
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
KNH2–KH: a metal amide–hydride solid solution†
Antonio
Santoru
 *ab,
Claudio
Pistidda
a,
Magnus H.
Sørby
c,
Michele R.
Chierotti
b,
Sebastiano
Garroni
d,
Eugenio
Pinatel
b,
Fahim
Karimi
a,
Hujun
Cao
a,
Nils
Bergemann
a,
Thi T.
Le
a,
Julián
Puszkiel
ae,
Roberto
Gobetto
b,
Marcello
Baricco
b,
Bjørn C.
Hauback
c,
Thomas
Klassen
a and
Martin
Dornheim
a
*ab,
Claudio
Pistidda
a,
Magnus H.
Sørby
c,
Michele R.
Chierotti
b,
Sebastiano
Garroni
d,
Eugenio
Pinatel
b,
Fahim
Karimi
a,
Hujun
Cao
a,
Nils
Bergemann
a,
Thi T.
Le
a,
Julián
Puszkiel
ae,
Roberto
Gobetto
b,
Marcello
Baricco
b,
Bjørn C.
Hauback
c,
Thomas
Klassen
a and
Martin
Dornheim
a
aNanotechnology Department, Helmholtz-Zentrum Geesthacht Max-Planck Straße 1, 21502, Geesthacht, Germany. E-mail: antonio.santoru@hzg.de
bDepartment of Chemistry and NIS centre, University of Torino, V. Giuria 7, 10125, Torino, Italy
cPhysics Department, Institute for Energy Technology (IFE), P.O. Box 40, NO-2027 Kjeller, Norway
dDepartment of Chemistry and Pharmacy, University of Sassari, V. Vienna 2, 07100, Sassari, Italy
eDepartment of Physicochemistry of Materials, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) y Centro Atómico Bariloche, Av. Bustillo km 9500, San Carlos de Bariloche, CP 8400, Argentina
First published on 31st August 2016
Abstract
We report for the first time the formation of a metal amide–hydride solid solution. The dissolution of KH into KNH2 leads to an anionic substitution, which decreases the interaction among NH2− ions. The rotational properties of the high temperature polymorphs of KNH2 are thereby retained down to room temperature.
The amides of alkaline and alkaline-earth metals were discovered and independently investigated by J. L. Gay-Lussac and H. Davy in the early 19th century.1,2 Further studies on their properties were performed towards the end of the same century, most systematically by A. W. Titherley.3 The determination of their crystal structures was possible only after the 1930s with the studies of Juza et al.,4–10 later continued by Jacobs et al.11,12
At that time this class of compounds was mostly used for organic synthesis. However, more recently, metal amide–metal hydride mixtures have been proven to be suitable for reversible hydrogen storage.13 Furthermore, metal amide–metal borohydride systems are regarded as potential solid state ionic conductors.14
The structural and thermal properties of light-weight amides prepared via the reaction of metal hydrides with ammonia have been systematically examined by in situ diffraction experiments.15 While the reaction mechanism and products reported in previous studies were confirmed for the amides of lithium and sodium,16–18 the formation of new K–N–H based intermediates was suggested for potassium amide. The same intermediates were proven to play a role in the desorption reactions of the K–Mg–N–H system.19 These intermediates are isolated here in the KNH2–KH system and, to the best of our knowledge, identified as the first metal amide–hydride solid solution.
The crystal structures of pristine potassium amide and potassium hydride have already been investigated via X-ray and neutron diffraction.7,8,12,20–22 Potassium hydride is known to crystallize in a cubic rock-salt type structure with space group (s.g.) Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and no polymorphic changes are expected in the temperature range from room temperature (RT) to 390 °C.21 For potassium amide, the stable polymorph at RT is monoclinic with the s.g. P21/m.7 Upon heating this phase transforms into a tetragonal structure in P4/nmm at 54 °C.12 The latter phase is stable only in a narrow temperature range; and already above 75 °C the stability of a cubic phase (Fmm) prevails.12,20,22 An increase of symmetry is therefore reached with the two phase transitions at a higher temperature. The explanation of the symmetry changes resides in the increasingly high orientational disorder of the amide anions, as proven by powder neutron diffraction (PND), quasielastic incoherent neutron scattering and orientation-dependent deuterium spin lattice relaxation.20,23,24 If the maximum temperature upon heating is kept below the decomposition temperature of KNH2 (ca. 340 °C), the two phase transitions and the associated rotational dynamics should be reversible upon cooling.
