Rudd, Br. J. Nutr., 2014, 111, 1313–1328 CrossRef CAS PubMed.
- A. Kobata, Proc. Jpn. Acad., Ser. B, 2010, 86, 731–747 CrossRef CAS PubMed.
- S. Warnke, J. Seo, J. Boschmans, F. Sobott, J. H. Scrivens, C. Bleiholder, M. T. Bowers, S. Gewinner, W. Schöllkopf, K. Pagel and G. von Helden, J. Am. Chem. Soc., 2015, 137, 4236–4242 CrossRef CAS PubMed.
- J. Wang, P. Cieplak and P. A. Kollman, J. Comput. Chem., 2000, 21, 1049–1074 CrossRef CAS.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- C. Baldauf, K. Pagel, S. Warnke, G. von Helden, B. Koksch, V. Blum and M. Scheffler, Chem. – Eur. J., 2013, 19, 11224–11234 CrossRef CAS PubMed.
- P. Chen, M. Marianski and C. Baldauf, ACS Macro Lett., 2016, 5, 55–59 CrossRef.
- D. J. Harvey, J. Am. Soc. Mass Spectrom., 2005, 16, 622–630 CrossRef CAS PubMed.
- D. J. Harvey, J. Mass Spectrom., 2000, 35, 1178–1190 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on materials, methods, experimental and computational procedures, and a movie illustrating charge migration are given. See DOI: 10.1039/c6cc06247d |
| ‡ Present address: University of Oxford, Department of Biochemistry, South Parks Road, OX1 3QU, Oxford, UK. |
| This journal is © The Royal Society of Chemistry 2016 |