
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An expeditious synthesis of early transition metal carbide nanoparticles on graphitic carbons†
Debora
Ressnig
ab,
Simona
Moldovan
c,
Ovidiu
Ersen
c,
Patricia
Beaunier
d,
David
Portehault
ab,
Clément
Sanchez
ab and
Sophie
Carenco
 *ab
*ab
aCollège de France, Laboratoire de Chimie de la Matière Condensée de Paris (LCMCP), 11 Place Marcelin Berthelot, F-75005 Paris, France
bSorbonne Universités, UPMC Université Paris 06, CNRS, Collège de France, Laboratoire de Chimie de la Matière Condensée de Paris (LCMCP), 4 Place Jussieu, F-75252 Paris Cedex 05, France. E-mail: sophie.carenco@upmc.fr
cInstitut de Physique et Chimie des Matériaux de Strasbourg (IPCMS), UMR 7504 CNRS-Université de Strasbourg (UdS), 23 rue du Loess, 67037 Strasbourg Cedex 08, France
dSorbonne Universités, UPMC Université Paris 06, UMR 7197-CNRS, Laboratoire de Réactivité de Surface (LRS), 4 Place Jussieu, F-75252 Paris Cedex 05, France
First published on 30th June 2016
Abstract
An expeditious synthesis of metal carbide nanoparticles onto various carbon supports is demonstrated. The procedure is versatile and readily yields TiC, VC, Mo2C and W2C nanoparticles on different types of carbons. The reaction is initiated at room temperature and proceeds within seconds. This novel synthetic route paves the way for a large variety of metal carbide–carbon nanocomposites that may be implemented in emerging nanotechnology fields.
Metal carbides combine the hardness and resistivity of ceramics with the electronic and optical properties of metals and therefore find widespread application as abrasives, cutting tools and more recently as catalytic materials, e.g. for the hydrogen evolution catalysis.1–4 The stabilization of nanocomposites has boosted research into these fields because higher performances result from the stabilization of nanoscale crystallites on tailored supports, in particular conductive ones.
Composites of metal carbide nanoparticles on a carbon phase are typically synthesized via the carbothermal reduction of a metal salt at temperatures between ∼700 °C and 1200 °C in the presence of a carbonizable carbon source: molecular precursors (e.g. glucose), polymers (cellulose (poly) ionic liquids) or gels (urea–glass route).5 In these routes, the formation of the metal carbide and the graphitic support is concerted. As a result, particles are often enclosed in an amorphous or even graphitic carbon shell, and the porosity of the carbon support is not easily controlled. This limits the scope of the composite to applications where accessibility to the metal carbide surface is not required (e.g. mechanical properties).
Thus, softer approaches to produce accessible nanoparticles at low temperature in organic solvents have been developed. They use strong reducing agents (e.g. sodium naphthalenide, potassium graphite, butyl lithium) in order to swiftly produce small amorphous nanoparticles. In most cases, metal nanoparticles are obtained instead of metal carbides,6–9 but in a few studies the latter were produced after a calcination step. Nelson et al. used crown ether to reduce metal halides which afforded amorphous particles that upon annealing at high temperatures (570–1200 °C) crystallized into metal carbides.10,11 A similar reaction between butyl lithium and metal chlorides followed by calcination 600–1000 °C afforded different metal carbides.12 The co-reduction of the metal chloride and a chlorinated carbon source by an alkali metal in autoclaves at elevated temperatures also produced metal carbides.13–15
The synthesis of crystalline metal carbide nanoparticles with a controlled size and accessible surface is still a challenge. Low temperatures are desirable in order to limit particle sintering and growth, but subsequent high calcination temperatures are still required in order to trigger the crystallization of the carbide phase as well as the graphitization of the carbon.
Altogether, an expeditious and straightforward synthetic route that could be applied to different sorts of carbon supports is desirable in order to expand and strengthen the current applications of transition metal carbide nanoparticles.