m and no polymorphic changes are expected in the temperature range from room temperature (RT) to 390 °C.21 For potassium amide, the stable polymorph at RT is monoclinic with the s.g. P21/m.7 Upon heating this phase transforms into a tetragonal structure in P4/nmm at 54 °C.12 The latter phase is stable only in a narrow temperature range; and already above 75 °C the stability of a cubic phase (Fmm) prevails.12,20,22 An increase of symmetry is therefore reached with the two phase transitions at a higher temperature. The explanation of the symmetry changes resides in the increasingly high orientational disorder of the amide anions, as proven by powder neutron diffraction (PND), quasielastic incoherent neutron scattering and orientation-dependent deuterium spin lattice relaxation.20,23,24 If the maximum temperature upon heating is kept below the decomposition temperature of KNH2 (ca. 340 °C), the two phase transitions and the associated rotational dynamics should be reversible upon cooling.
This is in agreement with the present in situ synchrotron radiation powder X-ray diffraction experiment (SR-PXD) on KNH2 (ESI†). When a 0.5KNH2 + 0.5KH mixture was investigated, the expected phase transformations of KNH2 took place, but the interaction of KNH2 and KH in the temperature range between 100 °C and 270 °C led to the formation of a new cubic structure (Fig. 1a).‡
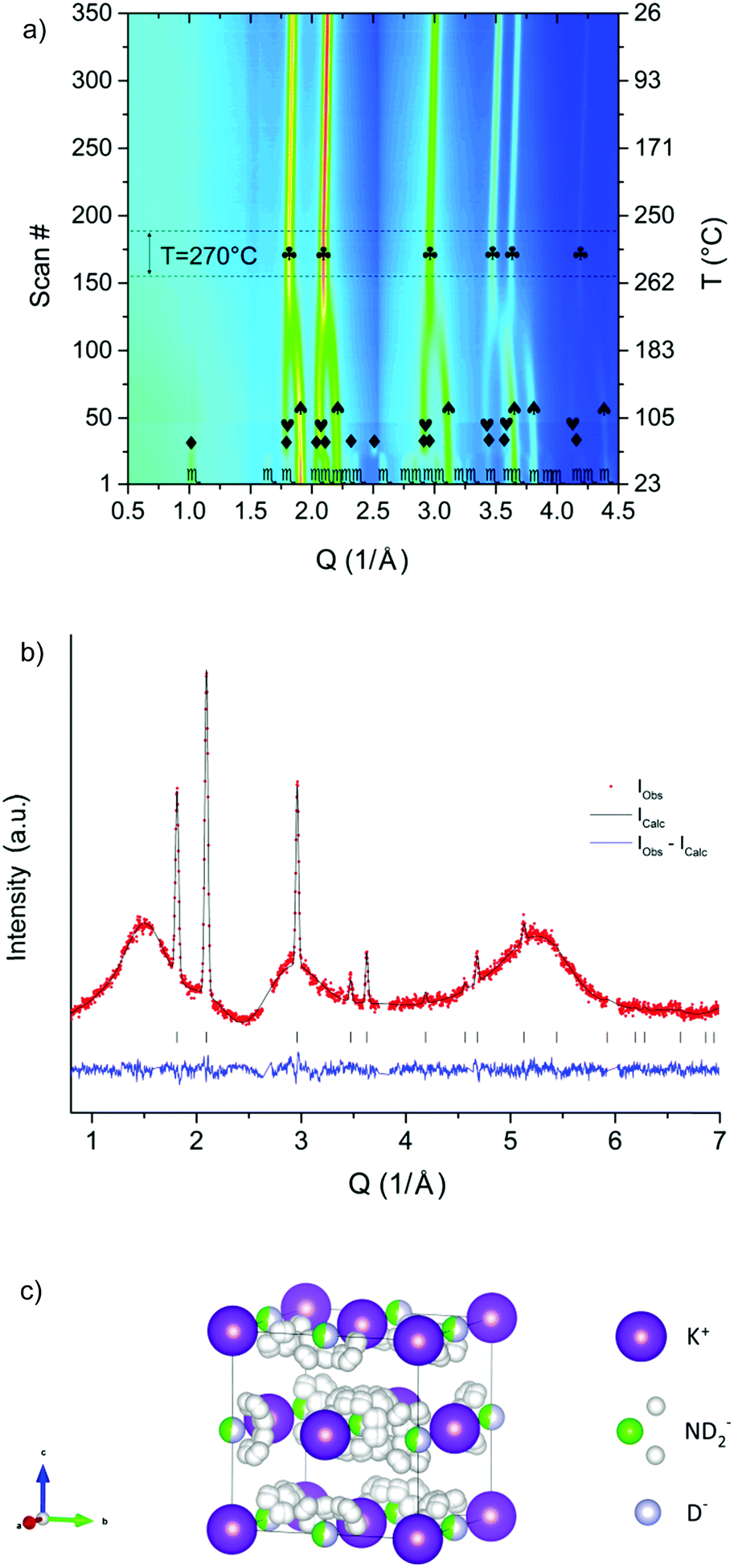 | ||
Fig. 1 (a) In situ SR-PXD experiment on the 0.5KNH2 + 0.5KH sample. 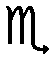 = KNH2 (P21/m), ◆ = KNH2 (P4/nmm), ❤ = KNH2 (Fmm), = KH (Fmm), ♣ = new phase (Fmm). (b) PND pattern of the nominal 0.5KND2 + 0.5KD mixture annealed up to 270 °C and kept under isothermal conditions during the data collection. Rwp (%) = 2.8 (corrected for background). The wavy