
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DNA-catalyzed glycosylation using aryl glycoside donors†
Anthony R.
Hesser‡
,
Benjamin M.
Brandsen‡
,
Shannon M.
Walsh
,
Puzhou
Wang
 and
Scott K.
Silverman
*
and
Scott K.
Silverman
*
Department of Chemistry, University of Illinois at Urbana-Champaign, 600 South Mathews Avenue, Urbana, Illinois 61801, USA. E-mail: scott@scs.illinois.edu
First published on 22nd June 2016
Abstract
We report the identification by in vitro selection of Zn2+/Mn2+-dependent deoxyribozymes that glycosylate the 3′-OH of a DNA oligonucleotide. Both β and α anomers of aryl glycosides can be used as the glycosyl donors. Individual deoxyribozymes are each specific for a particular donor anomer.
Deoxyribozymes are particular single-stranded sequences of DNA that have catalytic activity. They are identified by in vitro selection starting from entirely random sequence pools.1 DNA catalysts have been found for many chemical reactions, including RNA cleavage by transesterification, RNA ligation, and DNA and RNA hydrolysis.2 More recently, deoxyribozymes have been identified for creation or removal of many common post-translational modifications (PTMs) of peptides,3 including phosphorylation,4 dephosphorylation,5 and formation of dehydroalanine.6 Glycosylation is an important PTM for which de novo catalysts that allow sequence-selective peptide and protein modification will have broad utility.7 Toward the longer-term goal of peptide glycosylation deoxyribozymes, we previously sought DNA-catalyzed glycosylation using the sugar nucleotide UDP-GlcNAc as the glycosyl donor8 during the key in vitro selection step of each round, followed by capture via NaIO4 oxidation of the GlcNAc moiety and reductive amination with NH2-modified DNA. However, that approach was subverted by a UDP-GlcNAc-independent DNA-catalyzed DNA deglycosylation reaction.9 Here we report that both β and α anomers of aryl glycosides, which have been used as synthetic glycosyl donors by glycosyltransferase10 and mutant glycoside hydrolase11 protein enzymes, are effective glycosyl donors with DNA catalysts, using a DNA oligonucleotide 3′-OH group as the glycosyl acceptor.
The aryl glycoside donors used in this study are the derivatives of D-glucose (D-Glc) shown in Fig. 1, and the in vitro selection strategy is shown in Fig. 2. In each selection experiment, the 3′-OH glycosyl acceptor and the glycosyl donor were presented simultaneously to an N40 DNA pool (40 random nucleotides). The glycosyl donor was attached to a DNA oligonucleotide, which binds via Watson–Crick interactions to a fixed sequence of DNA adjacent to the N40 region. The DNA oligonucleotide with the 3′-OH was connected through a covalent loop to the N40 pool. In the key selection step, glycosylation of the 3′-OH enables subsequent separation by polyacrylamide gel electrophoresis (PAGE) of the DNA sequences that catalyzed the reaction. The PAGE-shifted DNA sequences are amplified by PCR and ligated to the 3′-OH DNA oligonucleotide to begin a new selection round.
