
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeed-mediated biomineralizaton toward the high yield production of gold nanoprisms†
Xi
Geng
,
Kristina L.
Roth
,
Megan C.
Freyman
,
Jianzhao
Liu
and
Tijana Z.
Grove
*
Department of Chemistry, Virginia Polytechnic Institute and State University, Blacksburg, VA 24060, USA. E-mail: tijana.grove@vt.edu
First published on 12th July 2016
Abstract
Gold nanotriangles (Au NTs) with tunable edge length were synthesized via a green chemical route in the presence of the designed consensus sequence tetratricopeptide repeat (CTPR) protein, halide anions (Br−) and CTPR-stabilized Ag seeds. The well-defined morphologies, tailored plasmonic absorbance from visible-light to the near infrared (NIR) region, colloidal stability and biocompatibility are attributed to the synergistic action of CTPR, halide ions, and CTPR-stabilized Ag seeds.
Over the past decade, tremendous attention and research efforts have been devoted to the synthesis of gold (Au) nanoplates. Because of their highly anisotropic structure and localized surface plasmon resonance (LSPR) properties, such nanostructures are exceptionally well suited for biomedical applications such as biosensing,1 diagnostics and therapeutics.2 The synthesis of planar Au nanoparticles (NPs) is typically achieved via cetyltrimethylammonium bromide/chloride (CTAB/CTAC)-based protocols.3–10 However, biomedical applications necessitate tedious and stringent purification processes for the complete removal of toxic cationic surfactants.11,12 A growing interest has thus been focused on the biocompatible and green synthetic approaches that mimic natural biomineralizaton process.13 In previous studies, plant extracts,14,15 amino acids,16 bovine serum albumin (BSA)17,18 amyloid fibril19 and other shape directing proteins20 have demonstrated dual functions as stabilizers and reducing agents to produce anisotropic Au nanoplates. In addition,21,22 Good's buffers have been reported to generate gold nanocrystals at ambient condition.23,24 However, one-pot synthetic strategies typically resulted in broad morphological distribution of NPs. Seed-mediated growth along with the addition of shape-directing halides has been invoked as the most potent tools for directing the anisotropic growth of noble metal NPs.9,10,25–28 The exquisite shape control is mainly realized through kinetic control as well as the preferential binding to the low index facets. As demonstrated in the present work, the incorporation of seed-mediated techniques into conventional biomineralizaton has the potential to provide unprecedented control over NP size and shape while maintaining biocompatibility requirements. To our knowledge, this is the first report of the synergistic action of protein, halide and protein-stabilized seeds for the efficient formation of triangular Au nanoprisms with narrow morphological distribution, excellent colloidal stability and low cytotoxicity.