background was originated from the quartz sample holder (see the ESI†). (c) Structural model of the cubic phase (s.g. Fmm) of composition K(ND2)0.46D0.54 obtained after the Rietveld refinement, taking into account the anionic substitution at the position (0.5 0.5 0.5) and the orientational disorder of amide anions. = KNH2 (P21/m), ◆ = KNH2 (P4/nmm), ❤ = KNH2 (Fmm), = KH (Fmm), ♣ = new phase (Fmm). (b) PND pattern of the nominal 0.5KND2 + 0.5KD mixture annealed up to 270 °C and kept under isothermal conditions during the data collection. Rwp (%) = 2.8 (corrected for background). The wavy background was originated from the quartz sample holder (see the ESI†). (c) Structural model of the cubic phase (s.g. Fmm) of composition K(ND2)0.46D0.54 obtained after the Rietveld refinement, taking into account the anionic substitution at the position (0.5 0.5 0.5) and the orientational disorder of amide anions. | ||
PND at 270 °C of a potassium deuteramide–potassium deuteride mixture confirmed the presence of a single cubic phase at this temperature (Fig. 1b).§
The structure was resolved assuming an ionic crystal with unaltered positions for the potassium cations and partially occupied sites at the original positions of amide and hydride anions. Indeed the formation of a solid solution is highly favorable due to the structural similarities between the two cubic polymorphs of KNH2 and KH (same space group, same cation, same charge for the anions, similar lattice constants).
The local symmetry of the deuteramide groups is compatible with the s.g. Fmm only assuming rotational dynamics and orientational disorder. In this case the restrictions imposed from both the s.g. and the rigid amide groups resulted in partially occupied sites (multiplicity = 192) for the deuterium atoms of each amide group (Fig. 1c).
