
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-dimensional self-assembly of perylene-diimide dyes by unidirectional transit of zeolite channel openings†
Gloria
Tabacchi
 a,
Gion
Calzaferri
b and
Ettore
Fois
*a
a,
Gion
Calzaferri
b and
Ettore
Fois
*a
aDepartment of Science and High Technology, University of Insubria, and INSTM, Via Valleggio 9, I-22100 Como, Italy. E-mail: ettore.fois@uninsubria.it
bDepartment of Chemistry and Biochemistry, University of Bern, Freiestrasse 3, CH-3012 Bern, Switzerland
First published on 26th July 2016
Abstract
Confined supramolecular architectures of chromophores are key components in artificial antenna composites for solar energy harvesting and storage. A typical fabrication process, based on the insertion of dye molecules into zeolite channels, is still unknown at the molecular level. We show that slipping of perylene diimide dyes into the one-dimensional channels of zeolite L and travelling inside is only possible because of steric-interaction-induced cooperative vibrational modes of the host and the guest. The funnel-like structure of the channel opening, larger at the entrance, along with a directionally asymmetric entrance–exit probability, ensures a favorable self-assembly process of the perylene units.
Insertion of dyes into the 1D channels of zeolite L (ZL) has led to an impressive number of functional composites.1–3 The principle of sequential insertion, which is based on single-file diffusion,4a is illustrated in Scheme 1. Transport inside can, at first glance, be understood as a 1D random walk, as pioneered by Einstein in his theory of Brownian motion.4b This model was later extended to porous materials and complex networks, where single-file diffusion processes take place.4,5 Such is the case for the encapsulation of perylene diimide (PDI) dyes in ZL (see Scheme 1). A 500 nm long ZL crystal, as a representative example, consists of 67
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 parallel channels, each formed by 666 unit cells 0.75 nm in length.6 Molecules that slip through the 0.71–0.75 nm wide channel openings (ESI,† Table S1),6 which widen to 1.26 nm at their largest extension before closing again in a periodic manner, lose much of their freedom of movement. Actually, the van der Waals diameter (vdW) of the PDIs slightly exceeds the diameter of the ZL channel opening.1 The question arises as to how these molecules can enter the channels and how they can travel inside, despite this steric constraint. A phenomenological understanding is given by a comparison of 17 differently substituted PDIs. 13 were successfully encapsulated under vacuum at high temperatures (180 °C to 260 °C, depending on the substituents),3a while 4 could not be inserted because of the hindrance of their substituents. Steric interactions between rigid host and guest partners, however, cannot explain why and how it is possible for the PDIs to enter the channels of ZL and to travel inside even if their width slightly exceeds the channel openings. Do molecular and reticular vibrations play a role in the insertion process? Herein, we address this issue, which, despite the boost of single molecule spectroscopy techniques,5e–h is very difficult to deal with experimentally.
000 parallel channels, each formed by 666 unit cells 0.75 nm in length.6 Molecules that slip through the 0.71–0.75 nm wide channel openings (ESI,† Table S1),6 which widen to 1.26 nm at their largest extension before closing again in a periodic manner, lose much of their freedom of movement. Actually, the van der Waals diameter (vdW) of the PDIs slightly exceeds the diameter of the ZL channel opening.1 The question arises as to how these molecules can enter the channels and how they can travel inside, despite this steric constraint. A phenomenological understanding is given by a comparison of 17 differently substituted PDIs. 13 were successfully encapsulated under vacuum at high temperatures (180 °C to 260 °C, depending on the substituents),3a while 4 could not be inserted because of the hindrance of their substituents. Steric interactions between rigid host and guest partners, however, cannot explain why and how it is possible for the PDIs to enter the channels of ZL and to travel inside even if their width slightly exceeds the channel openings. Do molecular and reticular vibrations play a role in the insertion process? Herein, we address this issue, which, despite the boost of single molecule spectroscopy techniques,5e–h is very difficult to deal with experimentally.