In this study, we demonstrate a protocol that does not require a separate annealing step and that allows carefully selecting the characteristics of a certain graphitic carbon for specific applications prior to metal carbide formation. In the first step, the carbon surface was charged with electrons by impregnation with a potassium melt, forming potassium graphite of controlled porosity and crystallinity. At room temperature, these were directly reacted with a chosen metal chloride in the absence of a solvent. The reaction was highly exothermic and fast, which allowed immediate functionalization of different carbons with early transition metal carbides, without applying external heat, long reaction times, or a secondary calcination step. We show here that this route was efficient for producing TiC on different graphitic carbon supports. Furthermore, we demonstrate that the approach can be widened to Mo2C, VC and W2C crystalline nanoparticles.
Titanium carbides deposited on regular graphite (g-C) were produced first. All reactions were carried out using standard Schlenk-line techniques (see the ESI†). In the first step, bronze colored KC8 was prepared using a standard procedure, by melting a stoichiometric ratio of potassium in the presence of graphite (SBET 9 m2 g−1).16 Liquid TiCl4 was subsequently added drop by drop to the KC8 powder, resulting in a dark solid. Each droplet produced an immediate and strongly exothermic reaction attested by the glowing of the solid and the warming of the reaction vessel. As a side effect a fraction of the titanium chloride evaporated. Therefore an excess amount of TiCl4 was added until no reaction was observed anymore on the dark solid. Excess TiCl4 was subsequently removed under vacuum to prevent the formation of metal oxides during the work-up. The overall reaction can be described as:
| 32Cgraph + 4K → 4KC8 | (1) |
| TiCl4(l) + 4KC8 → TiC(s) + 4KCl + 31Cgraph | (2) |
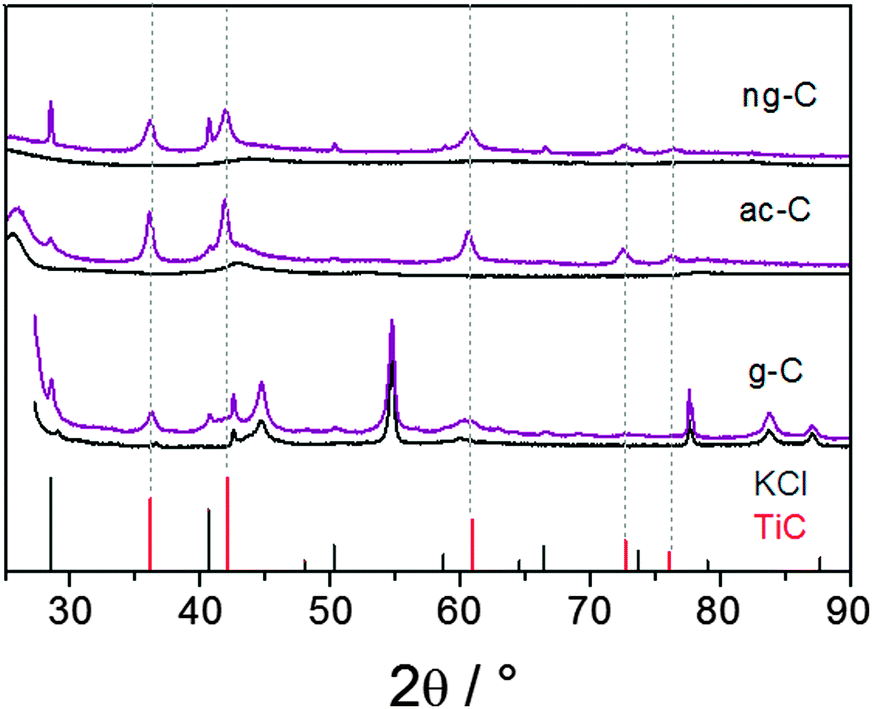 | ||
| Fig. 1 PXRD patterns of TiC/carbon nanocomposites (in purple) and patterns of the pristine carbons (in black). Bottom: Graphitic carbon, middle: acetylene black, top: macroporous carbon. Reference patterns: TiC [00-001-1222] in red, KCl [01-075-0296] in grey. | ||
The transmission electron microscopy (TEM) images of the TiC/g-C composite show that the graphite platelets are covered with nanoparticles (Fig. 2a and Fig. S1, S2, ESI†). Lattice fringes of 1.5 Å and 2.5 Å can be assigned to the distances d220 and d111 of TiC [00-001-1222], respectively (Fig. 1 and Fig. S2, ESI†). The corresponding energy-dispersive X-ray spectroscopy (EDX) confirms the presence of Ti in the nanoparticles (Fig. S2, ESI†). It also indicates that a small amount of oxygen is present after the work-up.