The final Rietveld refinement25,26 of the PND pattern (Fig. 1b) confirmed the structural model, i.e. the formation of a potassium amide–hydride solid solution. To the best of our knowledge, no similar cases have been reported so far for other alkaline metal amide–metal hydride mixtures.
The similarity of the chemical environment between the starting materials and the xKNH2 + (1 − x)KH samples at different compositions (x = 0.1, 0.5, 0.9) after annealing was verified by 1H magic angle spinning solid-state NMR (MAS SSNMR), see Fig. 2.¶
 | ||
| Fig. 2 (a) 1H (400.23 MHz) MAS SSNMR spectra of the starting reagents and xKNH2 + (1 − x)KH samples at different compositions (x = 0.1, 0.5, 0.9) after annealing recorded with a spinning speed of 32 kHz. Asterisks denote impurities. (b) 2D 1H (400.23 MHz) DQ MAS SSNMR spectrum of the KNH2 + KH (x = 0.5) after annealing recorded with a spinning speed of 32 kHz. The red line highlights the DQ correlation between the KH and KNH2 signals. | ||
No significant shifts are observed while the integral values perfectly reflect the composition of the solid solution. Furthermore, the same T11H value (46 s) for both signals supports the formation of a solid solution since it indicates that spin diffusion is active. This is only possible if they belong to the same phase or in the case of homogeneous samples on a nanometer scale.27 Direct evidence of the solid solution formation is provided by the 1H double-quantum (DQ) MAS SSNMR experiment (Fig. 2b). Indeed, the observed DQ correlation between the KNH2 (−3.2 ppm) and KH (4.8 ppm) signals implies that they are in close spatial proximity to each other (less than 5 Å). This is only possible if they are intimately related as in a solid solution.28,29 A similar correlation, although much weaker, is observed for the sample before annealing (ESI†), which can be explained with the formation of a small fraction of solid solution due to the fast rotation and slightly increased temperature during the NMR experiment.
The effect of the starting composition of the mixture on the final structure was studied by means of in situ SR-PXD. A linear relationship was found between the unit cell parameter of the cubic phase at T = 270 °C and the molar fraction of amide anions (Fig. 3 and Table 1) as expected from Vegard’s law.30 Therefore an almost ideal behavior is expected for the solid solution (ΔVmix ∼ 0 and ΔHmix ∼ 0).
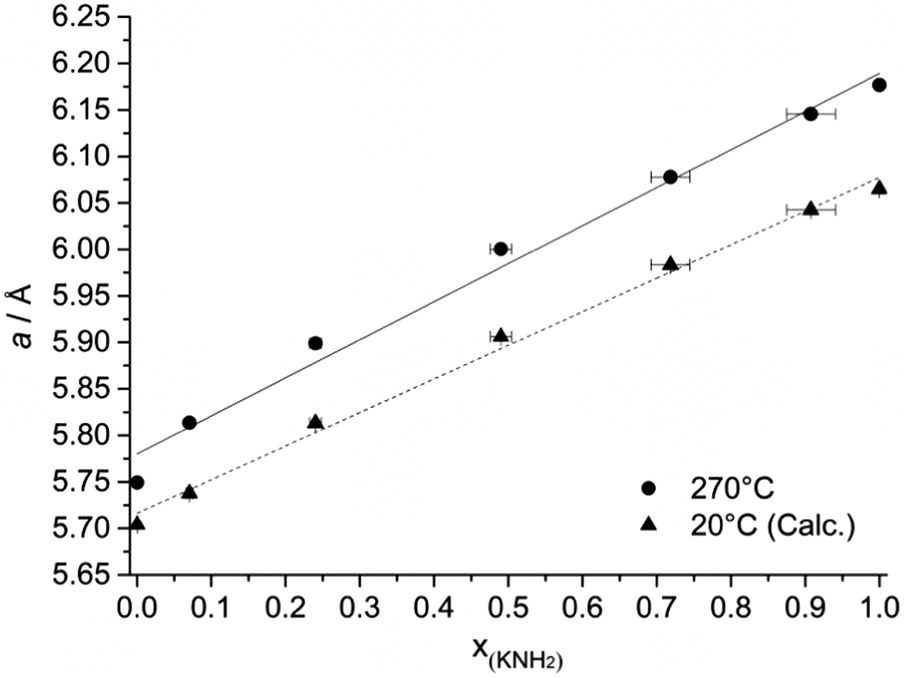 | ||
| Fig. 3 Unit cell parameters (dots) obtained by in situ SR-PXD experiments under isothermal conditions (T = 270 °C) as a function of the potassium amide content and the corresponding linear fit (continuous line). The values for T = 20 °C (triangles) were calculated using the thermal expansion coefficient of each phase and then fitted (dotted line). The error bars for molar fractions and cell parameters were calculated considering the errors of the Rietveld process. | ||
| x | x | ac/Å |
α/K−1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
ae/Å |
|---|---|---|---|---|