The details of the synthetic process are depicted in (Scheme S1, ESI†). Briefly, the CTPR protein (Fig. S1, ESI†) and sodium bromide were employed as the shape directing agents, while 3-(N-morpholino)propanesulfonic acid (MOPS) was used as a mild reducing agent.29 CTPR protein is a de novo protein sequence based on the tetratricopeptide repeat family.30 Repeat proteins have attracted attention of the biotechnology community for their modular structure and ease of engineering.31–35 Although not specifically designed for biomineralizaton, CTPR proteins have demonstrated remarkable utility in the synthesis of noble metal nanoparticles29,36 and nanoclusters.37 In a typical synthetic procedure, CTPR3 was first mixed with gold precursor, followed by the addition of aqueous NaBr solution. Upon injection of MOPS, the solution rapidly turn colorless indicating the conversion of Au(III) to Au(I). The overall reduction will proceed from one hour up to a couple of days depending upon the concentration of CTPR3 and NaBr. In the presence of 10 μM CTPR3 protein, but without bromide ion (Scheme S1-I, ESI†) small nanospheres (14.1 ± 5.6 nm) are produced (Fig. 1b). In comparison, a mixture of Au nanoprisms, five-fold penta-twinned Au NPs (decahedrons) and Au nanospheres are produced in the presence of both CTPR3 and NaBr (Scheme S1-ii, ESI† and Fig. 1c). The formation of anisotropic Au NPs is consistent with the broad shoulder and asymmetric shape of the corresponding UV-Vis spectrum (Fig. 1a, ii). In addition to Au NTs, the decahedrons with multi-fold twinning structure are also generated, as evidenced by the twin-grain boundaries and periodical fast Fourier transform (FFT) diffraction patterns (Fig. S4e and f, ESI†). Although the reaction condition depicted in Scheme S1-ii, ESI,† produces satisfactory yield of anisotropic Au NTs, their sizes and shapes are polydispersed (Fig. S5a, ESI†) due to the inherent drawbacks of one-pot synthesis such as unavoidable self-nucleation event and poor quality of seeds with diverse crystal structures.38 Aiming for high-yielding production of Au NTs with lower morphological polydispersity, CTPR3-stabilized Ag seeds were deliberately added to promote the growth of Au NTs (Scheme S1-iii, ESI† and Fig. 1e). In a general sense, the addition of Ag seed will induce the catalytic deposition of Au(0) atoms onto the planar-twinned Ag surface without causing disruptive galvanic