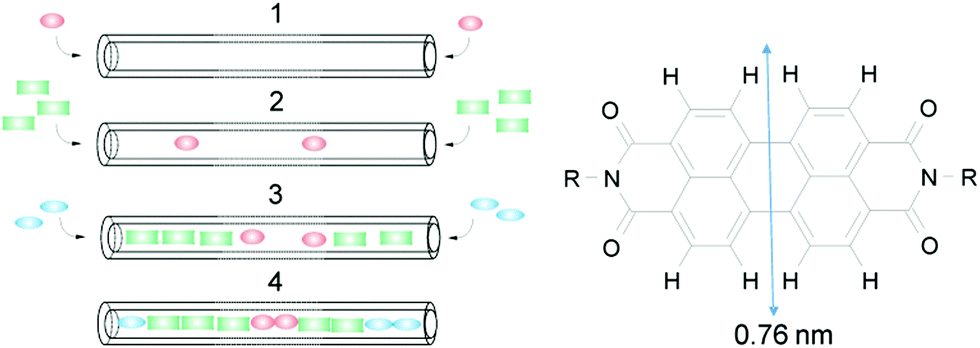 | ||
| Scheme 1 Left: Sequential loading, leading after steps 1, 2, and 3 to the organized 3 color composite 4. Right: PDI with vdW diameter.1 | ||
Progress in theoretical modeling of zeolites has been enormous, fortunately.7 We unraveled the interaction and orientation of dyes inside ZL and we explained how stopper molecules irreversibly modify ZL at the channel entrances.8 Now, to investigate the slipping of a PDI dye across those entrances we build a first-principles model featuring the channel openings of ZL (see the ESI†), as previously done for the ZL entrance functionalization.8e Then, we select a symmetric PDI (Scheme 1, R = CH3) and position it right outside the opening, with its longest molecular axis parallel to the channel axis, as depicted in Fig. 1 – for PDI, this is clearly the only possible way to cross the channel entrance. The high temperatures needed to obtain PDI–ZL adducts indicate that the PDI loading is an activated process, with timescales normally not accessible to molecular simulations. To explore how PDIs penetrate through the channel, we need to augment the finite temperature sampling of first principles molecular dynamics9a using the metadynamics approach.9b To achieve this aim, we use as the collective variable (CV) the average distance of the PDI core atoms from the ZL entrance ring,9c,dFig. 1a. After 5.0 ps equilibration at a temperature of 200 °C, the metadynamics simulation starts with the PDI molecule in front of the entrance, as in Fig. 1a, and evolves as featured in the Movie (ESI†), to end up with the free energy profile shown in Fig. 1b.
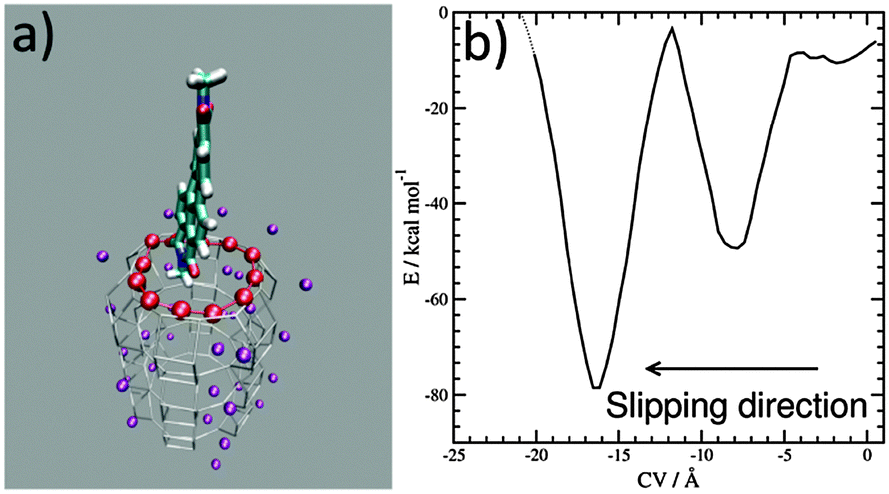 | ||
| Fig. 1 (a) Representation of the entrance ring of the channel and of the PDI. The Al and Si atoms (gray sticks) and the K+ cations (purple spheres) of ZL are also shown. The 12 O of the opening ring, involved in the CV definition, are highlighted as red spheres. The PDI is represented as sticks (C, cyan; O, red; N, blue; H, white). (b) Free energy profile (kcal mol−1) for the slipping of the PDI inside a channel as a function of the CV (Å). The free energy profile was calculated up to a value of −20.0 Å of the CV, and broadened with a Gaussian of 1.0 Å FWHM. | ||
Such a profile is characterized by a nearly flat initial region, followed by a deep well; then, a free energy increase occurs, followed by an even deeper well. This means that the transit of PDIs through the channel opening is a directionally asymmetric process:10 the entrance is favoured over the exit, and the molecule gets finally encapsulated into the porous matrix.