| a Nominal composition. b Refined composition (molar fraction) of KNH2. c Refined cell parameters at 270 °C. d Calculated linear thermal expansion coefficients. e Calculated cell parameters at 20 °C. Estimated standard deviations are given in parentheses. | ||||
| 0 | 0 | 5.74934(14) | 3.19(2) × 10−5 | 5.70390(14) |
| 0.1 | 0.070(5) | 5.81372(13) | 5.30(1) × 10−5 | 5.73769(13) |
| 0.3 | 0.240(8) | 5.8989(2) | 5.88(2) × 10−5 | 5.8134(2) |
| 0.5 | 0.490(14) | 6.0003 (2) | 6.32(1) × 10−5 | 5.9060(2) |
| 0.7 | 0.72(3) | 6.07768(12) | 6.83(1) × 10−5 | 5.98320(12) |
| 0.9 | 0.91(3) | 6.14553(14) | 6.83(2) × 10−5 | 6.04230(14) |
| 1 | 1 | 6.17667(8) | 7.38(1) × 10−5 | 6.06473 (8) |
It is noteworthy that, for the compositions x = 0.1, 0.3, 0.5, 0.7, the structure did not change during the cooling process down to RT, except for the thermal contraction of the unit cell volume (more details in Table 1 and in the ESI†).
It appears that the addition of potassium hydride can stabilize the cubic geometry, which retains the rotation of the amide anions even at RT. A similar behavior was previously reported for the sodium borohydride–sodium chloride systems, at different temperatures (T = −81 °C).31Ex situ PXD of annealed samples (x = 0.3, 0.5, 0.7, 0.9) collected with a Bragg Brentano diffractometer proves the coexistence of at least two different cubic structures. The composition x = 0.1, however, presented unchanged cubic phase (see the ESI†). At the composition x = 0.9, coexistence of the cubic and monoclinic phases was found. These results suggest the presence of a two phase field at RT. Nevertheless, even in the cases where phase segregation occurred, both cubic structures were proven to be unchanged even after several months, hence a complete transformation back to the pure monoclinic phase of KNH2 and cubic phase of KH did not occur.
It is noteworthy that the same structures are formed simply by mechanochemical treatments of the starting reactants. In some cases (x = 0.1, 0.3 and 0.9) manual grinding is enough to promote the formation of detectable amounts of K(NH2)xH(1−x) solid solution. These species are therefore easily formed and are expected to be possibly identified as reaction products or intermediates in future studies of amide-based systems containing potassium.
To the best of our knowledge, amide/hydride solid solutions were not reported so far. In this sense, the chemistry of the K-based amide/hydride system is interestingly peculiar and differs substantially from that of the other alkaline metal amides/hydrides.
The research leading to these results has received funding from the European Marie Curie Actions under ECOSTORE grant agreement no. 607040. The help of the beamline scientists Francisco Martinez Casado, Dorthe Haase, Olivier Balmes, (MAX-lab – Lund, Sweden), Martin Etter and Jozef Bednarcik (DESY – Hamburg, Germany) is thankfully acknowledged. A. S. thanks the research groups of Turin University, Sassari University and IFE for the fruitful discussion and Dr Klaus Taube, coordinator of the ECOSTORE project, for providing the essential networking opportunities.
