DOI:
10.1039/C6CC06102H
(Communication)
Chem. Commun., 2016,
52, 10992-10995
Increasing the Brønsted acidity of Ph2PO2H by the Lewis acid B(C6F5)3. Formation of an eight-membered boraphosphinate ring [Ph2POB(C6F5)2O]2†
Received
24th July 2016
, Accepted 5th August 2016
First published on 5th August 2016
Abstract
Autoprotolysis of the metastable acid (C6F5)3BOPPh2OH, prepared in situ by the reaction of the rather weak Brønsted acid Ph2PO2H with the strong Lewis acid B(C6F5)3, gave rise to the formation of the eight-membered ring [Ph2POB(C6F5)2O]2 and C6F5H. The conjugate base was isolated as stable sodium crown ether salt [Na(15-crown-5)][Ph2PO2B(C6F5)3].
Lewis acids can significantly increase the acidity of Brønsted acids.1 This principle is operative in the prototypical Lewis pair complex (C6F5)3BOH2, the adduct of the electron pair acceptor B(C6F5)3 and the electron pair donor H2O.2 In MeCN, the acidity of (C6F5)3BOH2 (pKa = 8.4) is very similar to that of HCl (pKa = 8.5).3 Thus, (C6F5)3BOH2 is a strong acid that readily protonates basic organic4 and organometallic compounds.2,5 Diphenylphosphinic acid, Ph2PO2H, is a rather weak acid. As it is well-known that B(C6F5)3 forms Lewis pair complexes with phosphine oxides,6 we were curious to study if B(C6F5)3 will also increase the Brønsted acidity of Ph2PO2H.
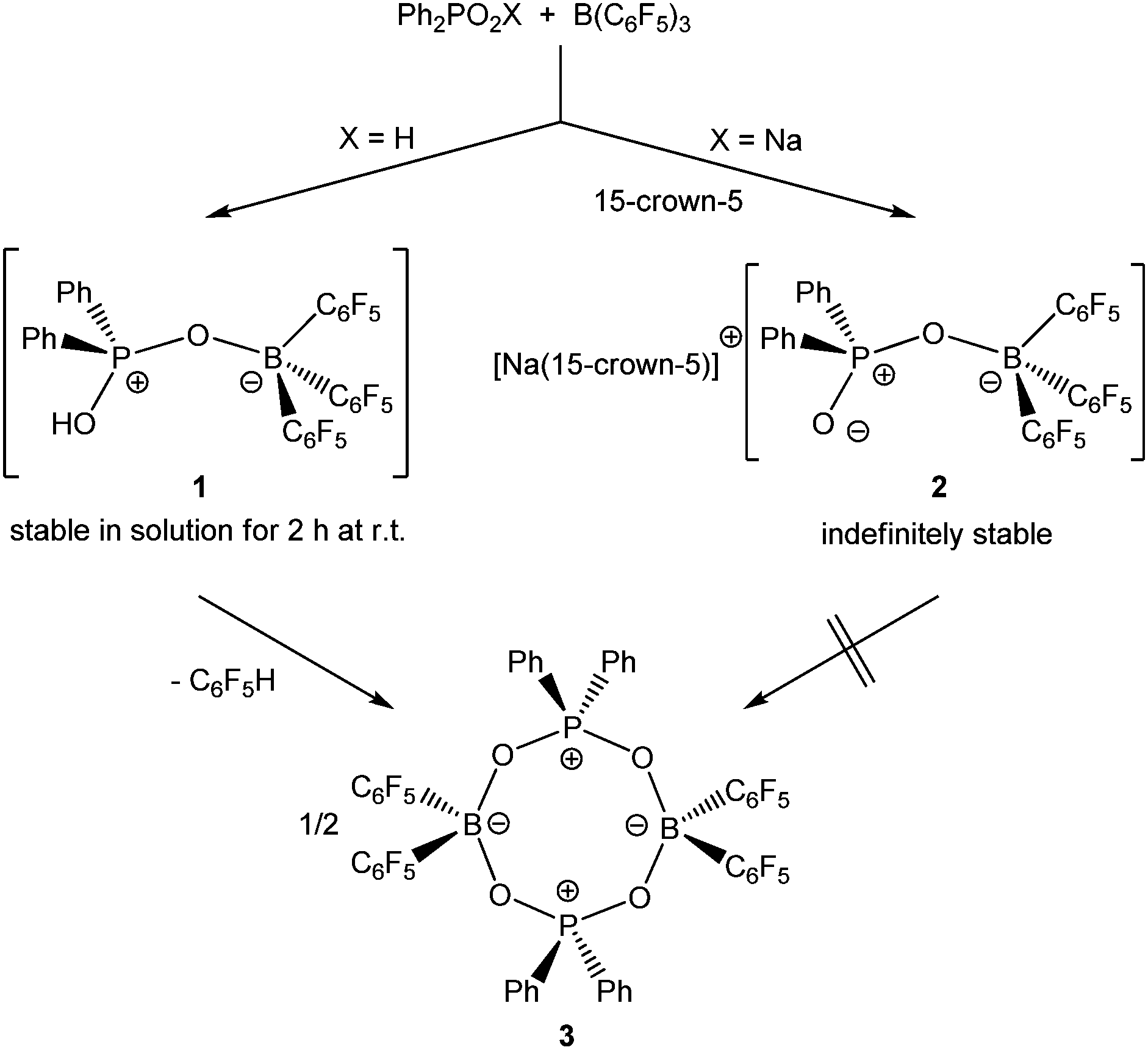
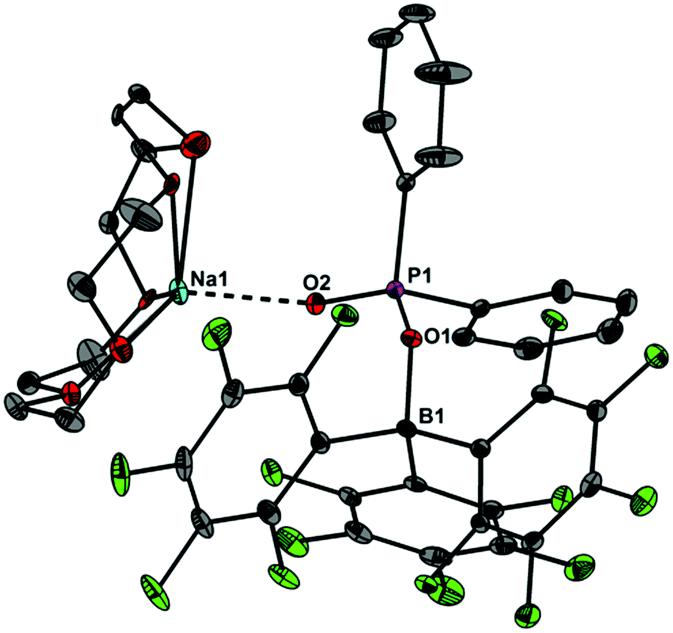
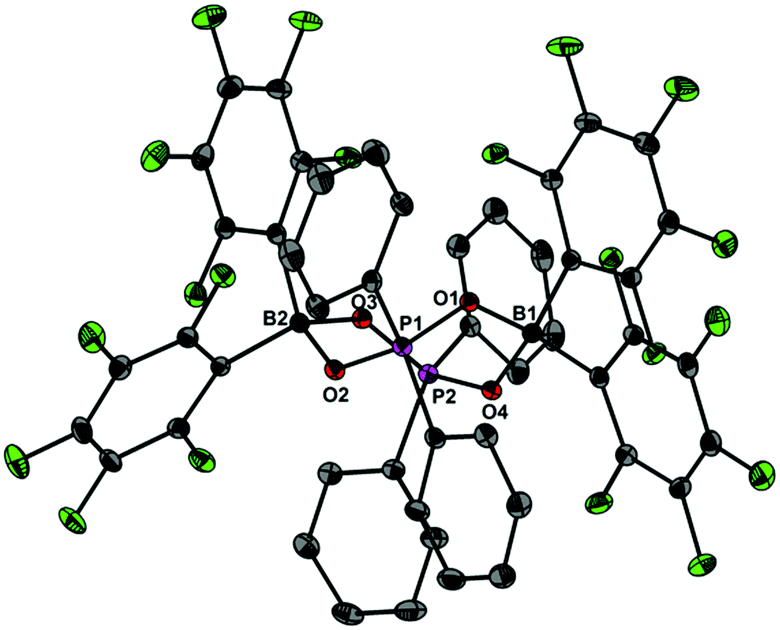
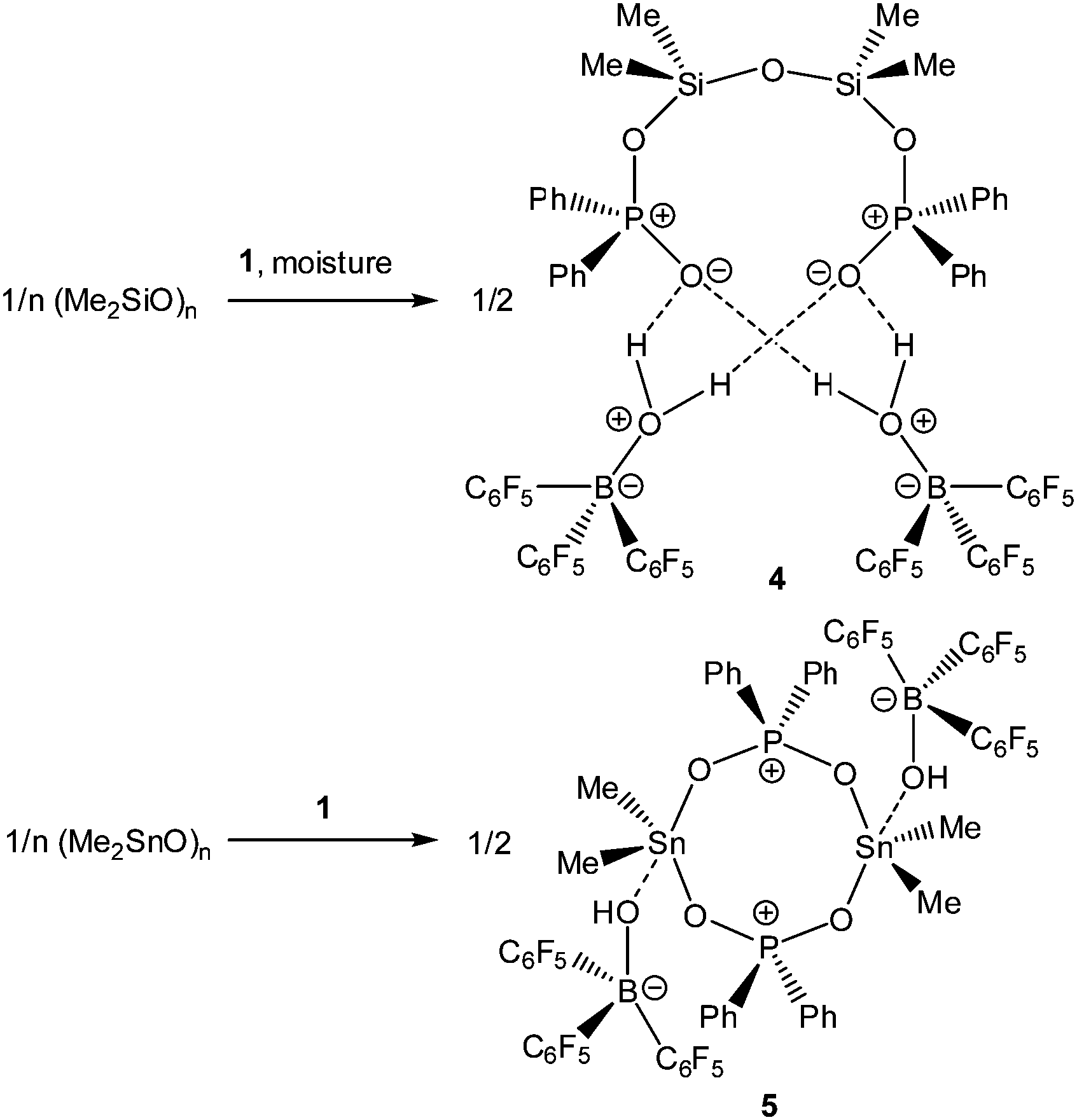
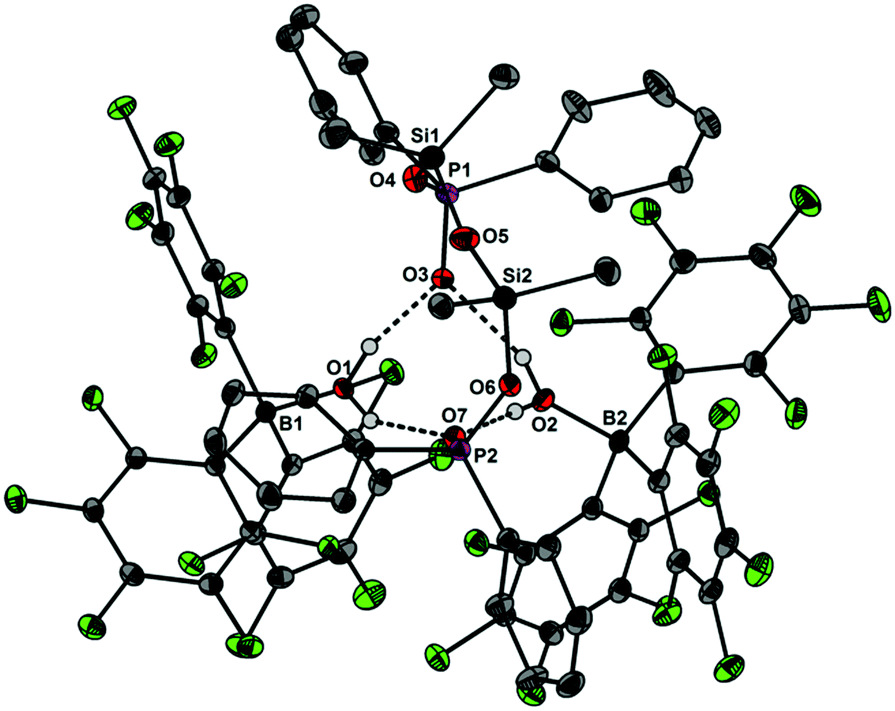
Upon dissolving Ph2PO2H and B(C6F5)3 in CDCl3, multinuclear NMR spectroscopy indeed indicates the formation of a single product that was assigned to (C6F5)3BOPPh2OH (1) (Scheme 1). The 31P NMR spectrum (CDCl3) of 1 shows signal at δ = 42.1 ppm that differs substantially from that of Ph2PO2H (33.9 ppm). The 11B NMR spectrum (CDCl3)3 of 1 exhibits a broad signal at δ = −1.3 ppm, which is significantly different from that of B(C6F5)3 (59.0 ppm). Solutions in CDCl3 show a limited stability and all attempts to isolate 1 by removal of the solvents failed. However, these solutions are stable at r.t. for 2 h; within this time NMR spectroscopy gave no evidence for the formation of other species. While the acid 1 could not be isolated, the reaction of Ph2PO2Na, B(C6F5)3 and 15-crown-5 provided the indefinitely stable, conjugate base [Na(15-crown-5)][Ph2PO2B(C6F5)3] (2), which was obtained as colourless crystals in 73% yield (Scheme 1). The 31P and 11B NMR spectra (THF-d8) gave signals at δ = 22.3 and −2.7 ppm. The molecular structure of 2 reveals that the [Na(15-crown-5)]+ ion and the [Ph2PO2B(C6F5)3]− ion are associated by a Na⋯O contact (Fig. 1). When a solution of 1 in CDCl3 was kept standing for a few hours or heated under reflux for a few minutes NMR spectroscopy indicates the formation of new species, which were identified as the eight-membered boraphosphinate ring [Ph2POB(C6F5)2O]2 (3) and C6F5H. On a preparative scale, 3 was isolated in 76% yield when a solution of 1 prepared in situ from Ph2PO2H and B(C6F5)3, in toluene was heated overnight under reflux (Scheme 1). This reactivity resembles the autoprotolysis of (C6F5)3BOH2 at elevated temperatures giving rise to the formation of [(C6F5)2BOH]3 and C6F5H.7 The eight-membered boraphosphinate ring 3 seems to be the first member of this compound class, however, we note the closely related series of cubic boraphosphonate cages in the literature comprising similar eight-membered ring subunits within the cage structure.8 The 31P and 11B NMR spectra (CDCl3) of 3 revealed signals at δ = 37.8 and 6.3 ppm, but no coupling information. The molecular structure of 3 comprises a strongly puckered B2P2O4 ring (puckering factor = 0.890), whereas isolobal eight-membered siloxane rings are usually almost planar (Fig. 2).9 The bond parameters of 3 are very similar to those of the cubic boraphosphonate cages.8 In a failed attempt to isolate 1 by crystallisation, a small crop of single crystals 4 was isolated, which turned out to be a hydrogen-bonded complex between two molecules of (C6F5)3BOH2 and the disiloxadiphosphinate [Ph2P(O)OSiMe2]2O. The formation of 4 can be rationalized by the accidental cleavage of silicon grease used to seal the joints and stopcocks (Scheme 2).10 The facile cleavage of siloxanes is remarkable and points to the high Brønsted acidity of 1. Variation of the stoichiometric ratio of the reactants gave no other product than 4. The O⋯O donor acceptor distances (2.542(5), 2.684(4), 2.681(4), 2.559(4) Å) are indicative of medium strength hydrogen bonding.11 The 31P, 29Si and 11B NMR spectra (THF-d8) of 4 show signals at δ = 32.4, −23.9 and 3.4 ppm. The molecular structure of 4 comprises a novel hydrogen bond motif featuring two BOH2 hydrogen bond donors and two PO hydrogen acceptors (Fig. 3). The hydrogen bond motif can be described as binary graph set R44(8)12 and is strongly reminiscent to that of (Ph3SiOH)4,13 in which four silanol groups serve as donors and acceptors.
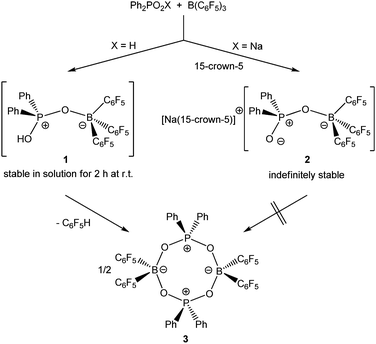 |
| | Scheme 1 Formation and reactivity of 1 and its stable sodium salt 2. | |
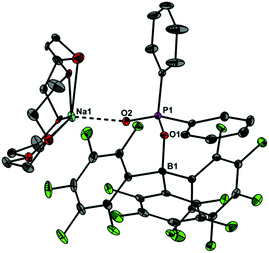 |
| | Fig. 1 Molecular structure of 2 showing 30% probability ellipsoids and the crystallographic numbering scheme. Selected bond parameters [Å, °]: B1–O1 1.508(2), P1–O1 1.544(1), P1–O2 1.482(1), Na1–O2 2.211(2), B1–O1–P1 134.5(1). | |
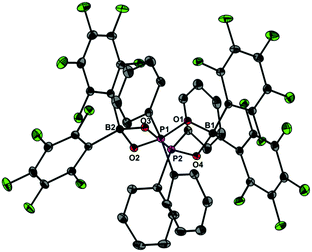 |