 | ||
| Fig. 1 Structures of glycosyl donors. Each is an aryl glycoside of D-glucose (1, β-D-Glc; 2, α-D-Glc). Compounds 1a, 1b, 1c, and 2b were evaluated in this study. | ||
 | ||
| Fig. 2 Key step of in vitro selection, in which DNA catalyzes glycosylation of a 3′-OH using an aryl glycoside donor, illustrated with a β-D-Glc donor. Glycosylation leads to a PAGE shift; the catalytically active DNA sequences are separated and PCR-amplified to enable the next selection round. The product is depicted here as the α-anomer, but the configurations for individual deoxyribozyme products in this study have not been assigned experimentally. See Fig. S1 and Table S1 (ESI†) for nucleotide details. | ||
The first selection experiment used the 2-chloro-4-nitrophenyl β-D-Glc glycosyl donor 1a. 2-Chloro-4-nitrophenyl β-D-glucuronic acid was prepared in two steps from 1-bromo-2,3,4-tri-O-acetyl-α-D-glucuronide methyl ester (see ESI†) and conjugated to a 5′-NH2-modified DNA oligonucleotide. This conjugate was stable (<0.5% degradation in 24 h) in both of our commonly used selection conditions,3 which are (A) 70 mM HEPES, pH 7.5, 1 mM ZnCl2, 20 mM MnCl2, 40 mM MgCl2, and 150 mM NaCl at 37 °C, and (B) 50 mM CHES, pH 9.0, 40 mM MgCl2, and 150 mM NaCl at 37 °C. Note that Mg2+ is present in both conditions A and B, whereas Zn2+ and Mn2+ cannot be used at the higher pH of conditions B due to precipitation and oxidation, respectively, although the higher pH could facilitate greater DNA-catalyzed reactivity. After 11 rounds in conditions A with 14 h incubation in the selection step of each round, the pool activity was 13% (see selection progression in Fig. S2A, ESI†). Individual deoxyribozymes were cloned from the round 11 pool, revealing two distinct sequences (Fig. S3A, ESI†): 11GV112 with rate constant kobs = 0.11 ± 0.01 h−1 (with Zn2+/Mn2+/Mg2+; n = 4, mean ± sd) and 55% yield at 48 h (Fig. 3A; the glycosylation product identity confirmed by MALDI mass spectrometry, Table S2, ESI†) and 11GV103 with kobs = 0.07 h−1 and 15% yield at 48 h (data not shown).§ The anomeric configurations of the glycosylation products were not assigned. For calibration regarding the kobs values of ∼0.1 h−1, “speed limits” of nucleic acid enzymes are generally considered to be ∼1 min−1;12 on that basis, there is room for improvement in kobs. Separately, after 8 rounds in conditions B, aberrantly migrating product bands were observed, and the selection experiment was discontinued.
 | ||
| Fig. 3 Glycosylation of a 3′-OH acceptor using the 11GV112 deoxyribozyme. (A) Metal ion dependence when using the 2-chloro-4-nitrophenyl β-D-Glc glycosyl donor 1a. In the PAGE image, representative timepoints are shown at t = 0, 12, and 48 h for the indicated metal ion combinations. The metal ion combinations not shown, including Mn2+ or Mg2+ alone, had no activity. S = substrate, P = product. Incubation conditions: 70 mM HEPES, pH 7.5, 150 mM NaCl, combinations of 1 mM ZnCl2, 20 mM MnCl2, and 40 mM MgCl2 as indicated, at 37 °C. The Zn2+ concentration was optimized; with 1a, the yield was lower at either 0.8 or 1.2 mM Zn2+ (data not shown). (B) Glycosyl donor dependence (in presence of all of Zn2+, Mn2+, and Mg2+). | ||
The 11GV112 deoxyribozyme was partially randomized and reselected (Fig. S2B, ESI†), providing a phylogeny that revealed many conserved nucleotides (Fig. S3B, ESI†). However, none of the reselected sequence variants had improved rate constant or yield in comparison with the parent sequence (data not shown). Evaluation of 11GV112 using various incubation conditions revealed dependence upon both Zn2+ and Mn2+ (kobs with Zn2+/Mn2+ 0.06 h−1), with trace activity in Zn2+/Mg2+ and no detectable activity in Zn2+ alone (Fig. 3A). 11GV112 was assayed with the β-series of glycosyl donors 1a, 1b, and 1c as well as the 4-nitrophenyl α-D-Glc donor 2b (Fig. 3B). Donor 1b supported glycosylation rate (kobs 0.15 h−1) and yield (44% at 48 h) comparable to that of 1a, whereas neither 1c nor 2b permitted any activity.