Notes and references
- J. L. Gay and L. J. Thenard, Ann. Phys., 1809, 32, 23–39 CrossRef.
- H. Davy, Philos. Trans. R. Soc. London, 1808, 98, 333–370 CrossRef.
- A. W. Titherley, J. Chem. Soc., Trans., 1894, 65, 504–522 RSC.
- R. Juza, Angew. Chem., 1964, 76, 290–300 CrossRef CAS.
- R. Juza, Z. Anorg. Allg. Chem., 1937, 231, 121–135 CrossRef CAS.
- R. Juza, K. Fasold and C. Haeberle, Z. Anorg. Allg. Chem., 1937, 234, 75–85 CrossRef CAS.
- R. Juza, H. Jacobs and W. Klose, Z. Anorg. Allg. Chem., 1965, 338, 171–178 CrossRef CAS.
- R. Juza and H. Liedtke, Z. Anorg. Allg. Chem., 1957, 290, 205–208 CrossRef CAS.
- R. Juza and K. Opp, Z. Anorg. Allg. Chem., 1951, 266, 313–324 CrossRef CAS.
- R. Juza, H. H. Weber and K. Opp, Z. Anorg. Allg. Chem., 1956, 284, 73–82 CrossRef CAS.
- H. Jacobs, Z. Anorg. Allg. Chem., 1971, 382, 97–109 CrossRef CAS.
- H. Jacobs and E. Von Osten, Z. Naturforsch., 1976, 31, 385–386 Search PubMed.
- P. Chen, Z. Xiong, J. Luo, J. Lin and K. L. Tan, Nature, 2002, 420, 302–304 CrossRef CAS PubMed.
- M. Matsuo, A. Remhof, P. Martelli, R. Caputo, M. Ernst, Y. Miura, T. Sato, H. Oguchi, H. Maekawa, H. Takamura, A. Borgschulte, A. Züttel and S.-i. Orimo, J. Am. Chem. Soc., 2009, 131, 16389–16391 CrossRef CAS PubMed.
- C. Pistidda, A. Santoru, S. Garroni, N. Bergemann, A. Rzeszutek, C. Horstmann, D. Thomas, T. Klassen and M. Dornheim, J. Phys. Chem. C, 2015, 119, 934–943 CAS.
- W. I. F. David, M. O. Jones, D. H. Gregory, C. M. Jewell, S. R. Johnson, A. Walton and P. P. Edwards, J. Am. Chem. Soc., 2007, 129, 1594–1601 CrossRef CAS PubMed.
- J. W. Makepeace, M. O. Jones, S. K. Callear, P. P. Edwards and W. I. F. David, Phys. Chem. Chem. Phys., 2014, 16, 4061–4070 RSC.
- H. Yamamoto, H. Miyaoka, S. Hino, H. Nakanishi, T. Ichikawa and Y. Kojima, Int. J. Hydrogen Energy, 2009, 34, 9760–9764 CrossRef CAS.
- A. Santoru, S. Garroni, C. Pistidda, C. Milanese, A. Girella, A. Marini, E. Masolo, A. Valentoni, N. Bergemann, T. T. Le, H. Cao, D. Haase, O. Balmes, K. Taube, G. Mulas, S. Enzo, T. Klassen and M. Dornheim, Phys. Chem. Chem. Phys., 2016, 18, 3910–3920 RSC.
- M. Müller, J. Senker, B. Asmussen, W. Press, H. Jacobs, W. Kockelmann, H. M. Mayer and R. M. Ibberson, J. Chem. Phys., 1997, 107, 2363–2373 CrossRef.
- V. G. Kuznetsov and M. M. Shkrabkina, J. Struct. Chem., 1962, 3, 532–537 CrossRef.
- M. Müller, B. Asmussen, W. Press, J. Senker, H. Jacobs, H. Büttner, W. Kockelmann and R. M. Ibberson, Phys. B, 1997, 234–236, 45–47 CrossRef.