 | ||
| Fig. 2 (a) TEM of TiC nanoparticles on graphite (scale bar 100 nm). Top inset shows the HRTEM image of a single TiC particle with lattice fringes of d220, white lines suggest the edges of the particle (scale bar 2 nm). Bottom inset shows the particle size distribution, calculated from over 100 nanoparticles. (b) STEM in a bright field (b, left) and HAADF (b, right) (scale bars: 50 nm). | ||
The bottom inset of Fig. 2a shows that the nanoparticles have an average diameter of 6.1 ± 1.2 nm. This is smaller than the size of the crystalline domains evaluated from the Scherrer equation, suggesting that larger TiC particles were not properly observed in TEM images. Larger nanoparticles may preferentially populate thicker graphitic domains, which TEM cannot image. Repeated TEM observations showed that some graphitic platelets are richly and others are barely populated with TiC particles. The inhomogeneity results from adding TiCl4 as micro-droplets to the solid powder.
Moreover, Fig. 2a suggests that the TiC nanoparticles are preferentially sitting on the edges and topographic steps of the graphite layer. Imaging using scanning TEM (STEM) with high-angle annular dark field (HAADF) detection provides Z-contrast imaging (Fig. 2b).17 White dots represent regions with high-Z (here, titanium). The accumulation of TiC particles on the edge of the graphite platelets is evident in this mode. Moreover, larger particles, suspected from XRD, are better observed in STEM-HAADF, though they represent a minority.
The TiC particles nucleate preferably at the edge defects of graphite. A similar preference for nucleation at graphite edges was previously seen in the reduction of metal salts with KC8 in solution.18
To demonstrate the generality of the synthetic approach, we extended the method to other carbons with different morphologies and crystallinities. These are: commercially available acetylene black (ac-C, ∼50% graphitic, SBET 63 m2 g−1) and a macroporous carbon with nanosized graphitic domains (ng-C, SBET 270 m2 g−1).19
Firstly, we observed that the potassium uptake of ac-C and ng-C is higher than g-C. We evaluated a close-to-maximum-loading by adding and intercalating potassium in small fractions until metallic potassium would remain unreacted in the vial. The determined potassium uptake rounds up to KC4 for ac-C and KC2 for ng-C. In both cases the initially black carbon powders were covered with a red brilliance after potassium insertion.
Both potassium-loaded carbons were treated with TiCl4 in the same way as described for g-C. The PXRD patterns of the products show the formation of TiC as the only crystalline Ti-containing product (Fig. 1, middle and top patterns). Traces of remaining KCl appear in the diffractograms despite intense washing over several days including treatment with ultrasound, in particular for the macroporous carbon (ng-C), thus suggesting some KCl inclusions in the carbon porosity. PXRD interpretation is straightforward thanks to the lower contribution of graphite. In both cases, reflections from TiC are detected and the absence of any TiO2 phase is confirmed. This strongly suggested that the reaction on g-C, discussed above, did not lead to TiO2 impurities either.