| | Fig. 2 Molecular structure of 3 showing 30% probability ellipsoids and the crystallographic numbering scheme. Selected bond parameters [Å, °]: B1–O1 1.501(3), B1–O4 1.507(3), B2–O2 1.506(3), B2–O3 1.513(3), P1–O1 1.538(2), P1–O2 1.537(2), P2–O3 1.534(2), P2–O4 1.539(2), B1–O1–P1 129.2(1), B1–O4–P2 129.1(1), B2–O2–P1 128.8(1), B2–O3–P2 134.2(1). | |
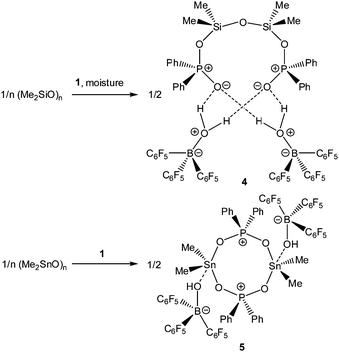 |
| | Scheme 2 Reactivity of 1 towards polymeric group 14 oxides (Me2SiO)n and (Me2SnO)n. | |
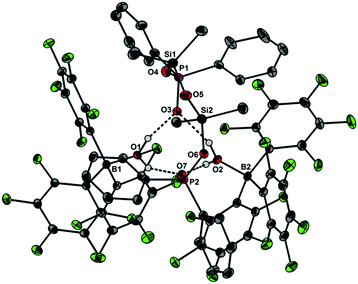 |
| | Fig. 3 Molecular structure of 4 showing 30% probability ellipsoids and the crystallographic numbering scheme. Selected bond parameters [Å, °]: B1–O1 1.562(5), B2–O2 1.556(5), P1–O3 1.492(3), P1–O4 1.558(4), P2–O6 1.567(3), P2–O7 1.502(3), Si1–O4 1.650(4), Si1–O5 1.613(4), Si2–O5 1.611(4), Si2–O6 1.668(3), P1–O4–Si1 149.7(2), P2–O6–Si2 143.8(2), Si1–O5–Si2 159.1(3), O1⋯O3 2.542(5), O1⋯O7 2.684(4), O2⋯O3 2.681(4), O2⋯O7 2.559(4). | |
To provide a quantitative description of the Brønsted acidity increase upon going from Ph2PO2H to 1 and to reveal the corresponding electronic structure changes we carried out DFT calculations of these acids and the conjugate bases with use of the Gaussian09 package.14 The optimized molecular geometries agree well with the experimental data for 2 (Fig. 1), [Ph2PO2]− and Ph2PO2H15 (Table S2, see ESI†). The difference in the dissociation enthalpies of Ph2PO2H and 1 (eqn (1) and (2)) ΔΔH = ΔH1 − ΔH2 is estimated at the M052X/6-31+G** level of theory as 34.0 kcal mol−1 (gas phase) and 14.1 kcal mol−1 (MeCN solution). These values are indicative of much higher Brønsted acidity of 1 as compared to that of the Ph2PO2H. Our calculations of atomic charges show that the O–H bond becomes more polar upon going from Ph2PO2H to 1 (Table S3, see ESI†). Calculated deformation electron densities (DED) reveal a weakening of the O–H covalent bonding upon coordination of B(C6F5)3 to Ph2PO2H (Fig. S35, see ESI†).