The first and nearly flat region of the curve (CV: from 0 Å to −5 Å, E ≈ −9 kcal mol−1) corresponds to the initial stages of the PDI encapsulation, Fig. 2a and b: only the relatively smaller and more polar part of the molecule, the diimide fragment, has passed through the channel entrance. In the first minimum region (CV ≈ −8 Å, E ≈ −48 kcal mol−1), roughly half of the molecule has slipped inside the channel: two of the PDI carbonyl groups interact with K+ cations of the first (and outermost) ZL cell, while the other two are still outside of the opening, Fig. 2d. In the second and deeper minimum (CV ≈ −16 Å, E ≈ −78 kcal mol−1), the PDI is fully encapsulated, and its carbonyl groups interact with K+ cations both in the 1st and the 2nd cell of the zeolite, Fig. 2f. Conversely, in the free energy maximum (CV ≈ −12 Å, E ≈ −4 kcal mol−1) the molecule moves from the 1st cell to the 2nd one and none of its carbonyl groups are involved in interactions with the zeolite K+ cations, Fig. 2e. The carbonyl–K+ interactions,8a therefore, trigger the slipping and may also rule the diffusion of PDIs inside the ZL channel system because of their substantial stabilizing effect: the interaction of a K+ cation with a PDI carbonyl oxygen amounts to 26.1 kcal mol−1. Another possible factor favouring the PDI inclusion could be a charge–π interaction, with the zeolite K+ cation positioned on top of the aromatic PDI plane. This interaction, although much weaker (9.9 kcal mol−1 (ESI,† Fig. S3)), may play a key role when the K+ cations are not bound to the PDI carbonyls, thus facilitating the transit of the molecule from the first to the second ZL cage (Fig. 2e).
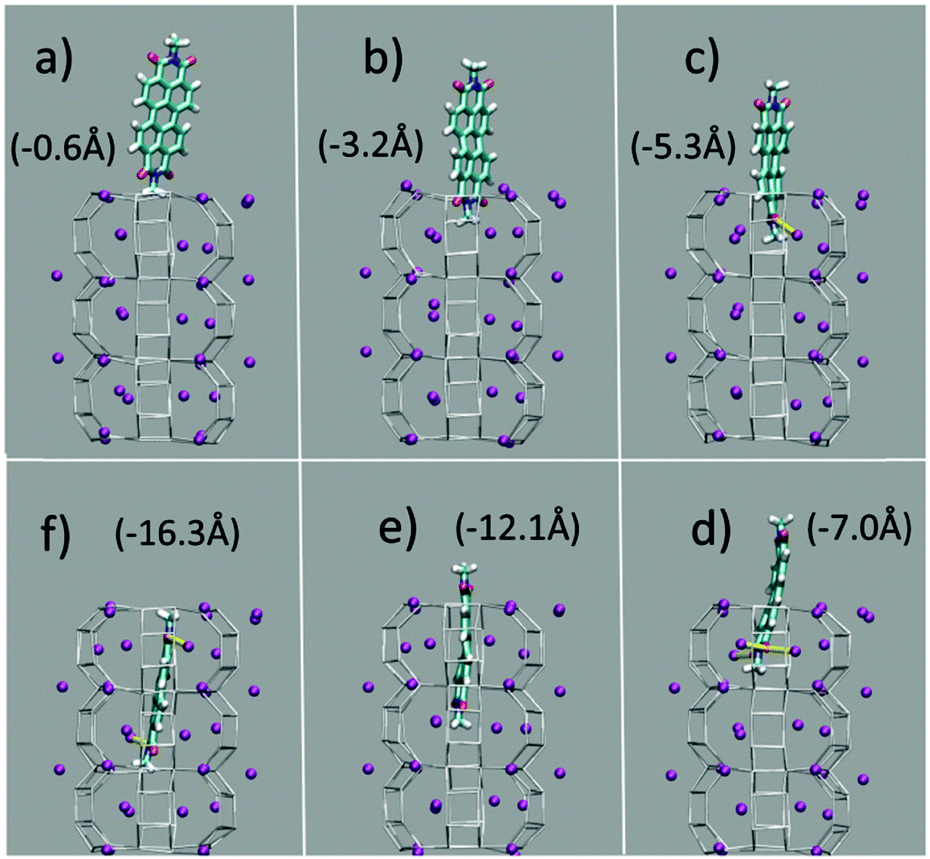 | ||
| Fig. 2 (a–f) Clockwise from the top left. They represent 6 relevant instantaneous configurations taken from the metadynamics simulation; the corresponding values of the CV parameter are in parentheses (see also the Movie, ESI†). Color code: The Si and Al of the framework are gray sticks. C, cyan; H, white; N, blue; carbonyl O, red; K+, purple. Coordination of K+ to the carbonyl oxygen atoms is highlighted by yellow sticks. | ||