etching, particularly under the condition of adequate amount of NaBr and CTPR3.39,40 Once 10 μl of CTPR3-Ag seeds36 with inherent stacking faults are introduced (Scheme S1-iii, ESI†), the majority of NPs are Au NTs (Fig. 1d and e). This morphological transition is accompanied by a pronounced red shift of the LSPR peak to 667 nm (Fig. 1a, iii). Interestingly, efficient formation of Au NTs was also realized using 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (Fig. S3, ESI†) implying that the choice of reducing agents potentially could be further extended to other biologically benign alternatives.
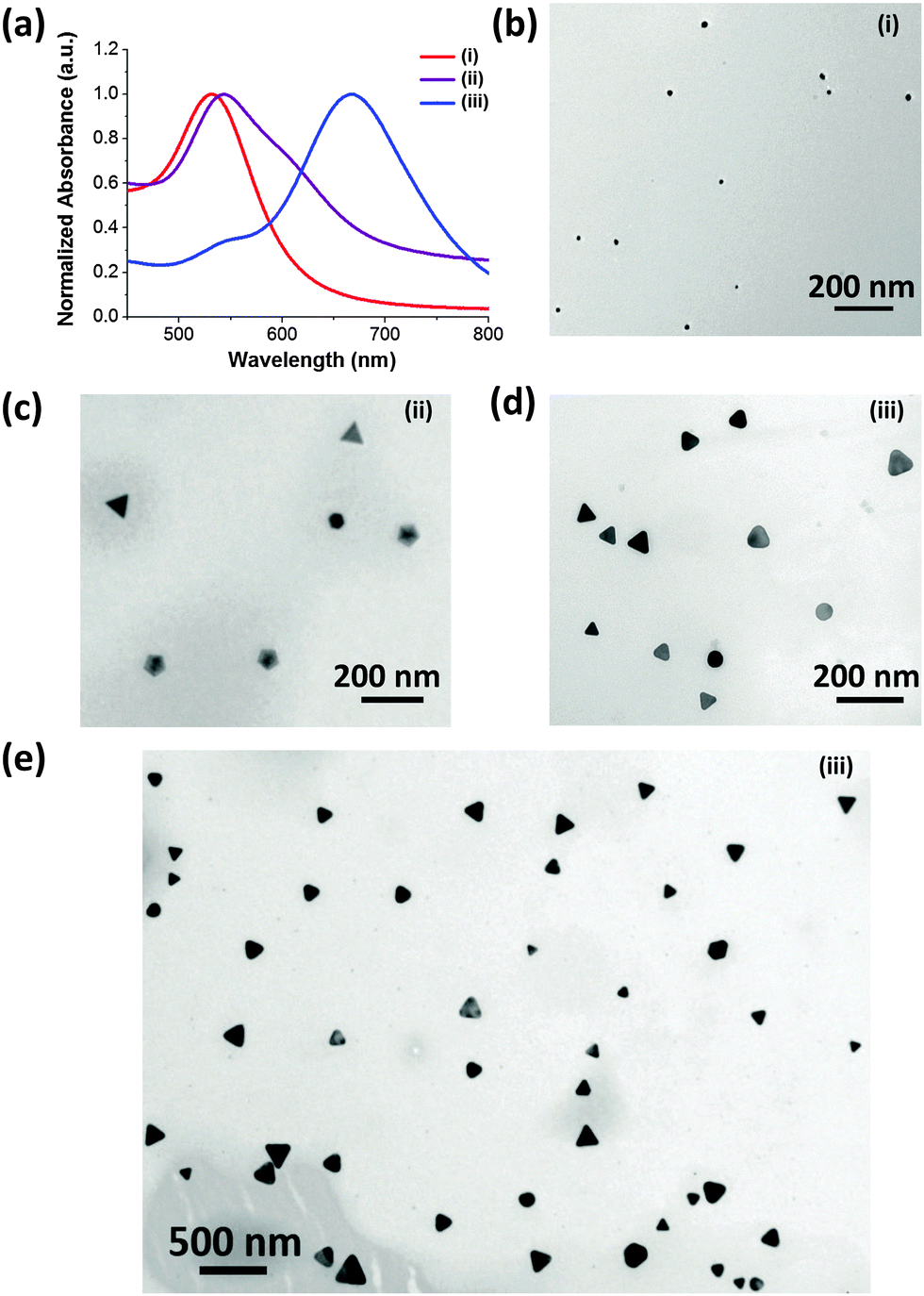 | ||
| Fig. 1 (a) UV-Vis extinction spectra and (b–e) TEM images of Au NPs prepared using varied experimental conditions: (i) CTPR3 only; (ii) CTPR3 and NaBr, (iii) CTPR3, NaBr, and Ag seeds. | ||
The single crystalline fcc structure Au NTs was identified by high resolution TEM (HRTEM) and hexagonal pattern of selected area electron diffraction (SAED) (Fig. S4a and b, ESI†). The measured interplanar spacing is 0.235 nm, consistent with the lattice parameter of Au(111) (Fig. S4a, ESI†). The composition of Au NTs was characterized using energy-dispersive X-ray spectroscopy (EDS) (Fig. S4c, ESI†). The smooth atomic force microscopy (AFM) profile is indicative of the planar top face of Au NTs (Fig. S4d, ESI†) with the measured thickness between 8–15 nm, mainly depending on the amount of Ag seeds added.
It has been demonstrated that the LSPR features for Au NTs are closely associated with their edge length, thickness, and the tips sharpness.13 As shown in the Fig. 2a, the LSPR of Au NTs can be conveniently tuned to span a broad range from visible-light to near infrared (NIR) region by simply adjusting the amount of Ag seeds added in the initial growth stage. The shift in dipole plasmon resonance agrees very well with the change in the NT edge-length observed in TEM images. The edge length of the Au NTs could be finely tailored from 61 ± 7 to 137 ± 33 nm (Fig. 2b–d). Notably, the yield of Au NTs is approximately 75% of the overall population without any purification process (Fig. S5b, ESI†), which will increase to 85% by harvesting after sedimentation process (Fig. 1e and Fig. S5c, ESI†). In sharp contrast, Au NTs with broader size distribution (41 ± 14 nm) are obtained using one pot biomineralizaton without adding Ag seed. Intriguingly, the addition of Ag seeds also bring great benefits by significantly reducing the use of NaBr and CTPR without compromising the overall yield. A strong dipole LSPR peak was identified for the Au NTs sample prepared in the presence of 2 μM CTPR3 and 5 mM NaBr (Fig. S6, ESI†).