These changes in the electronic structures explain the increased Brønsted acidity of 1. The ΔH1 − ΔH2 enthalpy change is equal to the ΔH3 − ΔH4 difference in the B–O bond dissociation energies in the [Ph2PO2B(C6F5)3]− anion and 1 (eqn (3) and (4)).
| | | Ph2PO2H ⇄ [Ph2PO2]− + H+ ΔH1 | (1) |
| | | (C6F5)3BOPPh2OH ⇄ [Ph2PO2B(C6F5)3]− + H+ ΔH2 | (2) |
| | | [Ph2PO2B(C6F5)3]− ⇄ [Ph2PO2]− + B(C6F5)3 ΔH3 | (3) |
| | | (C6F5)3BOPPh2OH ⇄ Ph2PO2H + B(C6F5)3 ΔH4 | (4) |
The B–O bond in [Ph
2PO
2B(C
6F
5)
3]
− is expected, therefore, to be stronger than that in
1. Indeed, the DED maps (Fig. S36, see ESI
†) demonstrate a higher B–O deformation density in the anion. This stabilization of [Ph
2PO
2B(C
6F
5)
3]
− also contributes to the higher Brønsted acidity of
1. To compare the acidities of Ph
2PO
2H and
1 with those of other acids we calculated
16 the p
Ka values in the gas phase and MeCN solution for a series of 15 compounds with tabulated experimental data in the ranges of p
Ka(gas) = 209–251 and p
Ka(MeCN) = 0–30 (Tables S4 and S5, see ESI
†). On the basis of the linear regressions between experimental and calculated p
Ka values (Fig. S37 and S38, see ESI
†) the expected p
Ka values for Ph
2PO
2H and
1 were found to be, respectively, 239.2 and 214.4 in the gas phase and 20.5 and 9.4 in MeCN solution. The gas-phase acidity of
1 appears to be stronger than that of CF
3SO
3H (p
Ka(gas) 219.6)
17 while in MeCN solution
1 is comparable with HCl and tosylic acid (p
Ka(MeCN) 8.5
3 and 8.6,
18 respectively).
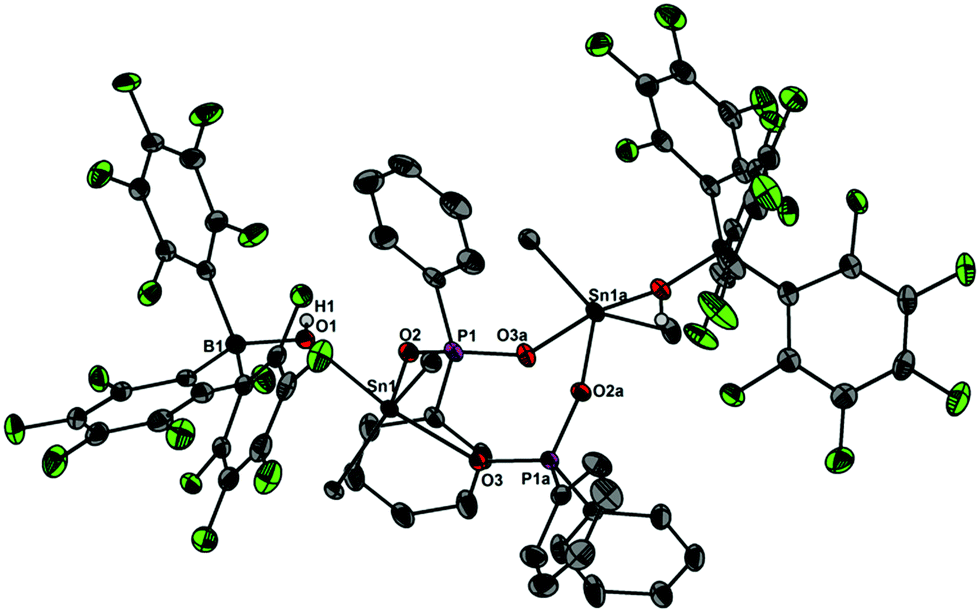
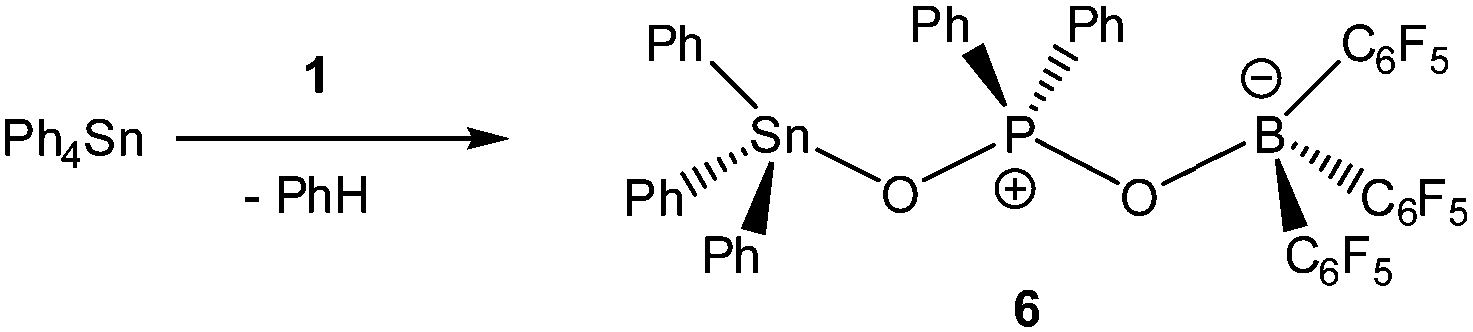
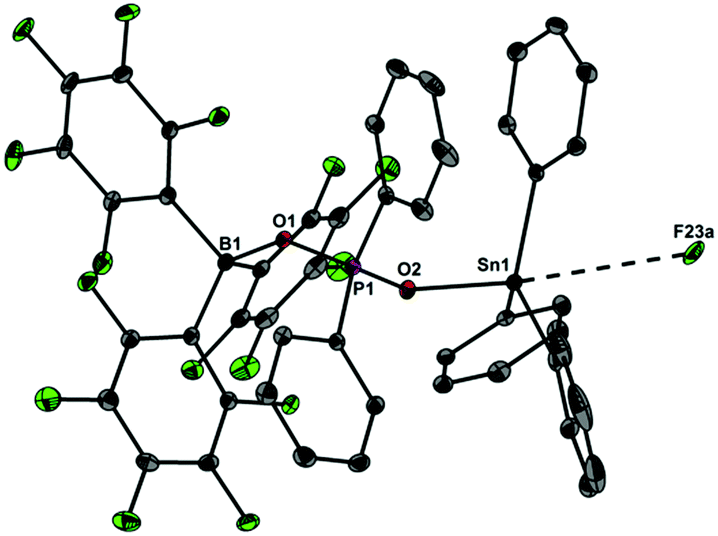
In light of the remarkable siloxane bond cleavage, we have started to elaborate the reactivity of 1 towards other element oxides. Indeed, the reaction of polymeric (Me2SnO)n with 1 rapidly occurred at r.t. and produced the eight-membered Sn2P2O4 heterocycle [Me2Sn(OPPh2O)2SnMe2][HOB(C6F5)3]2 (5) in 83% yield (Scheme 2). The 119Sn and 31P MAS NMR spectra show broad signals at δ = −180.5 ppm and 31.2 ppm. Freshly prepared solutions of phase-pure 5 (checked by powder diffraction) in