In a separate selection experiment, the 4-nitrophenyl α-D-Glc donor 2b was instead used. The 4-nitrophenyl glycoside (i.e., 2b rather than 2a) was used for synthetic simplicity; the key consideration is that in this experiment, 2b is the α rather than β anomer of the glycosyl donor. 4-Nitrophenyl α-D-glucuronic acid was prepared by TEMPO oxidation of 4-nitrophenyl α-D-glucose (see ESI†) and joined to 5′-NH2-modified DNA, providing a conjugate that was stable under selection conditions A (conditions B were not evaluated). After 16 selection rounds, the pool activity was 13% (Fig. S2C, ESI†), and individual deoxyribozymes were cloned, providing two distinct sequences (Fig. S3C, ESI†). The 16MJ132 deoxyribozyme had kobs = 0.080 ± 0.010 h−1 (with Zn2+/Mn2+/Mg2+; n = 4) and 28% yield at 48 h (Fig. 4), and 16MJ101 had kobs = 0.07 h−1 and 17% yield at 48 h (data not shown). Using the 4-nitrophenyl α-D-Glc donor 2b, 16MJ132 required both Zn2+ and Mn2+ (kobs with Zn2+/Mn2+ 0.07 h−1), and no activity was observed with the isomeric β-donor 1b. These patterns of metal dependence and glycosyl donor anomer dependence are analogous to those found for 11GV112.
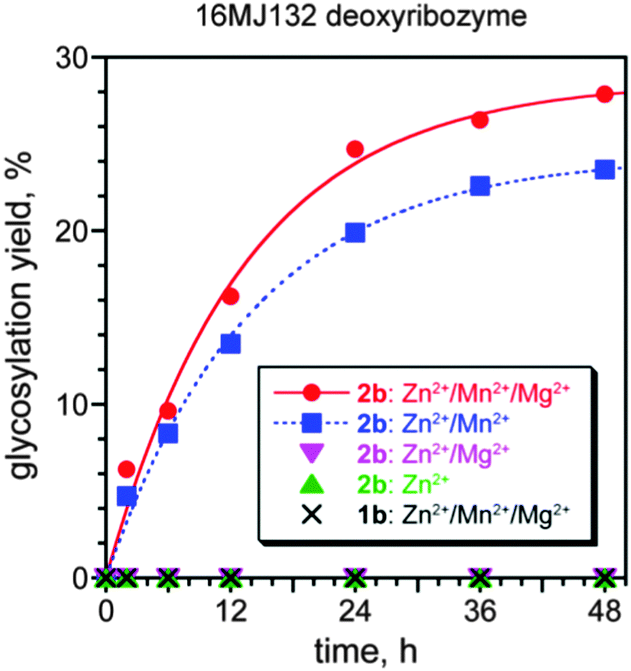 | ||
| Fig. 4 Glycosylation of a 3′-OH acceptor using the 16MJ132 deoxyribozyme. Shown are the metal ion dependence using the 4-nitrophenyl α-D-Glc glycosyl donor 2b, as well as assay with the 4-nitrophenyl β-D-Glc glycosyl donor 1b. Incubation conditions: 70 mM HEPES, pH 7.5, 150 mM NaCl, combinations of 0.4 mM ZnCl2, 20 mM MnCl2, and 40 mM MgCl2 as indicated, at 37 °C. The Zn2+ concentration was optimized; with 2b, similar kobs and yield were observed with 0.2–0.5 mM Zn2+, whereas the yield was lower at or above 0.6 mM Zn2+ (data not shown). The metal ion combinations not shown, including Mn2+ or Mg2+ alone, had no activity. | ||
In parallel with the above-described selection experiments in which a DNA 3′-OH was the glycosyl acceptor, we performed a separate selection that used a DNA-anchored CAAYAA hexapeptide as the intended glycosyl acceptor (see Fig. S4, ESI,† for structure) along with the 2-chloro-4-nitrophenyl β-D-Glc donor 1a. Although two new deoxyribozymes were identified (Fig. S3D, ESI†), neither DNA catalyst used the Tyr side chain as the acceptor. Instead, both of the new deoxyribozymes used a DNA nucleobase functional group within or very near to the 5′-end of the initially random N40 region as the nucleophile to attack the glycosyl donor, whereas the peptide moiety of the substrate was dispensable (Fig. S5, ESI†).¶ Similar branched products were observed in a separate selection with a CAASAA Ser-containing hexapeptide (data not shown). These observations indicate the need to improve our selection strategy to disallow the survival of DNA sequences that catalyze their own glycosylation rather than modification of the intended peptide substrate. In previous studies, we have initially found unanticipated DNA-catalyzed reactivity and then resolved the issue to achieve our initial goal; e.g., ref. 13 and 14.