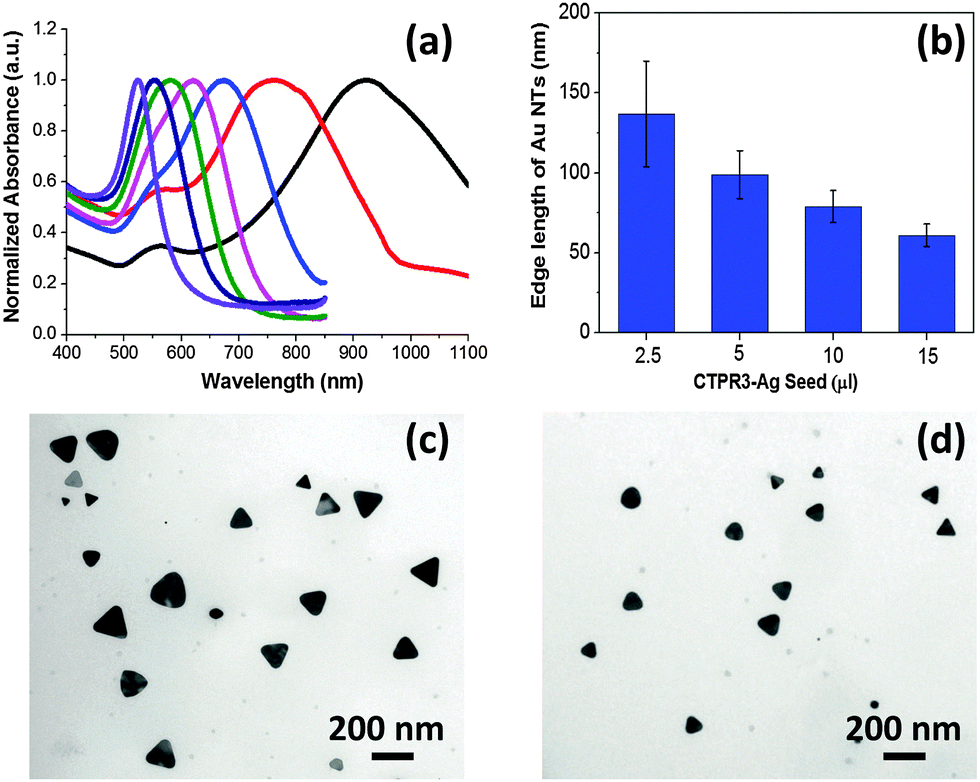 | ||
| Fig. 2 (a) UV-Vis extinction spectra of Au NPs prepared in the presence of 2.5 μl (black), 5 μl (red), 10 μl (light blue), 15 μl (magenta), 20 μl (green), 25 μl (dark blue), 50 μl (violet) Ag seeds. (b) Edge length of Au NTs prepared using varied amount of Ag seeds. TEM images of Au NTs prepared using (c) 2.5 μl and (d) 5 μl Ag seed. | ||
Proteins and peptides could be used as efficient capping agent to grant excellent colloidal stability and interesting physicochemical properties to the functional nanomaterials.41–43 We further explore the colloidal stability of the as-synthesized Au NTs and Au NTs in the fetal bovine serum (FBS). As shown in Fig. 3a, after Au NTs are incubated with FBS for 2 hours an increase in the hydrodynamic diameter of 30 nm is observed (Table S1, ESI†). The increase in diameter can be attributed to adsorption of proteins from the FBS solution. It is important to note that the colloidal stability of CTPR3-stabilized Au NTs is maintained during the centrifugation and re-dispersion processes and no agglomeration of NTs occurs after incubation with FBS. As expected, CTRP3-stabilized Au NTs have negative zeta potential due to the overall negative charge of the CTPR3. After incubation with FBS the zeta potential increases to −17.9 mV (Table S1, ESI†), which is in agreement with previous reports of −20 mV zeta potential for Au NPs after FBS incubation independent of the initial NP surface charge.11,44 The protein corona can also serve as protecting agent to lower the cytotoxicity of nanomaterials.45 To that end, mouse brain endothelial cells (MBECs) were incubated with the as-prepared CTPR-stabilized Au NTs for 6 hours and cell viability was measured using MTT assay. For Au NT concentration range 5–40 μg mL−1 no concentration dependent cell toxicity was observed (Fig. 3b).