The question then arises as to whether the porous inorganic framework is an active player in the slipping process.11 To find an answer, we take a closer look at the ZL structural changes as a function of the PDI penetration. The channel opening, defined by 2 sets of crystallographically different oxygens O1 and O2, is a 12-membered (12M) ring,6 which is slightly narrower along the O1–O1 directions (Fig. 3, left). Hence, the entrance process of a spherical object is controlled by the O1–O1 diameter. The average value of this quantity calculated for the entrance ring is larger compared to the ring positioned just below the entrance (Table 1, Fig. 3 and 4, and ESI,† Table S2). This result suggests a funnel-like structure of the ZL channel opening, which might well facilitate the PDI entrance, especially under the high temperature conditions adopted in the inclusion experiments.5d
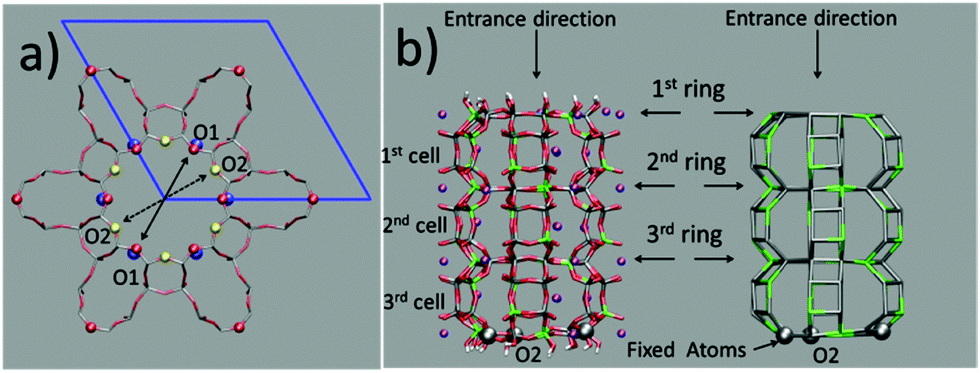 | ||
| Fig. 3 (a) Representation of the 12M channel section in the ab plane. The ZL framework atoms are represented as sticks (red for O and gray for T (Si/Al)), except for O1 (red spheres) and O2 (yellow spheres). The K+ facing the channel walls are blue spheres. The unit cell is represented as blue lines. The continuous arrow indicates one of the 3 O1–O1 and the dashed arrow one of the 3 O2–O2 diameters. (b) Left: All atom representation, parallel to the c axis, of the 3-cell model slab of the ZL. Al, green; Si, gray; O, red; H, white; K+, purple. The gray spheres represent the Si atoms held fixed along the simulation. A representation of only the Si and Al of the framework is on the right. The vertical arrow indicates the entrance direction aligned to a row of O2. The horizontal arrows indicate the 1st, 2nd and 3rd 12M rings. | ||
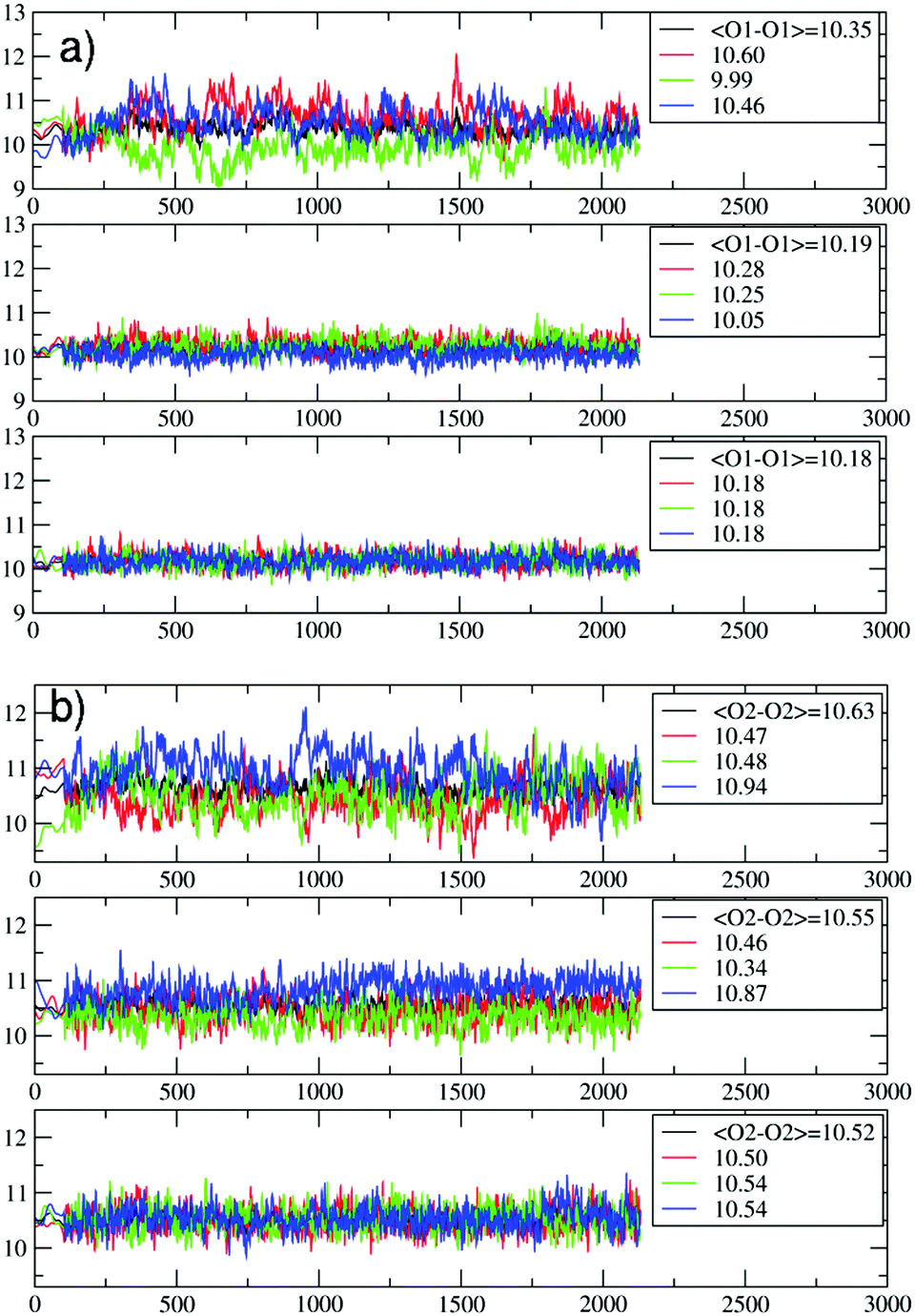 | ||
| Fig. 4 Evolution of the O1–O1 (a) and the O2–O2 (b) distances (in Å) during the PDI slipping in the metadynamics trajectory at 200 °C. From top to bottom, the distances refer to the 1st (the channel opening), the 2nd and the 3rd 12M ring (Fig. 3). In (b), the blue lines refer to the O2–O2 diameters along which the PDI slipping occurs (Fig. 5). | ||