CDCl3 shows four 119Sn NMR signals and three 31P NMR signals, which point to a reversible dynamic process that is not yet understood in full detail (see ESI†). Similar solution behaviour was observed for the related heterocycles [R2Sn(OPPh2O)2SnR2][O3SCF3]2 (R = Ph, t-Bu), which were obtained by the reaction of (Ph2SnO)n or (t-Bu2SnO)3 with Ph2PO2H and triflic acid.19 On a longer time scale (several weeks) 5 shows signs of irreversible decomposition in solution and in the solid-state. In both states, the same unassigned decomposition product with a 119Sn chemical shift of δ = 71.1 ppm slowly forms. The molecular structure of 5 contains a strongly puckered Sn2P2O4 ring (puckering factor = 0.888)9 that resembles that of the slightly less puckered [t-Bu2Sn(OPPh2O)2Snt-Bu2][O3SCF3]2 (puckering factor = 0.921) (Fig. 4).19 The spatial arrangement of the Sn atoms is distorted trigonal bipyramidal (geometrical goodness = 89.7°)20 and defined by a C2O3 donor set. The Sn–O bond lengths within the ring (2.040(2) and 2.156(2) Å) are shorter than that of the exocyclic HOB(C6F5)3 moiety (2.231(2) Å). The same trend was observed for [t-Bu2Sn(OPPh2O)2Snt-Bu2][O3SCF3]2,19 in which the endocyclic Sn–O bonds (2.045(3) and 2.173(4) Å) are shorter than the Sn–O bond length related with the triflate moiety (2.303(1) Å). It might be speculated that the longer Sn–O bonds are subject to electrolytic dissociation, which could explain the dynamic behaviour in solution. We finally studied the reactivity of 1 towards Ph4Sn, which proceeded with facile phenyl group cleavage providing Ph3SnOPPh2OB(C6F5)3 in 86% yield (Scheme 3). This reaction closely resembles the quantitative reaction of Ph4Sn with triflic acid giving rise to the formation Ph3SnO3SCF3.21 The 119Sn NMR spectrum (CDCl3) of 6 shows a doublet centred at δ = −59.6 ppm with a 2J(119Sn–O–31P) coupling of 146 Hz, which suggests that the Sn atoms are tetracoordinate in solution (Fig. 5). In the solid-state, 6 comprises a 1D coordination polymer with distorted trigonal bipyramidal Sn atoms (geometrical goodness = 51.6°)20 defined by a C3OF donor set.