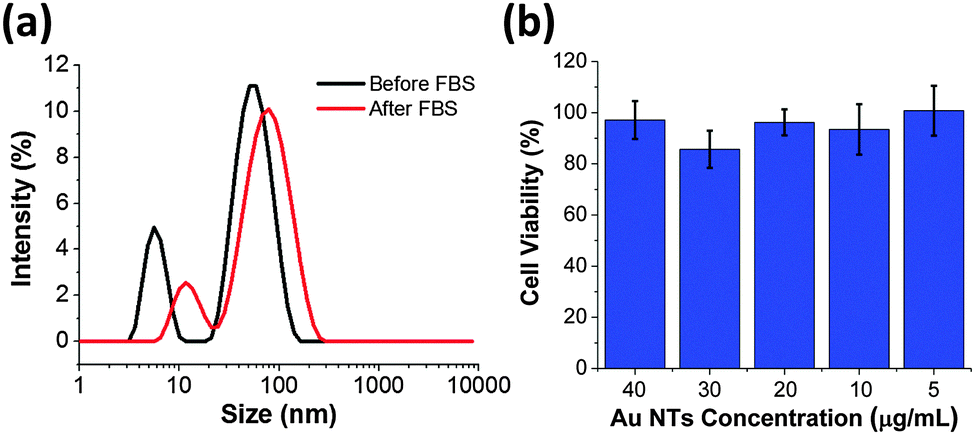 | ||
| Fig. 3 (a) DLS of Au NTs before and after incubation with fetal bovine serum (FBS). (b) Percent of viable MBEC cells determined from MTT assay after incubation with varied concentrations of Au NTs (5–40 μg). | ||
Even though the detailed growth mechanism is still elusive, we try to postulate a possible mechanism for the seed-mediated biomineralizaton. We have recently shown that the binding affinity of Au species to CTPR3 is driven mainly by metal–π interactions with tyrosine and tryptophan side chains and hydrogen bonding to asparagine side-chain.48,49 Moreover, the overall binding constant is ionic strength dependent. Interestingly, CTPR contains no cysteine or a specific gold binding sequence such as AG3.46,47 To elucidate the roles of CTPR3 and bromide we first performed syntheses at the fixed ratio of HAuCl4 and MOPS (0.2 mM: 40 mM), but either the concentration of CTPR3 or NaBr was adjusted. In the presence of 100 mM NaBr, the overall yield of anisotropic NPs increases as the concentration of CTPR3 increases. For instance, irregular sheet-like NPs are produced in the absence of CTPR3 (Fig. S7a, ESI†), limited number of Au NTs with rounded tips (<10%) are generated when concentration of CTPR3 is less than 1 μM (Fig. S7b, ESI†). But as concentration of CTPR3 reaches 4 μM, 35% of the NP population are Au NTs (Fig. S7c, ESI†). Further increase in CTPR3 concentration up to 20 μM only slightly improved the overall yield of Au NTs at the cost of extended reaction time (>2 weeks). On the other hand, increasing the concentration of NaBr will also facilitate the growth of anisotropic Au NPs (Fig. S8a and b, ESI†), which is consistent with the previous report.50 Nevertheless, when excess surface capping agent, either CTPR3 or NaBr is present, the reduction reaction will be dramatically inhibited and the free gold precursor will not be consumed efficiently to provide feedstock of Au(0) thereby hindering the growth of planar Au NTs. The yield of Au NTs declined and ill-defined NPs are formed after adding 300 mM NaBr (Fig. S8c, ESI†). The highest yield of Au NTs was reached in the presence of 10 μM CTPR3 and 100 mM NaBr. Under optimized synthetic conditions, approximately 70% of NPs are bounded with (111) facets, among which Au NTs accounted for 75% of the entire population (Fig. S5a, ESI†). The prevalence of Au NTs and decahedrons implies that the coexistence of protein and NaBr promotes the nucleation of seeds with either planar or penta-twinned defects.