| Ring | O1–O1 | O2–O2 |
|---|---|---|
| 1st | 10.338 (0.764) | 10.649 (0.795) |
| 2nd | 10.048 (0.735) | 10.547 (0.785) |
| 3rd | 10.067 (0.737) | 10.501 (0.780) |
| ∞ | 10.015 (0.732) | 10.475 (0.778) |
The temperature effects on the 12M ring opening are actually remarkable, and can be appreciated by looking at the variation of the O1–O1 and O2–O2 diameters along the 200 °C trajectory, depicted in Fig. 4. All distances show large temperature induced oscillations, of the order of 0.1 nm. As 200 °C corresponds to 329 cm−1, these oscillations should be ascribable mainly to the bending motion of the Si–O–Si (Al–O–Si) and O–Si–O (O–Al–O) angles (typical frequencies below 400 cm−1) and to collective vibrational modes of lower energy.12 What is to notice is that the oscillations do not depend solely on thermal effects, but are correlated with and driven by the slipping PDI. During the passage across the 1st and 2nd 12M rings, the O2–O2 distances which the PDI is aligned to show both the greatest average values and the largest amplitude oscillations, indicating that the dye, due to its size, has jostled its way to slip inside. This fact clearly emerges by observing that the structural changes of the 1st and 2nd rings along the process are indeed quite different from those of the 3rd and innermost ring, Fig. 4. Because the PDI never crossed that ring in the sampled configurations, the thermal oscillations are equally distributed among its O1–O1 and O2–O2 distances (Fig. 4a and b, bottom panels). On the other hand, in the first two rings, which are crossed by the transiting molecule, a large elongation of one O2–O2 diameter implies a shortening of the other diameters, hence an elliptic deformation of the ring. We therefore conclude that the channel gets locally deformed through a PDI-induced redistribution of the thermally activated – and otherwise random – oscillations of the zeolite framework structure (Fig. 5).