- M. Müller, B. Asmussen, W. Press, J. Senker, H. Jacobs, H. Büttner and H. Schober, J. Chem. Phys., 1998, 109, 3559–3567 CrossRef.
- J. Senker, Solid State Nucl. Magn. Reson., 2004, 26, 22–35 CrossRef CAS PubMed.
- H. M. Rietveld, Acta Crystallogr., 1967, 22, 151–152 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- K. Gaglioti, M. R. Chierotti, F. Grifasi, R. Gobetto, U. J. Griesser, D. Hasa and D. Voinovich, CrystEngComm, 2014, 16, 8252–8262 RSC.
- D. Braga, L. Chelazzi, F. Grepioni, E. Dichiarante, M. R. Chierotti and R. Gobetto, Cryst. Growth Des., 2013, 13, 2564–2572 CAS.
- M. R. Chierotti and R. Gobetto, CrystEngComm, 2013, 15, 8599–8612 RSC.
- L. Vegard, Z. Phys., 1921, 5, 17–26 CrossRef CAS.
- J. E. Olsen, P. Karen, M. H. Sørby and B. C. Hauback, J. Alloys Compd., 2014, 587, 374–379 CrossRef CAS.
- U. Bösenberg, C. Pistidda, M. Tolkiehn, N. Busch, I. Saldan, K. Suarez-Alcantara, A. Arendarska, T. Klassen and M. Dornheim, Int. J. Hydrogen Energy, 2014, 39, 9899–9903 CrossRef.
- B. C. Hauback, H. FjellvåG, O. Steinsvoll, K. Johansson, O. T. Buset and J. Jørgensen, J. Neutron Res., 2000, 8, 215–232 CrossRef CAS.
- V. Favre-Nicolin and R. Cerny, J. Appl. Crystallogr., 2002, 35, 734–743 CrossRef CAS.
- A. C. L. A. R. B. V. Dreele, Los Alamos National Laboratory Report LAUR 86–748, 2000.
- B. Toby, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Powder X-ray and neutron diffractograms, Rietveld refinements, thermal decomposition, and experimental details. See DOI: 10.1039/c6cc05777b |
| ‡ The in situ SR-PXD experiments were performed at the diffraction beamline I711, MAX Lab (Lund, Sweden) and at the diffraction beamline P02, DESY (Hamburg, Germany) (monochromatic beams of λ ≈ 0.99 Å and ≈ 0.2 Å were employed respectively). The in situ cells and the procedure used are described elsewhere.19,32 |
| § PND was performed at the PUS instrument at the JEEP II reactor at IFE, Norway.33 Neutrons with λ = 1.5539 Å were provided by a focussing Ge(511) monochromator at 90° take-off angle. The data was collected in the range 2θ = 10–130° (Δ2θ = 0.05°) by 2 detector banks each with 7 vertically stacked position sensitive detectors. The sample was contained in an argon-filled quartz tube with 6 mm diameter placed inside an in-house built furnace. The structure solution was carried out using the software “FOX”.34 The Rietveld refinement was performed by means of the software GSAS35 and the EXPGUI graphic interface.36 |
| ¶ Solid-state NMR experiments were run on a Bruker AVANCE II 400 instrument operating at 400.23 MHz for 1H and equipped with a 2.5 mm probe. The 1H MAS spectra were recorded at the spinning speed of 32 kHz with the DEPTH sequence (π/2 − π–π) for the suppression of the probe background signal (1H 90° = 2.5 μs; scans = 16; relaxation delay = 53 s). The 2D 1H DQ MAS experiments were performed at 32 kHz with the back-to-back (BABA) recoupling pulse sequence with excitation time durations of one rotor period (1H 90° = 2.5 μs; 32 scans; t1 increments = 46; relaxation delay = 53 s). 1H scale was calibrated with adamantane (1H signal at 1.87 ppm) as external standards. |
| This journal is © The Royal Society of Chemistry 2016 |