Herein we postulate that at the initial stage of the one-pot synthesis (Scheme S1-ii, ESI†), Au nuclei with planar or penta-twinning structures are produced. The former will assemble into triangle-like pattern at the localized domain. This phenomenon was previously observed in the synthesis of Au nanoplates using extract of lemongrass and BSA.14,51 Au NPs with corrugated and stepped edges are observed after 12 and 24 h consistent with the continuous deposition of gold atoms onto the triangle analogues (Fig. S9, ESI†). Interaction between CTPR3 and Au(III) is stronger at higher ionic strengths, thus as the concentration of NaBr increases more Au(III) ions will be complexed with the protein influencing the reduction kinetics.49 At intermediate NaBr concentration there will be less free Au(III) ions accessible to the (111) faces with lower surface energy and chemical reactivity as compared with the stepped edges bearing high-density defects. CTPR3 NH2 or NH3+ pendant groups may also interact with Br− and enhance the steric hindrance analogous to the zipping mechanism of CTAB.52 The (111) faces are further passivated by the adsorption of Br− and CTPR3 via the surfactant templating or face-blocking process.53 This scenario is supported by the compositional distribution of Br, O and N enriched on the (111) facets of Au NTs as shown in the Fig. S10 (ESI†). Eventually, the corrugated and stepped face gradually diminished whilst the Au NTs and decahedrons bounded with (111) faces are generated. Unfortunately, the broad distribution of crystallinity of nuclei along with the concomitant self-nucleation of isotropic NPs restrict the precise control over the size and shape. Once CTPR3-stabilized Ag seeds with innate planar twinning structure are added (Scheme S1-iii, ESI†), the initial self-nucleation process will be appreciably circumvented. It has been well recognized that the twinned crystal seeds prompt the lateral growth of 2D planar nanostructures by providing low-energy re-entrant groves.13 Consequently, the as-reduced Au(0) will be predominantly deposited onto the facets – other than (111) – of planar twinned Ag nanocrystals in a rapid manner thereby improving the yield along with the quality of the Au NTs. As observed in STEM-EDX experiments, the trace amount of Ag element is evenly distributed over the entire area of the Au NTs rather than accumulated in the core (Fig. S10 and S11, ESI†). This observation is consistent with the dissolution and diffusion of Ag into the Au NTs, particularly when exposed to the light and under the condition of high halide concentration.
In summary, we have developed a facile and high-yielding green methodology for the syntheses of anisotropic Au nanoprisms at ambient condition, in which NaBr, CTPR as well as CTPR-stabilized Ag seeds have imposed synergistic effects upon the morphology of the Au NPs. Indeed, since the CTPR sequence was not specifically designed or selected for binding Au, it is tempting to propose that the physico-chemical properties of biomolecules (e.g. pI, number of aromatic side-chains, etc.) are more important for efficient synthesis of anisotropic Au nanostructures than the actual specific binding motifs. Thus this synthetic strategy can be further extended to a vast diversity of biomolecules as long as physico-chemical properties are optimized. One can then easily envision “designer coronas” where specific molecular recognition moieties can be incorporated into the engineered proteins or peptides to match a biomedical application.54 Furthermore, this work demonstrates that the incorporation of seed-mediated growth into conventional biomineralizaton strategy yields noble metal NPs with unprecedented control over size and shape. Obtained Au NTs with well-defined morphologies exhibited tailored plasmonic absorbance ranging from visible to NIR region. Reported NPs are colloidally stable and biocompatible thus holding great promises for versatile biosensing and biomedical applications.
The authors would like to thank Dr Guoliang Liu for insightful comments, suggestions, and discussion on this research work. Authors acknowledge ICTAS Nanoscale Characterization and Fabrication Lab (NCFL) for the use of AFM and TEM. This work was in part supported by the JFC ICTAS grant number 119106 to TZG.
Notes and references
- S. R. Beeram and F. P. Zamborini, ACS Nano, 2010, 4, 3633–3646 CrossRef CAS PubMed.
- B. Pelaz, V. Grazu, A. Ibarra, C. Magen, P. del Pino and J. M. de la Fuente, Langmuir, 2012, 28, 8965–8970 CrossRef CAS PubMed.
- J. E. Millstone, S. Park, K. L. Shuford, L. Qin, G. C. Schatz and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 5312–5313 CrossRef CAS PubMed.
- J. E. Millstone, G. S. Métraux and C. A. Mirkin, Adv. Funct. Mater., 2006, 16, 1209–1214 CrossRef CAS.
- T. K. Sau and C. J. Murphy, J. Am. Chem. Soc., 2004, 126, 8648–8649 CrossRef CAS PubMed.