 | ||
| Fig. 5 The elliptic deformation of the 12M ring during the PDI transit. Left: Projection in the ab plane. Right: Lateral view of the same configuration. O1 and O2 atoms are represented as red and yellow spheres respectively; the red lines are guides to the eye. The configuration, featuring the largest elliptic deformation of the 12M ring opening, is extracted from the region of the maximum in the free energy profile. In this configuration, the maximum diameter of the 1st ring is 11.997 Å and the minimum is 9.686 Å. | ||
We have shown that the size and structure of PDI greatly influence the ZL framework oscillations, which are essential for the dye encapsulation. A closer inspection of the trajectory reveals that the vibrations of the PDI molecule also actively contribute to this process. Initially, the PDI enters the channel with its short axis oriented along one of the O1–O1 diameters, as shown in Fig. 2a and b, because the K+ sites facing the channel wall are located just below the O1 sites, Fig. 3. Due to the strong interaction with the PDI's carbonyl groups, these K+ cations draw the molecule to the entrance; however, then the dye reorients, and eventually slips inside the channel through the widest passage, i.e., along one of the O2–O2 diameters, Fig. 2c and d.
Such a molecular reorientation process, instrumental in the PDI insertion, is very far from being a rigid rotation of the aromatic backbone: it occurs, indeed, via an out-of-plane distortion of the molecule, Fig. 2c. This distortion corresponds just to the lowest energy normal mode of the PDI (ESI,† Fig. S4). Altogether, our results therefore indicate that both ZL and PDIs exploit their low energy vibrational modes, cooperatively, for reaching the innermost and deepest free energy minimum, thus accomplishing the encapsulation process.
In conclusion, the answer to the question of how PDIs can slip into ZL channels and travel inside despite their apparently too large width can be summarized by the following points: (i) the funnel-like shape of the channel opening does facilitate the initial phase of the PDI entrance; (ii) the stabilizing interactions among the PDI carbonyl oxygens and the ZL K+ cations provide the main energetic driving force; (iii) the cooperative vibrational modes of the host and the guest play so well to enable these processes, via a local elliptic deformation of the channel; and (iv) the asymmetry in the free energy profile clearly indicates that the PDI entrance process is favoured over the PDI exit.
Overall, the self-assembly of PDI–ZL adducts is possible, at high temperatures, because of the thermodynamically induced unidirectional motion of PDIs. This motion is combined with cooperative vibrational modes, which allow overcoming the barrier imposed by the structure of the channels. It is probable that similar mechanisms could govern the build-up and behaviour of other classes of host–guest composites.13
The present work was supported by the Italian MIUR through the projects ImPACT (FIRBRBFR12CLQD), INFOCHEM (PRIN2010CX2TLM_006), and Insubria University FAR2014. The CINECA supercomputing center (Italy) is acknowledged for computing time (ISCRA project DENARI, HP10B6JTDP).
Notes and references
- (a) G. Calzaferri, S. Huber, H. Maas and C. Minkowski, Angew. Chem., Int. Ed., 2003, 42, 3732 CrossRef CAS PubMed; (b) G. Calzaferri, M. Pauchard, H. Maas, S. Huber, A. Khatyr and T. Schaafsma, J. Mater. Chem., 2002, 12, 1 RSC.