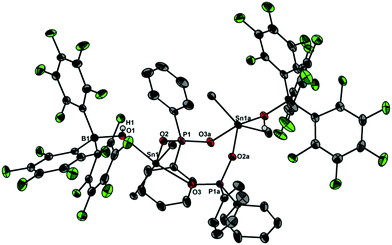 |
| | Fig. 4 Molecular structure of 5 showing 30% probability ellipsoids and the crystallographic numbering scheme. Selected bond parameters [Å, °]: B1–O1 1.512(3), P1–O2 1.538(2), P1–O3a 1.515(2), Sn1–O1 2.231(2), Sn1–O2 2.040(2), Sn1–O3a 2.156(2), B1–O1–Sn1 133.5(2), P1–O2–Sn1 139.5(1), P1–O3a–Sn1a 138.1(1). | |
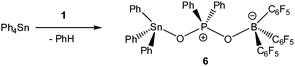 |
| | Scheme 3 Phenyl group cleavage in Ph4Sn using 1. | |
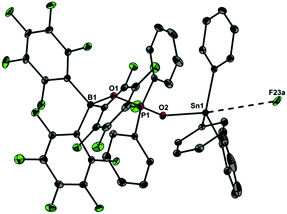 |
| | Fig. 5 Molecular structure of 6 showing 30% probability ellipsoids and the crystallographic numbering scheme. Selected bond parameters [Å, °]: B1–O1 1.527(2), P1–O1 1.521(1), P1–O2 1.527(1), Sn1–O2 2.058(1), Sn1–F23a 3.392(3), B1–O1–P1 139.9(1), P1–O2–Sn1 142.29(7). | |
The Brønsted acidity of Ph2PO2H was significantly increased upon addition of the Lewis acid B(C6F5)3 giving rise to (C6F5)3BOPPh2OH (1) in solution. Unlike its conjugate base [Na(15-crown-5)][Ph2PO2B(C6F5)3] (2), the acid 1 is thermally unstable and undergoes autoprotolysis and formation of the boraphosphinate ring [Ph2POB(C6F5)2O]2 (3) and C6F5H. Despite its limited life span, 1 can be used for synthetic purposes, as was demonstrated for two examples from organotin chemistry. The stable water adduct (C6F5)3BOH2 is known to bind up to two additional water molecules via hydrogen bonding, e.g. (C6F5)3BOH2·2H2O,22 which adversely affects the stoichiometric control of protonation reactions. Moreover, the various related anions, e.g. [HOB(C6F5)3]−, [HO{B(C6F5)3}2]− and [O{B(C6F5)3}2]2−,2,4 suggest that hydroxide and oxide ions may be also transferred upon protonation. These adverse properties have not been observed for 1. We are currently investigating if the acidity of other Brønsted acids, such sulfinic and sulfonic acids, may be also increased by applying the same concept.
The Deutsche Forschungsgemeinschaft (DFG) is gratefully acknowledged for financial support. The theoretical part of this work was supported by the Russian Science Foundation (Project 14-13-00832). We thank Dr Daniel Himmel (Universität Freiburg) for an invaluable discussion about pKa value calculations.
Notes and references
-
Superacid Chemistry, ed. G. A. Olah, G. K. S. Prakash, A. Molnár and J. Sommer, John Wiley & Sons, Inc., Hoboken, New Jersey, 2nd edn, 2009 Search PubMed.
- A. A. Danopoulos, J. R. Galsworthy, M. L. H. Green, S. Cafferkey, L. H. Doerrer and M. B. Hursthouse, Chem. Commun., 1998, 2529 RSC.
- C. Bergquist, B. M. Bridgewater, C. J. Harlan, J. R. Norton, R. A. Friesner and G. Parkin, J. Am. Chem. Soc., 2000, 122, 10581 CrossRef CAS.
-
(a) R. Duchateau, R. A. van Santen and G. P. A. Yap, Organometallics, 2000, 19, 809 CrossRef CAS;
(b) R. T. Stibrany and P. Brant, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 644 CAS;
(c) M. J. Drewitt, M. Niedermann and M. C. Baird, Inorg. Chim. Acta, 2002, 340, 207 CrossRef CAS;
(d) A. Di Saverio, F. Focante, I. Camurati, L. Resconi, T. Beringhelli, G. D'Alfonso, D. Donghi, D. Maggioni, P. Mercandelli and A. Sironi, Inorg. Chem., 2005, 44, 5030 CrossRef CAS PubMed;
(e) F. Focante, I. Camurati, L. Resconi, S. Guidotti, T. Beringhelli, G. D'Alfonso, D. Donghi, D. Maggioni, P. Mercandelli and A. Sironi, Inorg. Chem., 2006, 45, 1683 CrossRef CAS PubMed;
(f) F. Focante, P. Mercandelli, A. Sironi and L. Resconi, Coord. Chem. Rev., 2006, 250, 170 CrossRef CAS.