- Y. Huang, A. R. Ferhan, Y. Gao, A. Dandapat and D.-H. Kim, Nanoscale, 2014, 6, 6496–6500 RSC.
- L. Scarabelli, M. Coronado-Puchau, J. J. Giner-Casares, J. Langer and L. M. Liz-Marzán, ACS Nano, 2014, 8, 5833–5842 CrossRef CAS PubMed.
- L. Chen, F. Ji, Y. Xu, L. He, Y. Mi, F. Bao, B. Sun, X. Zhang and Q. Zhang, Nano Lett., 2014, 14, 7201–7206 CrossRef CAS PubMed.
- J. E. Millstone, W. Wei, M. R. Jones, H. Yoo and C. A. Mirkin, Nano Lett., 2008, 8, 2526–2529 CrossRef CAS PubMed.
- T. H. Ha, H.-J. Koo and B. H. Chung, J. Phys. Chem. C, 2007, 111, 1123–1130 CAS.
- A. M. Alkilany, P. K. Nagaria, C. R. Hexel, T. J. Shaw, C. J. Murphy and M. D. Wyatt, Small, 2009, 5, 701–708 CrossRef CAS PubMed.
- A. M. Alkilany and C. J. Murphy, J. Nanopart. Res., 2010, 12, 2313–2333 CrossRef CAS PubMed.
- J. E. Millstone, S. J. Hurst, G. S. Métraux, J. I. Cutler and C. A. Mirkin, Small, 2009, 5, 646–664 CrossRef CAS PubMed.
- S. S. Shankar, A. Rai, B. Ankamwar, A. Singh, A. Ahmad and M. Sastry, Nat. Mater., 2004, 3, 482–488 CrossRef CAS PubMed.
- B. Liu, J. Xie, J. Y. Lee, Y. P. Ting and J. P. Chen, J. Phys. Chem. B, 2005, 109, 15256–15263 CrossRef CAS PubMed.
- Y. Shao, Y. Jin and S. Dong, Chem. Commun., 2004, 1104–1105, 10.1039/B315732F.
- J. Xie, J. Y. Lee and D. I. C. Wang, J. Phys. Chem. C, 2007, 111, 10226–10232 CAS.
- L. Au, B. Lim, P. Colletti, Y.-S. Jun and Y. Xia, Chem. – Asian J., 2010, 5, 123–129 CrossRef CAS PubMed.
- C. Li, S. Bolisetty and R. Mezzenga, Adv. Mater., 2013, 25, 3694–3700 CrossRef CAS PubMed.
- J. Xie, J. Y. Lee, D. I. C. Wang and Y. P. Ting, Small, 2007, 3, 672–682 CrossRef CAS PubMed.
- N. Goswami, K. Zheng and J. Xie, Nanoscale, 2014, 6, 13328–13347 RSC.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962–975 CrossRef CAS PubMed.
- J. Xie, J. Y. Lee and D. I. C. Wang, Chem. Mater., 2007, 19, 2823–2830 CrossRef CAS.
- S. Saverot, X. Geng, W. Leng, P. J. Vikesland, T. Z. Grove and L. R. Bickford, RSC Adv., 2016, 6, 29669–29673 RSC.
- N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001, 17, 6782–6786 CrossRef CAS.
- W. Niu, L. Zhang and G. Xu, Nanoscale, 2013, 5, 3172–3181 RSC.
- M. L. Personick and C. A. Mirkin, J. Am. Chem. Soc., 2013, 135, 18238–18247 CrossRef CAS PubMed.
- S. E. Lohse, N. D. Burrows, L. Scarabelli, L. M. Liz-Marzán and C. J. Murphy, Chem. Mater., 2014, 26, 34–43 CrossRef CAS.
- X. Geng and T. Z. Grove, RSC Adv., 2015, 5, 2062–2069 RSC.
- E. R. G. Main, K. Stott, S. E. Jackson and L. Regan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5721–5726 CrossRef CAS PubMed.
- S. H. Mejias, J. Lopez-Andarias, T. Sakurai, S. Yoneda, K. P. Erazo, S. Seki, C. Atienza, N. Martin and A. L. Cortajarena, Chem. Sci., 2016 10.1039/C6SC01306F.