Finally, recalling our earlier effort to use free UDP-GlcNAc as a glycosyl donor,9 we sought DNA-catalyzed glycosylation using DNA-anchored UDP-Glc (prepared by conjugating UDP-glucuronic acid to NH2-modified DNA). Initial assays revealed instability of UDP-Glc under selection conditions A and B, requiring the use of milder variations (see Fig. S6, ESI,† for details of conditions). No catalytic activity was observed in three selections with DNA-anchored UDP-Glc. This finding emphasizes the value of aryl glycosides for such selection experiments. RNA-catalyzed nucleotide synthesis by glycosylation of nucleobases with sugar pyrophosphates has been reported,15 but such glycosyl donors are similarly anticipated to be rather unstable under common selection conditions, and their synthesis is less straightforward than for aryl glycosides.
In summary, we have demonstrated that both β and α anomers of aryl glycosides can be effective glycosyl donors for DNA catalysts that glycosylate the 3′-OH group of a DNA oligonucleotide substrate. A deoxyribozyme identified with one anomer is inactive with the other anomer, suggesting but not requiring mechanistic differences between the two varieties of DNA enzyme. Glycosylated oligonucleotides (i.e., oligonucleotide–carbohydrate conjugates) have practical applications;16 with continued development, deoxyribozymes may provide an alternative synthetic route that avoids various complications associated with carbohydrate synthetic chemistry. Many challenges remain in order to achieve our long-term goal of identifying peptide-glycosylating deoxyribozymes. In particular, the new DNA catalysts reported here function only with a DNA 3′-OH glycosyl acceptor and also require that the aryl glycoside donor is DNA-anchored. Both reaction partners must be addressed via further selection efforts, which are ongoing in our laboratory.
This work was supported by a grant to S. K. S. from the National Institutes of Health (R01GM065966). B. M. B. was partially supported by NIH T32GM070421. S. M. W. was partially supported by an NIH predoctoral fellowship (F31GM115147). Mass spectrometry was performed at the UIUC School of Chemical Sciences Mass Spectrometry Laboratory on an instrument purchased with support from NIH grant S10RR027109A.
Notes and references
- (a) G. F. Joyce, Annu. Rev. Biochem., 2004, 73, 791–836 CrossRef CAS PubMed; (b) G. F. Joyce, Angew. Chem., Int. Ed., 2007, 46, 6420–6436 CrossRef CAS PubMed.
- (a) K. Schlosser and Y. Li, Chem. Biol., 2009, 16, 311–322 CrossRef CAS PubMed; (b) S. K. Silverman, Acc. Chem. Res., 2009, 42, 1521–1531 CrossRef CAS PubMed; (c) S. K. Silverman, Angew. Chem., Int. Ed., 2010, 49, 7180–7201 CrossRef CAS PubMed; (d) M. Chandra, A. Sachdeva and S. K. Silverman, Nat. Chem. Biol., 2009, 5, 718–720 CrossRef CAS PubMed; (e) D. J. Parker, Y. Xiao, J. M. Aguilar and S. K. Silverman, J. Am. Chem. Soc., 2013, 135, 8472–8475 CrossRef CAS PubMed.
- S. K. Silverman, Acc. Chem. Res., 2015, 48, 1369–1379 CrossRef CAS PubMed.
- (a) S. M. Walsh, A. Sachdeva and S. K. Silverman, J. Am. Chem. Soc., 2013, 135, 14928–14931 CrossRef CAS PubMed; (b) S. M. Walsh, S. N. Konecki and S. K. Silverman, J. Mol. Evol., 2015, 81, 218–224 CrossRef CAS PubMed.
- J. Chandrasekar and S. K. Silverman, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5315–5320 CrossRef CAS PubMed.