- (a) S. Fibikar, G. Luppi, V. Martínez-Junza, M. Clemente-Léon and L. De Cola, ChemPlusChem, 2015, 80, 62 CrossRef CAS; (b) A. Bertucci, H. Lülf, D. Septiadi, A. Manicardi, R. Corradini and L. De Cola, Adv. Healthcare Mater., 2014, 3, 1812 CrossRef CAS PubMed; (c) P. Li, Y. Wang, H. Li and G. Calzaferri, Angew. Chem., Int. Ed., 2014, 53, 2904 CrossRef CAS PubMed; (d) J. M. Beierle, R. Roswanda, P. M. Erne, A. C. Coleman, W. R. Browne and B. L. Feringa, Part. Part. Syst. Charact., 2013, 30, 273 CrossRef CAS; (e) M. Tsotsalas, K. Kopka, G. Luppi, S. Wagner, M. P. Law, M. Schäfers and L. De Cola, ACS Nano, 2010, 4, 342 CrossRef CAS PubMed; (f) B. Schulte, M. Tsotsalas, M. Becker, A. Studer and L. De Cola, Angew. Chem., Int. Ed., 2010, 49, 6881 CrossRef CAS PubMed; (g) S. Hashimoto, K. Samata, T. Shoji, N. Taira, T. Tomita and S. Matsuo, Microporous Mesoporous Mater., 2009, 117, 220 CrossRef CAS; (h) F. Cucinotta, Z. Popovic, E. A. Weiss, G. M. Whitesides and L. De Cola, Adv. Mater., 2009, 21, 1142 CrossRef CAS; (i) L.-Q. Dieu, A. Devaux, I. López-Duarte, M. V. Martínez-Díaz, D. Brühwiler, G. Calzaferri and T. Torres, Chem. Commun., 2008, 1187 RSC; (j) G. Calzaferri and K. Lutkouskaya, Photochem. Photobiol. Sci., 2008, 7, 879 RSC; (k) H. Manzano, L. Gartzia-Rivero, J. Bañuelos and I. López-Arbeloa, J. Phys. Chem. C, 2013, 117, 13331 CrossRef CAS; (l) W. Insuwan, K. Rangsriwatananon, J. Meeprasert, S. Namuangruk, Y. Surakhot, N. Kungwan and S. Jungsuttiwong, Microporous Mesoporous Mater., 2014, 197, 348 CrossRef CAS; (m) L. Gartzia-Rivero, J. Bañuelos and I. López-Arbeloa, Int. Rev. Phys. Chem., 2015, 34, 515 CrossRef CAS; (n) D. Lencione, M. H. Gehlen, L. N. Trujillo, R. C. F. Leitao and R. Q. Albuquerque, Photochem. Photobiol. Sci., 2016, 15, 398 RSC.
- (a) P. Cao, O. Khorev, A. Devaux, L. Sägesser, A. Kunzmann, A. Ecker, R. Häner, D. Brühwiler, G. Calzaferri and P. Belser, Chem. – Eur. J., 2016, 22, 4046 CrossRef CAS PubMed; (b) L. Gigli, R. Arletti, G. Tabacchi, E. Fois, J. G. Vitillo, G. Martra, G. Agostini, S. Quartieri and G. Vezzalini, J. Phys. Chem. C, 2014, 118, 15732 CrossRef CAS; (c) M. Busby, A. Devaux, C. Blum, V. Subramaniam, G. Calzaferri and L. De Cola, J. Phys. Chem. C, 2011, 115, 5974 CrossRef CAS.
- (a) J. Kärger, D. M. Ruthven and D. N. Theodorou, Diffusion in Nanoporous Materials, Wiley-VCH Verlag, Weinheim, Germany, 2012 Search PubMed; (b) A. Einstein, Ann. Phys., 1905, 17, 549 ( Ann. Phys. , 1906 , 19 , 371 ) CrossRef CAS.
- (a) F. Feil, S. Naumov, J. Michaelis, R. Valiullin, D. Enke, J. Kärger and C. Bräuchle, Angew. Chem., Int. Ed., 2012, 51, 1152 CrossRef CAS PubMed; (b) A. R. Dutta, P. Sekar, M. Dvoyashkin, C. R. Bowers, K. J. Ziegler and S. Vasenkof, Chem. Commun., 2015, 51, 13346 RSC; (c) H. Jobic, Phys. Chem. Chem. Phys., 2016, 18, 17190, 10.1039/c6cp00410e; (d) S. Pagliara, S. L. Dettmer and U. F. Keyser, Phys. Rev. Lett., 2014, 113, 048102 CrossRef PubMed; (e) W. E. Moerner, Y. Shechtman and Q. Wang, Faraday Discuss., 2015, 184, 9 RSC; (f) S. H. Parekh and K. F. Domke, Chem. – Eur. J., 2013, 19, 11822 CrossRef CAS PubMed; (g) S. Ito, Y. Taga, K. Hiratsuka, S. Takei, D. Kitagawa, S. Kobatake and H. Miyasaka, Chem. Commun., 2015, 51, 13756 RSC; (h) Z. Ristanović, J. P. Hofmann, M.-I. Richard, T. Jiang, G. A. Chahine, T. U. Schülli, F. Meirer and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 7496 CrossRef PubMed.
- (a) International Zeolite Association, http://www.iza-structure.org; (b) T. Ohsuna, B. Slater, F. Gao, J. Yu, Y. Sakamoto, G. Zhu, O. Terasaki, D. E. W. Vaughan, S. Qiu and C. R. A. Catlow, Chem. – Eur. J., 2004, 10, 5031 CrossRef CAS PubMed; (c) L. Gigli, R. Arletti, S. Quartieri, F. Di Renzo and G. Vezzalini, Microporous Mesoporous Mater., 2013, 177, 8 CrossRef CAS; (d) J. M. Newsam, J. Phys. Chem., 1989, 93, 7689 CrossRef CAS.
- V. Van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell and C. R. A. Catlow, Chem. Soc. Rev., 2015, 44, 7044 RSC.
- (a) E. Fois, G. Tabacchi and G. Calzaferri, J. Phys. Chem. C, 2010, 114, 10572 CrossRef CAS; (b) E. Fois, G. Tabacchi and G. Calzaferri, J. Phys. Chem. C, 2012, 116, 16784 CrossRef CAS; (c) E. Fois, G. Tabacchi, A. Devaux, P. Belser, D. Brühwiler and G. Calzaferri, Langmuir, 2013, 29, 9188 CrossRef CAS PubMed; (d) X. Zhou, T. A. Wesolowski, G. Tabacchi, E. Fois, G. Calzaferri and A. Devaux, Phys. Chem. Chem. Phys., 2013, 15, 159 RSC; (e) G. Tabacchi, E. Fois and G. Calzaferri, Angew. Chem., Int. Ed., 2015, 54, 11112 CrossRef CAS PubMed.
- (a) R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471 CrossRef CAS PubMed; (b) A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562 CrossRef CAS PubMed; (c) G. Tabacchi, S. Silvi, M. Venturi, A. Credi and E. Fois, ChemPhysChem, 2016, 17, 1913, DOI:10.1002/cphc.201501160; (d) http://www.cpmd.org Copyright IBM Corp. 1990–2015, MPI für Festkörperforschung Stuttgart 1997–2001.
- C. Ceriani, E. Fois, A. Gamba, G. Tabacchi, O. Ferro, S. Quartieri and G. Vezzalini, Am. Mineral., 2004, 89, 102 CrossRef CAS.
- (a) R. M. Barrer and D. E. W. Vaughan, Trans. Faraday Soc., 1971, 67, 2129 RSC; (b) P. Demontis, E. S. Fois, G. B. Suffritti and S. Quartieri, J. Phys. Chem., 1990, 94, 4329 CrossRef CAS; (c) J.-R. Hill and J. Sauer, J. Phys. Chem., 1995, 99, 9536 CrossRef CAS; (d) E. Fois, A. Gamba, E. Spanò and G. Tabacchi, J. Mol. Struct., 2003, 644, 55 CrossRef CAS; (e) E. Fois, A. Gamba, G. Tabacchi, S. Quartieri and G. Vezzalini, Phys. Chem. Chem. Phys., 2001, 3, 4158 RSC; (f) L. Giussani, E. Fois, E. Gianotti, G. Tabacchi, A. Gamba and S. Coluccia, ChemPhysChem, 2010, 11, 1757 CrossRef CAS PubMed; (g) S. E. Boulfelfel, P. I. Ravikovitch and D. S. Sholl, J. Phys. Chem. C, 2015, 119, 15643 CrossRef CAS; (h) A. Ghysels, S. L. C. Moors, K. Hemelsoet, K. De Wispelaere, M. Waroquier, G. Sastre and V. Van Speybroeck, J. Phys. Chem. C, 2015, 119, 23721 CrossRef CAS; (i) A. J. O'Malley, C. R. A. Catlow, M. Monkenbusch and H. Jobic, J. Phys. Chem. C, 2015, 119, 26999 CrossRef; (j) J. J. Gutiérrez-Sevillano, S. Calero, S. Hamad, R. Grau-Crespo, F. Rey, S. Valencia, M. Palomino, S. R. G. Balestra and A. R. Ruiz-Salvador, Chem. – Eur. J., 2016, 22, 10036, DOI:10.1002/chem.201600983.
- (a) P. Bornhauser and G. Calzaferri, J. Phys. Chem., 1996, 100, 2035 CrossRef CAS; (b) A. M. Bieniok and H.-B. Bürgi, J. Phys. Chem., 1994, 98, 10735 CrossRef CAS.
- E. Roduner and S. G. Radhakrishnan, Chem. Soc. Rev., 2016, 45, 2768 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Computational set-up, tables and figures related to the computations, further details and a movie of the penetration process. See DOI: 10.1039/c6cc05303c |
| This journal is © The Royal Society of Chemistry 2016 |