- T. Z. Grove, C. O. Osuji, J. D. Forster, E. R. Dufresne and L. Regan, J. Am. Chem. Soc., 2010, 132, 14024–14026 CrossRef CAS PubMed.
- N. A. Carter and T. Z. Grove, Biomacromolecules, 2015, 16, 706–714 CrossRef CAS PubMed.
- R. N. Parker and T. Z. Grove, Biochem. Soc. Trans., 2015, 43, 856–860 CrossRef CAS PubMed.
- A. L. Cortajarena, F. Yi and L. Regan, ACS Chem. Biol., 2008, 3, 161–166 CrossRef CAS PubMed.
- X. Geng, W. Leng, N. A. Carter, P. J. Vikesland and T. Z. Grove, J. Mater. Chem. B, 2016, 4, 4182–4190 RSC.
- P. Couleaud, S. Adan-Bermudez, A. Aires, S. H. Mejías, B. Sot, A. Somoza and A. L. Cortajarena, Biomacromolecules, 2015, 16, 3836–3844 CrossRef CAS PubMed.
- C. Gao, J. Goebl and Y. Yin, J. Mater. Chem. C, 2013, 1, 3898–3909 RSC.
- C. Gao, Z. Lu, Y. Liu, Q. Zhang, M. Chi, Q. Cheng and Y. Yin, Angew. Chem., Int. Ed., 2012, 51, 5629–5633 CrossRef CAS PubMed.
- Z. Qian and S.-J. Park, Chem. Mater., 2014, 26, 6172–6177 CrossRef CAS.
- Z. Luo, K. Zheng and J. Xie, Chem. Commun., 2014, 50, 5143–5155 RSC.
- X.-R. Song, N. Goswami, H.-H. Yang and J. Xie, Analyst, 2016, 141, 3126–3140 RSC.
- Q. Yao, X. Yuan, Y. Yu, Y. Yu, J. Xie and J. Y. Lee, J. Am. Chem. Soc., 2015, 137, 2128–2136 CrossRef CAS PubMed.
- S. Ritz, S. Schöttler, N. Kotman, G. Baier, A. Musyanovych, J. Kuharev, K. Landfester, H. Schild, O. Jahn, S. Tenzer and V. Mailänder, Biomacromolecules, 2015, 16, 1311–1321 CrossRef CAS PubMed.
- G. Caracciolo, S. Palchetti, V. Colapicchioni, L. Digiacomo, D. Pozzi, A. L. Capriotti, G. La Barbera and A. Laganà, Langmuir, 2015, 31, 10764–10773 CrossRef CAS PubMed.
- C.-L. Chen and N. L. Rosi, Angew. Chem., Int. Ed., 2010, 1924–1942 CrossRef CAS PubMed.
- B. D. Briggs and M. R. Knecht, J. Phys. Chem. Lett., 2012, 3, 405–418 CrossRef CAS PubMed.
- J. Yu, M. L. Becker and G. A. Carri, Langmuir, 2012, 28, 1408–1417 CrossRef CAS PubMed.
- K. L. Roth, X. Geng and T. Z. Grove, J. Phys. Chem. C, 2016, 120, 10951–10960 CAS.
- H. Li, J. Jo, J. Wang, L. Zhang and I. Kim, Cryst. Growth Des., 2010, 10, 5319–5326 CAS.
- Z.-H. Xue, B.-B. Hu, S.-X. Dai and Z.-L. Du, Mater. Chem. Phys., 2010, 123, 278–283 CrossRef CAS.
- A. Swami, A. Kumar, M. D'Costa, R. Pasricha and M. Sastry, J. Mater. Chem., 2004, 14, 2696–2702 RSC.
- J. Xiao and L. Qi, Nanoscale, 2011, 3, 1383–1396 RSC.
- N. S. Abadeer and C. J. Murphy, J. Phys. Chem. C, 2016, 120, 4691–4716 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of CTPR protein structure, experimental procedures and TEM images, etc. See DOI: 10.1039/c6cc04708d |
| This journal is © The Royal Society of Chemistry 2016 |