- J. Chandrasekar, A. C. Wylder and S. K. Silverman, J. Am. Chem. Soc., 2015, 137, 9575–9578 CrossRef CAS PubMed.
- (a) S. I. van Kasteren, H. B. Kramer, D. P. Gamblin and B. G. Davis, Nat. Protoc., 2007, 2, 3185–3194 CrossRef CAS PubMed; (b) K. W. Moremen, M. Tiemeyer and A. V. Nairn, Nat. Rev. Mol. Cell Biol., 2012, 13, 448–462 CrossRef CAS PubMed; (c) M. Dalziel, M. Crispin, C. N. Scanlan, N. Zitzmann and R. A. Dwek, Science, 2014, 343, 37 CrossRef CAS PubMed.
- (a) L. L. Lairson, B. Henrissat, G. J. Davies and S. G. Withers, Annu. Rev. Biochem., 2008, 77, 521–555 CrossRef CAS PubMed; (b) M. R. Bond and J. A. Hanover, J. Cell Biol., 2015, 208, 869–880 CrossRef CAS PubMed.
- C. Höbartner, P. I. Pradeepkumar and S. K. Silverman, Chem. Commun., 2007, 2255–2257 RSC.
- R. W. Gantt, P. Peltier-Pain, W. J. Cournoyer and J. S. Thorson, Nat. Chem. Biol., 2011, 7, 685–691 CrossRef CAS PubMed.
- (a) P. Bojarová and V. Křen, Trends Biotechnol., 2009, 27, 199–209 CrossRef PubMed; (b) P. M. Danby and S. G. Withers, ACS Chem. Biol., 2016, 11 DOI:10.1021/acschembio.1026b00340.
- (a) G. M. Emilsson, S. Nakamura, A. Roth and R. R. Breaker, RNA, 2003, 9, 907–918 CrossRef CAS PubMed; (b) R. R. Breaker, G. M. Emilsson, D. Lazarev, S. Nakamura, I. J. Puskarz, A. Roth and N. Sudarsan, RNA, 2003, 9, 949–957 CrossRef CAS PubMed.
- A. Sachdeva and S. K. Silverman, Org. Biomol. Chem., 2012, 10, 122–125 Search PubMed.
- B. M. Brandsen, T. E. Velez, A. Sachdeva, N. A. Ibrahim and S. K. Silverman, Angew. Chem., Int. Ed., 2014, 53, 9045–9050 CrossRef CAS PubMed.
- (a) P. J. Unrau and D. P. Bartel, Nature, 1998, 395, 260–263 CrossRef CAS PubMed; (b) K. E. Chapple, D. P. Bartel and P. J. Unrau, RNA, 2003, 9, 1208–1220 CrossRef CAS PubMed; (c) P. J. Unrau and D. P. Bartel, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15393–15397 CrossRef CAS PubMed; (d) M. W. Lau, K. E. Cadieux and P. J. Unrau, J. Am. Chem. Soc., 2004, 126, 15686–15693 CrossRef CAS PubMed.
- (a) T. L. Sheppard, C.-H. Wong and G. F. Joyce, Angew. Chem., Int. Ed., 2000, 39, 3660–3663 CrossRef CAS; (b) T. S. Zatsepin and T. S. Oretskaya, Chem. Biodiversity, 2004, 1, 1401–1417 CrossRef CAS PubMed; (c) H. Yan and K. Tram, Glycoconjugate J., 2007, 24, 107–123 CrossRef CAS PubMed; (d) Y. Singh, P. Murat and E. Defrancq, Chem. Soc. Rev., 2010, 39, 2054–2070 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc04329a |
| ‡ A. R. H. and B. M. B. are co-first-authors of this manuscript. |
| § Each deoxyribozyme in this study was named as, for example, 11GV112, where 11 is the round number, GV1 is the systematic alphabetic designation for the particular selection, and 12 is the clone number. |
| ¶ We speculate that the conditions B selection experiment with the DNA oligonucleotide acceptor and 1a donor provided analogous branched products rather than 3′-OH glycosylation, although the products were not investigated in detail. |
| This journal is © The Royal Society of Chemistry 2016 |