The Scherrer size of the TiC crystallites is in the same range as that for graphite (Table 1). As with g-C, the average size estimated from TEM observations is slightly under-evaluated due to the difficulty in observing particles in the thicker regions. The TiC distribution on ac-C and ng-C divides in richer and less populated areas, as shown by STEM in bright field and HAADF mode (Fig. S3 and S4, ESI†). Optimizing the procedure in a way that allows for spraying smaller droplets would certainly improve the distribution of the particles on the carbon.
| Carbon | TiC size/nm | Raman (ID/IG) pristine/modified | TiC wt%max calc.a | |
|---|---|---|---|---|
| TEM | Scherrer | |||
| a “TiC wt%max calc.” is the maximum loading of TiC on each carbon support. It was calculated by assuming that TiCl4 and the potassium-loaded carbon (KC8 for graphite, KC4 for acetylene-black, KC2 for macroporous carbon) were introduced in a stoichiometric ratio. For example, for the reaction with graphite, one mole of TiC forms along with 31 carbon atoms (see eqn (2)), which gives a TiC weight ratio of 14%. This is the higher TiC loading because the only carbon introduced comes from the reducing agent. | ||||
| g-C | 6.1 ± 1.2 | 14 | 0.37/0.34 | 14 |
| ac-C | 3.9 ± 1.1 | 15 | 1.85/1.94 | 25 |
| ng-C | 7.2 ± 1.6 | 10 | 1.45/1.36 | 42 |
Most importantly, complementary analyses of pristine and modified carbons with nitrogen sorption, SEM and PXRD analysis, FTIR and Raman spectroscopy corroborate the conservation of the carbon structure, porosity and morphology (Fig. S5–S8, ESI†).
We therefore think that this method can be applied to functionalize a broad variety of sp2 carbons. Limitations arise when light elements are introduced into the graphitic network and disturb the electronic configuration of the graphitic pattern. For example, it was not possible to charge graphite's nitrogen-rich analogue g-C3N4 with potassium.
Next, the expandability of the protocol to other transition metals was explored. Acetylene black was selected as a support because of its many technological uses and lower crystallinity. VCl4 like titanium chloride is liquid at room temperature and therefore the most suitable precursor for further elaborations.
The reaction of ac-C with VCl4 was more intense than with TiCl4. Snapshots of a movie of the reaction are presented in Fig. S11 (ESI†). PXRD is consistent with the formation of VC [01-089-2719], with a maximum crystallite size of 28 nm (Fig. 3, top pattern). As for TiC, the smaller nanoparticles of VC are easier observed by TEM on ac-C (Fig. 4). Lattice fringes of 2.4 Å match with the d220 distance of the VC structure (Fig. 4b, inset).
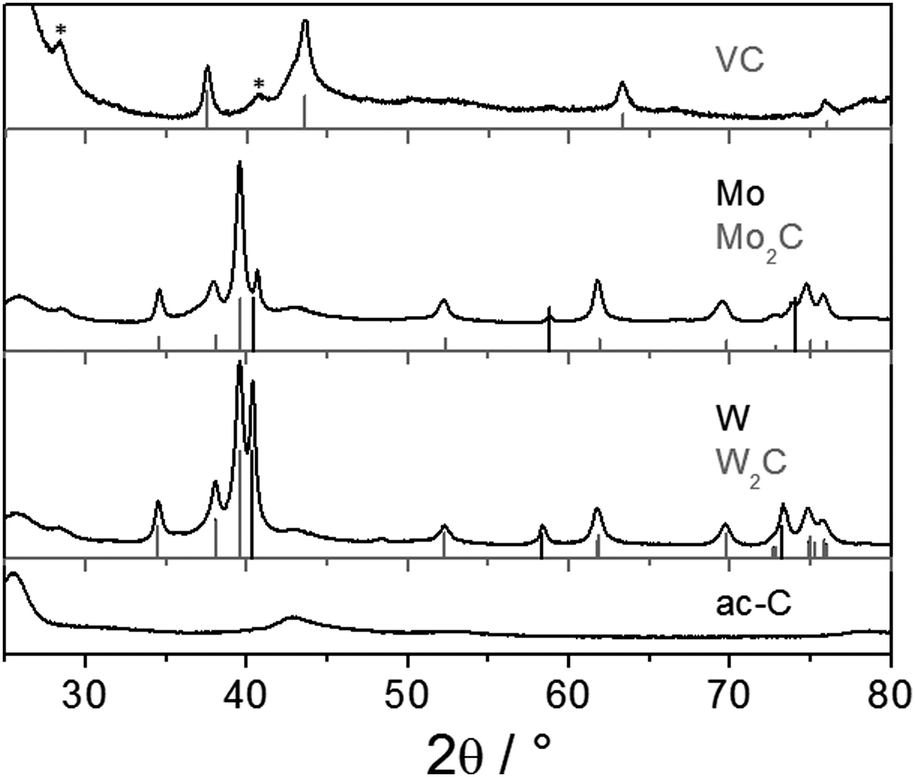 | ||
| Fig. 3 PXRD patterns of ac-C/metal carbide composites with vanadium (top), molybdenum (middle) and tungsten (bottom). The reference patterns are: VC [01-089-2719] (in grey), Mo [00-001-1205] (in black), Mo2C [04-004-8327] (in grey), W [00-004-0806] (in black), and W2C [01-089-2371] (in grey). Stars assign reflections of KCl. | ||
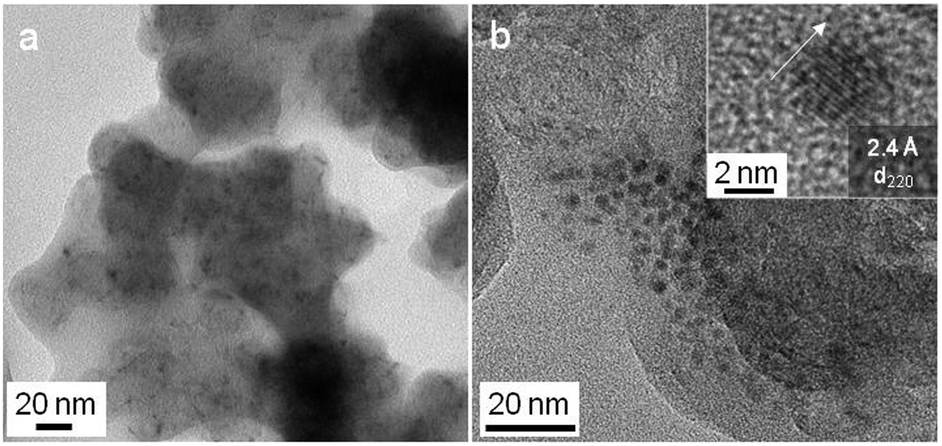 | ||
| Fig. 4 TEM (a) and HRTEM (b, inset) of the VC/ac-C composite. | ||
Other transition metal chlorides (e.g. MoCl5) are solid at room temperature and do not react when mixed with potassium graphites. A straightforward approach to initiate the reaction is to heat the vessel that contains the potassium graphite/metal chloride mixture using a heat gun to the boiling point of the metal chloride (e.g. 268 °C for MoCl5).
Fig. 3 (middle and bottom) shows PXRD patterns obtained from the reaction of potassium-loaded ac-C with MoCl5 or WCl6 initiated by sublimation. Both reactions yielded the metal rich hexagonal carbides ε-W2C and Mo2C. Traces of the pure metals are also detected. Those were reaction byproducts of the strong reduction, because in the absence of potassium-loaded ac-C, simple sublimation and recrystallization of the metal chlorides did not yield the decomposition of the metal chloride precursors. TEM shows the presence of small and crystalline metal carbide nanoparticles on ac-C (Fig. S8 and S9, ESI†).
Note that even if the approach can be widened to several metal carbides, complementary experiments with specific transition metal chlorides such as FeCl2 and NiCl2 yielded the metal and not the metal carbide. Hence, the method has its limitations, presumably related to the respective stability of the carbides versus the corresponding metals.
In summary, we present a versatile, expeditious method to functionalize the surface of different types of graphitic carbons with metal carbide nanoparticles via the formation of potassium graphites. Potassium graphites are reactive precursors that can be easily prepared on the gram scale and safely stored under inert conditions. The functionalization itself is exothermic and very fast (timescale of a few seconds) and no external heating is required. The structure and morphology of the graphitic carbons are retained during functionalization, which increases the control in the composite design. Work is underway to extend this protocol to more sophisticated carbons and to other metal carbide structures, as well as to test the activity of the composite in electrochemical reactions. Besides, upscaling of the reaction to a 10 g scale will require heat management, which could be done by diluting the graphite in inert KCl salts that would be washed with water.
Collège de France and Fondation du Collège de France are acknowledged for financial support. J. M. Krafft is acknowledged for Raman analyses.
Notes and references
- J. Han, J. Duan, P. Chen, H. Lou, X. Zheng and H. Hong, Green Chem., 2011, 13, 2561 RSC.
- Q. S. Gao, C. X. Zhang, S. H. Xie, W. M. Hua, Y. H. Zhong, N. Ren, H. L. Xu and Y. Tang, Chem. Mater., 2009, 21, 5560 CrossRef CAS.
- R. Michalsky, Y.-J. Zhang and A. A. Peterson, ACS Catal., 2014, 4, 1274 CrossRef CAS.
- C. Giordano and M. Antonietti, Nano Today, 2011, 6, 366 CrossRef CAS.
- C. Giordano and T. Corbiere, Colloid Polym. Sci., 2013, 291, 1297 CAS.
- K. L. Tsai and J. L. Dye, J. Am. Chem. Soc., 1991, 113, 1650 CrossRef CAS.
- C. Schöttle, P. Bockstaller, R. Popescu, D. Gerthsen and C. Feldmann, Angew. Chem., Int. Ed., 2015, 54, 9866 CrossRef PubMed.
- S. Carenco, M. Demange, J. Shi, C. Boissière, C. Sanchez, P. Le Floch and N. Mézailles, Chem. Commun., 2010, 46, 5578 RSC.
- G. P. Boldrini, D. Savoia, E. Tagliavini, C. Trombini and A. Umani-Ronchi, J. Organomet. Chem., 1985, 280, 307–312 CrossRef CAS.
- J. A. Nelson and M. J. Wagner, Chem. Mater., 2002, 14, 1639 CrossRef CAS.
- J. A. Nelson and M. J. Wagner, Chem. Mater., 2002, 14, 4460 CrossRef CAS.
- Y.-H. Chang, C.-W. Chiu, Y.-C. Chen, C.-C. Wu, C.-P. Tsai, J.-L. Wang and H.-T. Chiu, J. Mater. Chem., 2002, 12, 2189–2191 RSC.
- Z. Ju, N. Fan, X. Ma, J. Li, X. Ma, L. Xu and Y. Qian, J. Phys. Chem. C, 2007, 111, 16202 CAS.
- S. Dyjak, M. Norek, M. Polanski, S. Cudzilo and J. Bystrzycki, Int. J. Refract. Met. Hard Mater., 2013, 38, 87 CrossRef CAS.
- Y. Gu, L. Chen, Z. Li, Y. Qian and W. Zhang, Carbon, 2004, 42, 235–238 CrossRef CAS.
- D. E. Bergbraiter and J. M. Killough, J. Am. Chem. Soc., 1978, 100, 2126–2134 CrossRef.
- S. Carenco, S. Moldovan, L. Roiban, I. Florea, D. Portehault, K. Vallé, P. Belleville, C. Boissière and L. Rozes, et al. , Nanoscale, 2016, 8, 1260–1279 RSC.
- A. Fürstner, Angew. Chem., Int. Ed., 1993, 32, 164 CrossRef.
- D. Ressnig, T. Corbiere, T. Lunkenbein, U. Braun, M. G. Willinger and M. Antonietti, J. Mater. Chem. A, 2014, 2, 18076–18081 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional TEM, SEM, Raman, and N2 sorption results. See DOI: 10.1039/c6cc04157d |
| This journal is © The Royal Society of Chemistry 2016 |