-
(a) G. S. Hill, L. Manojlovic-Muir, k. W. Muir and R. J. Puddephatt, Organometallics, 1997, 16, 525 CrossRef CAS;
(b) L. Doerrer and M. L. H. Green, J. Chem. Soc., Dalton Trans., 1999, 4325 RSC;
(c) D. Neculai, H. W. Roesky, A. M. Neculai, J. Magull, B. Walfort and D. Stalke, Angew. Chem., Int. Ed., 2002, 41, 4294 CrossRef CAS;
(d) G. Schatte, T. Chivers, H. M. Tuononen, R. Suontamo, R. Laitinen and J. Valkonen, Inorg. Chem., 2005, 44, 443 CrossRef CAS PubMed;
(e) J. Sánchez-Nieves, L. M. Frutos, P. Royo, O. Castaño and E. Herdtweck, Organometallics, 2005, 24, 2004 CrossRef;
(f) J. Sánchez-Nieves, L. M. Frutos, P. Royo, O. Castaño, E. Herdtweck and M. E. G. Mosquera, Inorg. Chem., 2010, 49, 10642 CrossRef PubMed.
-
(a) M. A. Beckett, D. S. Brassington, S. J. Coles and M. B. Hursthouse, Inorg. Chem. Commun., 2000, 3, 530 CrossRef CAS;
(b) M. A. Beckett, D. S. Brassington, M. E. Light and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 2001, 1768 RSC;
(c) A. J. P. Cardenas, B. J. Culotta, T. H. Warren, S. Grimme, A. Stute, R. Fröhlich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2011, 50, 7567 CrossRef CAS PubMed.
-
(a) J. Tian, S. Wang, Y. Feng, J. Li and S. Collins, J. Mol. Catal. A: Chem., 1999, 144, 137 CrossRef CAS;
(b) T. Beringhelli, G. D'Alfonso, D. Donghi, D. Maggioni, P. Mercandelli and A. Sironi, Organometallics, 2003, 22, 1588 CrossRef CAS;
(c) A. Y. Timoshkin and G. Frenking, Organometallics, 2008, 27, 371 CrossRef CAS.
-
(a) K. Diemert, U. Englert, W. Kuchen and F. Sandt, Angew. Chem., Int. Ed., 1997, 36, 241 CrossRef CAS;
(b) M. G. Walawalkar, R. Murugavel, H. W. Roesky and H.-G. Schmidt, Organometallics, 1997, 16, 516 CrossRef CAS;
(c) M. G. Walawalkar, R. Murugavel, H. W. Roesky and H.-G. Schmidt, Inorg. Chem., 1997, 36, 4202 CrossRef CAS;
(d) J. Mortier, I. D. Gridnev and P. Guénot, Organometallics, 2000, 19, 4266 CrossRef CAS;
(e) J. Tönnemann, R. Scopelliti, K. O. Zhurov, L. Menin, S. Dehnen and K. Severin, Chem. – Eur. J., 2012, 18, 9939–9945 CrossRef PubMed;
(f) Q. Liu, N. D. Contrella, A. S. Filatov and R. F. Jordan, Organometallics, 2015, 34, 254 CrossRef CAS.
- J. Beckmann, D. Dakternieks, A. E. K. Lim, K. F. Lim and K. Jurkschat, THEOCHEM, 2006, 761, 177 CrossRef CAS.
- I. Haiduc, Organometallics, 2004, 23, 3 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 49 Search PubMed.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed., 1995, 34, 1555 CrossRef CAS.
- K. F. Bowes, C. Glidewell and J. N. Low, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, o409 Search PubMed.
-
M. J. Frisch, et al., Gaussian09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed. See ESI† for the computational details.
-
(a) J. Guo, W.-K. Wong and W.-Y. Wong, Polyhedron, 2005, 24, 927 CrossRef CAS;
(b) K. A. Lyssenko, G. V. Grintselev-Knyazev and M. Yu. Antipin, Mendeleev Commun., 2002, 12, 128 CrossRef.
- pKa(gas) were calculated on the basis of the M052X/6-31+G** sums of the electronic and thermal Gibbs free energies; pKa(MeCN) were estimated at the same level of theory using the SMD solvation model: A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed . See ESI† for the details of calculations.
- pKaexp(gas) were obtained from the gas-phase acidity (GA) values: J. E. Bartmess, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, NIST, Gaithersburg, MD, 20899, http://webbook.nist.gov.
- A. Kütt, I. Leito, I. Kaljurand, L. Soovali, V. M. Vlasov, L. M. Yagupolskii and I. A. Koppel, J. Org. Chem., 2006, 71, 2829 CrossRef PubMed.
- J. Beckmann, D. Dakternieks, A. Duthie and C. Mitchell, Organometallics, 2003, 22, 2161 CrossRef CAS.
- U. Kolb, M. Beuter and M. Dräger, Inorg. Chem., 1994, 33, 4522 CrossRef CAS.
-
(a) M. Schmeisser, P. Sartori and B. Lippsmeier, Chem. Ber., 1970, 103, 868 CrossRef CAS;
(b) J. Beckmann, E. Lork and O. Mallow, Main Group Met. Chem., 2012, 35, 183 CAS.
- X. Wang and P. P. Power, Angew. Chem., Int. Ed., 2011, 50, 10965 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, NMR spectra, X-ray crystallography, X-ray powder diffraction, computational details, additional references. CCDC 1480495 and 1411098–1411113. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc06102h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence