
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
“Water-in-salt” electrolytes enable the use of cost-effective aluminum current collectors for aqueous high-voltage batteries†
R.-S.
Kühnel
*,
D.
Reber
,
A.
Remhof
,
R.
Figi
,
D.
Bleiner
and
C.
Battaglia
Empa, Swiss Federal Laboratories for Materials Science and Technology, 8600 Dübendorf, Switzerland. E-mail: ruben-simon.kuehnel@empa.ch
First published on 4th August 2016
Abstract
The extended electrochemical stability window offered by highly concentrated electrolytes allows the operation of aqueous batteries at voltages significantly above the thermodynamic stability limit of water, at which the stability of the current collector potentially limits the cell voltage. Here we report the observation of suppressed anodic dissolution of aluminum in “water-in-salt” electrolytes enabling roll-to-roll electrode fabrication for high-voltage aqueous lithium-ion batteries on cost-effective light-weight aluminum current collectors using established lithium-ion battery technology.
Batteries based on aqueous electrolytes promise a cost advantage over those based on non-aqueous electrolytes because exposure of the battery components to an ambient atmosphere during the manufacturing process and possibly even during operation does not harm battery operation. In addition aqueous batteries offer intrinsically improved operational safety due to the absence of flammable organic solvents and can be operated within a wider temperature range (e.g., traditional lithium-ion batteries can only be operated up to a maximum temperature of <60 °C).
In the past, the low electrochemical stability window of water (thermodynamically only 1.23 V) limited the operating voltage and thus the specific energy of aqueous batteries. Recently, relatively large stability windows of up to 3 V have been reported for highly concentrated, “water-in-salt”, aqueous solutions of lithium bis(trifluoromethylsulfonyl)imide (LiTFSI, see the inset of Fig. 1a for an illustration of the structure of the TFSI anion) providing an opportunity for the development of high-voltage aqueous batteries.1
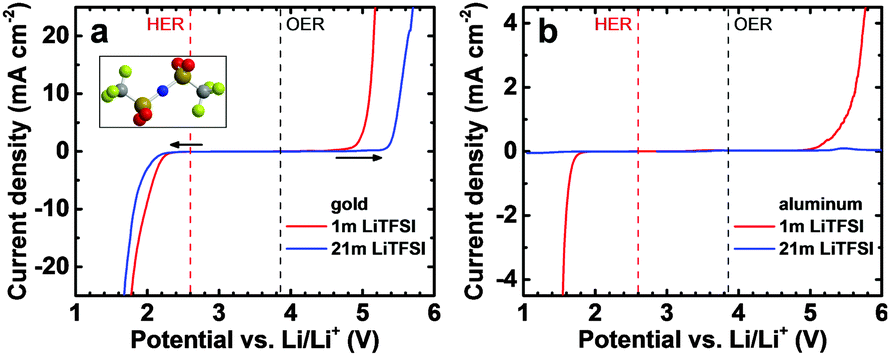 | ||
| Fig. 1 Electrochemical stability window of 1 m and 21 m aqueous LiTFSI solutions on (a) gold and (b) aluminum electrodes. Linear sweep voltammetry experiments at a scan rate of 10 mV s−1 were carried out for the cathodic and anodic limits in two separate cells. The thermodynamic potentials for the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) at pH = 7 are also shown by the vertical dashed lines. The inset in (a) shows a ball-and-stick model of the atomic structure of a TFSI anion. | ||
The first lab-scale batteries utilizing highly concentrated LiTFSI salt in water as the electrolyte were fabricated with stainless steel current collectors.1,2 However, aluminum (Al) is the preferred current collector material on the cathode side of non-aqueous lithium-ion batteries due to its much lower density, higher electronic conductivity, low cost, and the ability to be processed into thin foils by rolling. The potential-pH (Pourbaix) diagram of Al indicates thermodynamic stability, i.e. passivation due to the air-formed aluminum oxide layer, only in a small pH range (e.g., from about pH = 4 to pH = 8.5 for a concentration of soluble Al3+ species of 0.1 mM).3 This small stability window has prevented the use of Al as the current collector in traditional aqueous batteries (examples of typical electrolyte solutions: lead–acid batteries:4 37 wt% H2SO4 solution (pH < 0); nickel–metal hydride batteries:5 6 M KOH solution (pH > 14)). Aqueous LiTFSI solutions are near neutral in the pH value, well inside the passive region of the Pourbaix diagram of Al. However, the passivating oxide film can be prone to localized break down, so called pitting, in the presence of certain anions, with chloride being the most prominent example.6 In fact, Al has been shown to also suffer from pitting corrosion in solutions of TFSI− in polar organic solvents when relatively low anodic potentials of 3.55 V vs. Li/Li+ are applied.7,8 The oxidative stability of the highly concentrated aqueous solutions of LiTFSI exceeds 4 V vs. Li/Li+, opening the question of stability of Al against anodic dissolution, i.e., pitting corrosion, at these high potentials.
Here, we show that anodic Al dissolution in aqueous electrolytes is strongly suppressed at high LiTFSI concentrations, rendering Al a promising material for the current collector in aqueous batteries. Electrochemical tests using Al working electrodes are complemented by cycling a LiMn2O4-activated carbon prototype cell with Al current collectors. Our results represent an important step towards the realization of a cost-competitive aqueous battery relevant to stabilization and load leveling of electricity grids transporting a growing fraction of intermittent renewable electricity.9–11
Electrolytes were prepared by dissolving LiTFSI salt (99.95%, Sigma-Aldrich) in high purity H2O (18 MΩ, Milli-Q water purification system). All electrochemical tests were carried out in glass cells (volume: 3 mL, eDAQ) in three electrode configuration versus a Ag/AgCl reference electrode (eDAQ) using a multi-channel potentiostat (Bio-Logic VMP3). All potentials were converted to the Li/Li+ scale. A gold disc working electrode (diameter: 1 mm) and a platinum coated titanium rod counter electrode, used for the experiments shown in Fig. 1 and 2, were obtained from eDAQ. The Al foil (99.9% and 99.3% (for the battery tests)) and LiMn2O4 (>99%) were supplied by MTI and Sigma-Aldrich, respectively. Inductively coupled plasma optical emission spectrometry (Varian Vista Pro Radial) was performed on selected electrolyte samples.
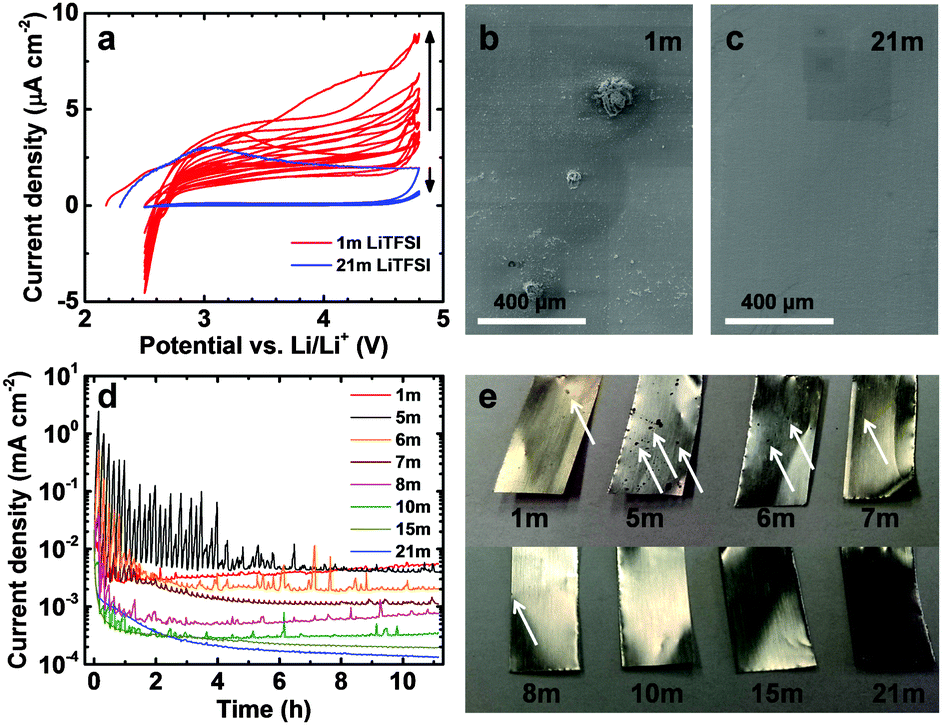 | ||
| Fig. 2 Stability of Al electrodes against anodic dissolution in aqueous LiTFSI solutions of various concentrations. (a) Cyclic voltammetry carried out between 2.5 and 4.8 V vs. Li/Li+ at a scan rate of 0.5 mV s−1 and (d) chronoamperometry carried out at 4.8 V vs. Li/Li+. The electrodes were relaxed for 1 min after every 9 min of polarization at 4.8 V. (b and c) SEM images of Al electrodes taken after the cyclic voltammetry experiment. (e) Photographs of Al electrodes taken after the chronoamperometry experiment. | ||
To distinguish between electrolyte stability and Al–electrolyte interaction, we first carried out linear sweep voltammetry on inert gold (Au) electrodes shown in Fig. 1a. The electrochemical stability window (ESW) of aqueous LiTFSI solutions increases with increasing concentration of the electrolyte from 1.4 V for the 1 m to 2.2 V for the 21 m solution when considering a relatively small threshold current density of 50 μA cm−2. For a larger cut-off current density of 250 μA cm−2, the ESWs increase to 2.3 and 3.0 V for the 1 m and 21 m solution, respectively (see arrows). Additional threshold current density–ESW pairs can be found in Table S1 (ESI†). The increase in electrochemical stability with concentration and the deviation from thermodynamic potentials is especially pronounced for the oxygen evolution reaction (OER), while smaller overpotentials are found for the hydrogen evolution reaction (HER).
In a second step, we investigated the electrochemical stability of aluminum electrodes (Fig. 1b). The ESWs of aqueous LiTFSI solutions turn out to be wider on Al than on Au with the kinetics of HER and OER being generally slower. Using the 50 μA cm−2 threshold current density, we obtain very large apparent ESWs of 3.2 and 4.2 V for the 1 m and 21 m LiTFSI solutions, respectively. The reduction in HER and OER kinetics is especially pronounced for the 21 m solution, where the current density remains remarkably low even outside the 4.2 V stability window. The slower kinetics on Al than on Au are due to the oxide passivation layer naturally forming on Al in air and possibly also due to higher overpotentials.12
For the use as the current collector in a high-voltage aqueous battery, Al has to be stable against anodic dissolution in the operating voltage range of the battery. To test the stability of Al in aqueous LiTFSI solutions, we carried out cyclic voltammetry (CV) with bare Al working electrodes. Our CV experiments (Fig. 2a) show low anodic stability of Al in aqueous solutions of LiTFSI at electrolyte concentration typical for lithium-ion batteries (1 m) as can be seen by the increasing current density with the cycle number and the hysteresis of the current response, in line with the results reported for polar organic solvents.13,14 Interestingly, the current density is lower and decreases with the cycle number for the 21 m electrolyte, indicating passivation and good stability of Al in this electrolyte. Between 2.5 and 2.7 V vs. Li/Li+, negative currents are observed for the 1 m cell. We assign these currents to the reduction of electrolyte oxidation products formed at high potentials as well as at the onset of hydrogen evolution.
Scanning electron microscopy (SEM) images shown in Fig. 2b and c of the polarized Al electrodes after the CV experiment further confirm the suppression of anodic Al dissolution at high LiTFSI concentration. While several large (up to 150 μm in size) corrosion pits are apparent on the Al foil polarized in the 1 m LiTFSI solution, the electrode polarized in the 21 m solution showed practically no signs of Al dissolution.
As the aluminum current collectors spend only a short time at high potentials during a CV measurement, we applied a harsher testing protocol simulating the situation in which a battery is stored fully charged, i.e., at high potential difference, for an extended period of time. Al electrodes were polarized for 9 min at 4.8 V vs. Li/Li+, a potential just below the strong increase in current density visible in Fig. 1a. To further increase the harshness of the experimental conditions, we relaxed the electrode potential for 1 min at open-circuit after each 9 min period to reduce transport limitations at the electrode–electrolyte interface. This process was repeated for a total polarization time of 10 h. This protocol simulates the conditions in a battery, where the electrolyte is not flowing, more realistically than a rotating disc experiment, typically employed to eliminate mass transport limitations. Inspection of Fig. 2d reveals that the higher the LiTFSI concentration in the electrolyte, the lower the current density after 10 h (note the logarithmic scale of the ordinate), illustrating the strong dependence of the Al dissolution process on the concentration. The current density in the quasi steady-state after 10 h decreased from 5.5 to 0.1 μA cm−2. During the first hours of the experiment, current spikes related to strong Al dissolution at defect sites are observed stochastically during many 9 min polarization steps especially for low LiTFSI concentrations. In contrast, no such current spikes are observed for LiTFSI concentrations ≥15 m, reflecting the high stability of Al at these concentrations. The values of the current maxima are higher for the electrolytes with concentrations up to 8 m than for the 1 m solution due to the higher room temperature conductivity of the former electrolytes (data not shown). After some time, the currents reach constant values for all electrolytes, occasionally interrupted by smaller current peaks. The absence of current spikes for the 15 m solution indicates that a threshold concentration for the water–LiTFSI system dividing pronounced Al dissolution and good passivation exists between 10 and 15 m (see discussion on ion solvation and free H2O molecules below).
During the course of this rather harsh experiment, the Al foils, with a thickness of 16 μm, polarized in the LiTFSI electrolytes of lower concentration were perforated (Fig. 2e, white arrows point to selected corrosion pits), while the foils remained much less affected in the highly concentrated electrolytes, confirming the strong suppression of anodic Al dissolution in highly concentrated aqueous electrolytes. A closer examination of the Al foils via SEM (Fig. S1, ESI†) reveals that slight pitting takes place at high LiTFSI concentrations under these very harsh conditions. However, the diameter and depth of the corrosion pits are much smaller at high than at low to medium LiTFSI concentrations. The pit diameter goes through a maximum for the 5 m electrolyte as expected from the highest average current densities observed in Fig. 2d for this electrolyte. The typical maximum pit diameter strongly decreases from >500 μm for 5 m (Fig. S1b, ESI†) to below 1.5 μm for 21 m (Fig. S1f, ESI†). The pit depth also strongly decreases. For 5 m, the foil is perforated by pits in many locations, while for 21 m only very superficial attacks are observed. As expected, the amount of electrolyte decomposition products present on the Al foils strongly decreases with increasing LiTFSI concentration, in line with the ESW. While for the 1 m solution, localized electrolyte decomposition actually seems to be the dominant process taking place at 4.8 V vs. Li/Li+ (see the large cluster of electrolyte decomposition products on top of/adjacent to the corrosion pit in Fig. S1a, ESI†) hardly any decomposed electrolyte could be detected for the 21 m electrolyte.
It has been established that the availability of solvent molecules is essential for anodic Al dissolution in TFSI-based electrolytes to take place.14 For example, Al shows very good stability in TFSI-based ionic liquids. However, upon addition of a small amount of a polar solvent, Al becomes much less stable.13 The high stability in solvent-free electrolytes could be related to steric hindrance of Al3+ complexation by TFSI anions only or to necessary presence of polar solvent molecules for the initiation of pit formation at defect sites in the oxide layer. Above 10 m, the presence of free H2O molecules in the LiTFSI–H2O system is dramatically changing as illustrated by changes in Li+ coordination:1 At 10 m, the primary solvation sheath consists in average of close to four H2O molecules per Li+, typical for diluted aqueous electrolytes, and very little TFSI−. For concentrations >10 m, the number of TFSI anions in the Li+ solvation sheath increases and mixed coordination dominates. At 21 m, less than 2.5 H2O molecules per Li+ are present in the primary solvation sheath. At the same time, the fraction of free H2O strongly decreases1 from about 40% for the 10 m electrolyte to about 15% for the 21 m electrolyte, and the H2O molecules become less mobile due to coordination to Li+ becoming increasingly dominant.14 This leads to a low probability for free H2O to reach the Al–electrolyte interface and hence results in strongly suppressed anodic Al dissolution.
To confirm that the high stability of Al found during experiments with Al foil working electrodes also translates to good stability in a battery, we assembled a prototype battery containing a LiMn2O4 (LMO) cathode coated on an Al foil current collector. We choose LMO, the archetypical example of a spinel oxide cathode material, despite its relatively low cycling stability (LMO is well known to suffer from capacity decay due to a Jahn–Teller distortion accompanied with anisotropic volume change induced by locally present excess Mn3+ during the discharge process, especially at high rates),15 because cathode materials from the other two main classes of cathode materials, olivines and layered transition metal oxides, either have relatively low redox potentials, e.g., 3.5 V vs. Li/Li+ for LiFePO416 as an example of the former class, or show a basic reaction with water, e.g., LiNixCoyMnzO217 as an example of the latter class, driving the pH value of the slurry and potentially of the aqueous electrolyte outside the stability range of Al. An oversized activated carbon pellet was used as negative electrode to minimize the influence of the anode on the cycling stability of the battery. Fig. 3a shows the results from cyclic voltammetry between 3.7 and 4.6 V vs. Li/Li+ at a scan rate of 100 μV s−1. For both the 1 m and the 21 m electrolytes, two pairs of LMO delithiation and lithiation peaks are clearly visible. The delithiation and lithiation potentials are shifted by about 250 mV to more positive potentials for the 21 m compared to the 1 m electrolyte due to the much higher Li+ activity of the 21 m electrolyte as illustrated in ref. 1. As expected from our previous experiments, the LMO cycling stability is much higher in the 21 m electrolyte, as evidenced by the decreasing/stable lithiation peak intensities for the 1 m (see arrow) and 21 m electrolyte in Fig. 3a, respectively. Furthermore, with the cycle number accelerating parasitic side reactions that we mainly ascribe to anodic Al dissolution take place during the CV measurement in the 1 m electrolyte as evidenced by the (increasing) anodic current especially above 4.2 V. To investigate the long-term stability of LMO in the electrolytes we carried out constant current cycling experiments at a current density of 1 C (Fig. 3b). The results reveal a strong dependence of cycling stability on the LiTFSI concentration. The battery with the 21 m electrolyte solution shows a capacity retention typical for LMO of 83% after 100 cycles, while much more pronounced capacity fading and generally lower Coulombic efficiency is observed for 1 m LiTFSI, although the 1 m cell was cycled in a lower potential range derived from CV. During the 20th cycle, the 1 m cell prematurely failed, seen by the abrupt drop in capacity, due to total loss of contact between the active material and the remaining intact part of the Al current collector.
 | ||
| Fig. 3 Cycling of LiMn2O4 coated on Al current collectors in 1 m and 21 m aqueous LiTFSI solutions. (a) Cyclic voltammetry carried out between 3.7 and 4.6 V vs. Li/Li+ at a scan rate of 100 μV s−1 and (b) constant current cycling at 1 C carried out between 3.7 and 4.35 V and 3.95 and 4.6 V vs. Li/Li+ in 1 m and 21 m LiTFSI, respectively. Selected potential profiles are shown in (c). An oversized activated carbon electrode was used as the counter electrode. The composition of the LiMn2O4 electrodes was 87% active material, 5% carbon black, and 8% PVdF binder. The typical mass loading was 4 to 5 mg cm−2. | ||
The effect of anodic Al dissolution in the 1 m cell taking place at the same potentials as LMO delithiation is also seen in the potential profiles in Fig. 3c, where the potential plateaus become increasingly wider after the 9th cycle (see arrows).
Visual inspection of the LMO electrodes cycled in 1 m LiTFSI (not shown) reveals that the Al current collector shows strong signs of Al attack and dissolution, while the electrode cycled in the 21 m electrolyte appears virtually unaffected. We further recovered the electrolyte solution of the 21 m cell to determine its Al and Mn contents, after dilution and acidification with 1% v/v HNO3, via inductively coupled plasma optical emission spectrometry. The recovered electrolyte contained Al and Mn in concentrations of 0.2 and 0.1 μg mL−1, respectively, with the estimated detection limit being 0.1 μg mL−1 for both elements. The detection of some Mn in the electrolyte is in line with the instability of LMO. The additional Al concentration of 0.1 μg mL−1 (0.1 μg mL−1 of Al was already present in the electrolyte before the cell test) corresponds to a total mass loss after 100 cycles of 0.006% of the Al current collector immersed in the 21 m electrolyte and to an irreversible charge consumption of 0.004% per cycle with respect to the average discharge capacity assuming transfer of three electrons per Al atom. This rate of Al dissolution may be acceptable from an application point of view, assuming it is not accelerating with increasing cycle number.
So far, the discussion focused on the use of Al as the current collector for the cathode side. Interestingly, Al current collectors can also be employed at the anode side of high-voltage aqueous batteries based on highly concentrated neutral pH electrolytes. In non-aqueous lithium-ion batteries, copper is the standard current collector material for the anode side, but represents a major cost driver. Al is not an option in this case as lithium alloys with Al at about 0.3 V vs. Li/Li+,18 a potential more positive than the potentials of lithium insertion into graphite, the standard anode material of lithium-ion batteries. The alloy formation is accompanied by a volume change and hence pulverization of the Al current collector. However, the hydrogen evolution reaction limits the anode potential of high-voltage aqueous batteries to potentials more positive than the lithium–Al alloy formation potential making the use of Al as the current collector for the anode side possible.
The ability to use cost-effective light-weight Al as the current collector material on cathode and anode sides for future high-voltage aqueous batteries allows benefitting from electrode fabrication know-how obtained for non-aqueous lithium-ion batteries. Future research should focus on identifying and optimizing (high-capacity) anode and cathode materials that utilize the full potential window of highly concentrated aqueous LiTFSI electrolytes.
Notes and references
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Science, 2015, 350, 938 CrossRef CAS PubMed.
- L. Suo, F. Han, X. Fan, H. Liu, K. Xu and C. Wang, J. Mater. Chem. A, 2016, 4, 6639 CAS.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, Pergamon Press, London, 1966 Search PubMed.
- A. J. Salkind, A. G. Cannone and F. A. Trumbure, in Handbook of batteries, ed. D. Linden and T. B. Reddy, McGraw-Hill, New York, 3rd edn, 2002, ch. 23 Search PubMed.
- M. Fetcenko, in Handbook of batteries, ed. D. Linden and T. B. Reddy, McGraw-Hill, New York, 3rd edn, 2002, ch. 30 Search PubMed.
- Z. Szklarska-Smialowska, Corros. Sci., 1999, 41, 1743 CrossRef CAS.
- L. J. Krause, W. Lamanna, J. Summerfield, M. Engle, G. Korba, R. Loch and R. Atanasoski, J. Power Sources, 1997, 68, 320 CrossRef CAS.
- L. Péter and J. Arai, J. Appl. Electrochem., 1999, 29, 1053 CrossRef.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928 CrossRef CAS PubMed.
- G. Crabtree, Nature, 2015, 526, S92 CrossRef CAS PubMed.
- J. O. G. Posada, A. J. R. Rennie, S. Perez Villar, V. L. Martins, J. Marinaccio, A. Barnes, C. F. Glover, D. A. Worsley and P. J. Hall, Renewable Sustainable Energy Rev., 2016 DOI:10.1016/j.rser.2016.02.024.
- P. Quaino, F. Juarez, E. Santos and W. Schmickler, Beilstein J. Nanotechnol., 2014, 5, 846 CrossRef CAS PubMed.
- R.-S. Kühnel, M. Lübke, M. Winter, S. Passerini and A. Balducci, J. Power Sources, 2012, 214, 178 CrossRef.
- D. W. McOwen, D. M. Seo, O. Borodin, J. Vatamanu, P. D. Boyle and W. A. Henderson, Energy Environ. Sci., 2014, 7, 416 CAS.
- X. Li, Y. Xu and C. Wang, J. Alloys Compd., 2009, 479, 310 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CrossRef CAS.
- I. Doberdò, N. Löffler, N. Laszczynski, D. Cericola, N. Penazzi, S. Bodoardo, G.-T. Kim and S. Passerini, J. Power Sources, 2014, 248, 1000 CrossRef.
- A. S. Baranski and W. R. Fawcett, J. Electrochem. Soc., 1982, 129, 901 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6cc03969c |
| This journal is © The Royal Society of Chemistry 2016 |