
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe pKa of Brønsted acids controls their reactivity with diazo compounds†
Na
Fei
,
Basilius
Sauter
and
Dennis
Gillingham
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, CH-4056, Basel, Switzerland. E-mail: dennis.gillingham@unibas.ch
First published on 23rd May 2016
Abstract
We study the O-alkylation of phosphate groups by alkyl diazo compounds in a range of small molecules and biopolymers. We show that the relatively high pKa of phosphate in comparison to the other naturally occurring Brønsted acids can be exploited to control alkylation selectivity. We provide a simple protocol for chemical modification of some of the most important instances of phosphates in natural compounds including in small molecule metabolites, nucleic acids, and peptides.
The phosphate group and its esters are ubiquitous in Nature, serving critical roles in nucleic acids, cell membranes, metabolism, signal transduction, and numerous secondary metabolites.1,2 Protein phosphorylation is the most important post-translation modification of proteins, and yet, despite more than sixty years of study, most instances remain poorly understood.3 It is a difficult problem to study: signal transduction networks are complicated and locating phosphorylation sites in proteins in time and space is analytically challenging.4–7 Chemical methods to build or modify phosphate esters have been essential in studying their biology but the two main approaches have limitations: phosphorus(III) chemistry is water-sensitive (although recent efforts are addressing this issue)8 and phosphate ester activation leads to side-reactions. For example, one challenge in selective inositol phosphate syntheses9,10 is their proclivity for phosphate ester migration once they are activated.11 Diazo based esterification would offer a solution to such problems since it operates by O-alkylation rather than acyl transfer. Diazo esterification is used often in organic synthesis and has recently been explored by the Raines lab as a labelling reaction in chemical biology. They have developed techniques for the mild aqueous generation of diazo compounds12 and shown how these can be used for the reversible labelling of carboxylate groups in proteins.13,14
We demonstrate here that pH control of the reaction medium not only reduces acid-mediated hydrolysis of diazo compounds, judged by diazo compound half-life, (see the ESI† page 5), but also allows toggling of O-alkylation selectivity amongst Brønsted acids (see Path B in Fig. 1).
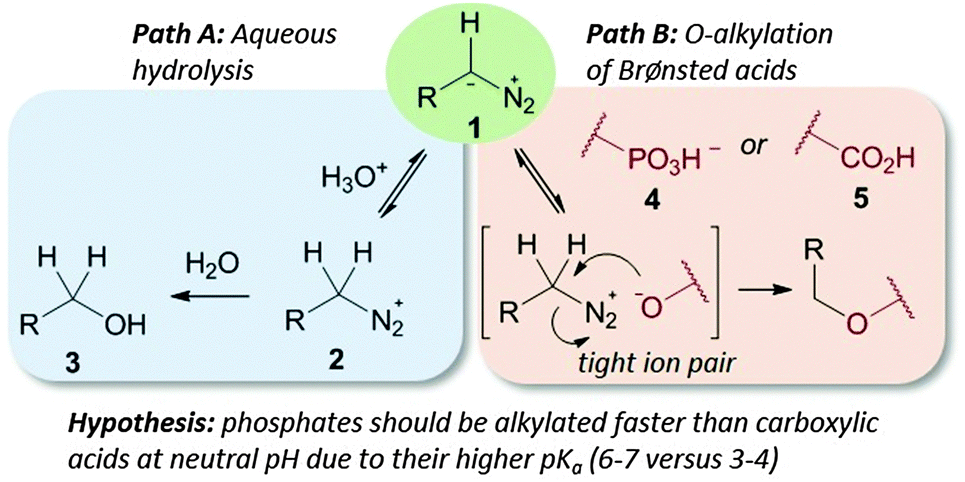 | ||
| Fig. 1 Diazo stability in water is controlled by its pKa (Path A); Brønsted acids capable of protonating diazo compounds lead to O-alkylation (Path B). | ||
Despite the importance of the phosphate esters and their known reactivity with diazo compounds no head-to-head comparison of their reactivity with that of carboxylates or a detailed look at the reactivity difference between phosphate mono- and diesters has been attempted. There are a handful of cases of phosphate ester alkylation by diazo compounds.15–21 For example, nucleotide monophosphates were first modified by phenyldiazomethane in 1955,22 and caged inositol and nucleoside monophosphates are often made through reactions of phosphates with diazo compounds.23,24 For example, a diazo-substituted coumarin25 has been employed as a photocaging reagent of DNA and RNA before delivery into zebrafish. While these reports highlight the potential of phosphate alkylation, they did not explore the reaction scope, mechanism, or compare of reactions with different Brønsted acids. In our own work on metal-catalysed DNA and RNA alkylation by diazo compounds,26–28 we sometimes found low levels of phosphate alkylation in product streams. In this paper we take a close look at phosphate monoester alkylation and compare it to carboxylate and phosphate diester alkylation under neutral pH aqueous conditions.
For reactions of diazo compounds in water the pKa of its protonated form (the diazonium ion) is critical.13 If the conjugate base (1 in Fig. 1) is too basic it will be protonated quickly by water and decompose unselectively through hydrolysis (Path A in Fig. 1).29 When used to perform transformations in water diazo compounds akin to diazomethane must therefore be used in enormous excess since hydrolysis will always predominate (for example diazomethane has a pKa of 10).29 If too weakly basic diazo compounds will not deprotonate the Brønsted acids (required for Path B in Fig. 1) to initiate reaction – compounds in this class include diazomalonates and donor–acceptor substituted diazo esters. These stable diazo compounds have long lifetimes in water and hence can be used to perform aqueous metal-carbene reactions. Their weak basicity, however, renders them of little value in Brønsted acid alkylation. Quantifying the middle ground between these two extremes is essential for controlling selectivity in diazo-type esterifications.
The first step in a productive diazo-type O-alkylation is protonation by the Brønsted acid (see Path B in Fig. 1).30 It should then be possible to exploit pKa differences between acids to control O-alkylation selectivity. To test this hypothesis we examined product ratios of CMP versus benzoic acid alkylation in aqueous buffer at different pHs using commercially available trimethylsilyldiazomethane (1b in Fig. 2) as the diazo compound. Indeed at low pH benzoic acid alkylation is preferred, but as the pH is raised phosphate alkylation begins to dominate (see Fig. 2). This is consistent with benzoic acid being largely benzoate at pHs higher than six, while a large fraction of the phosphate remains protonated.
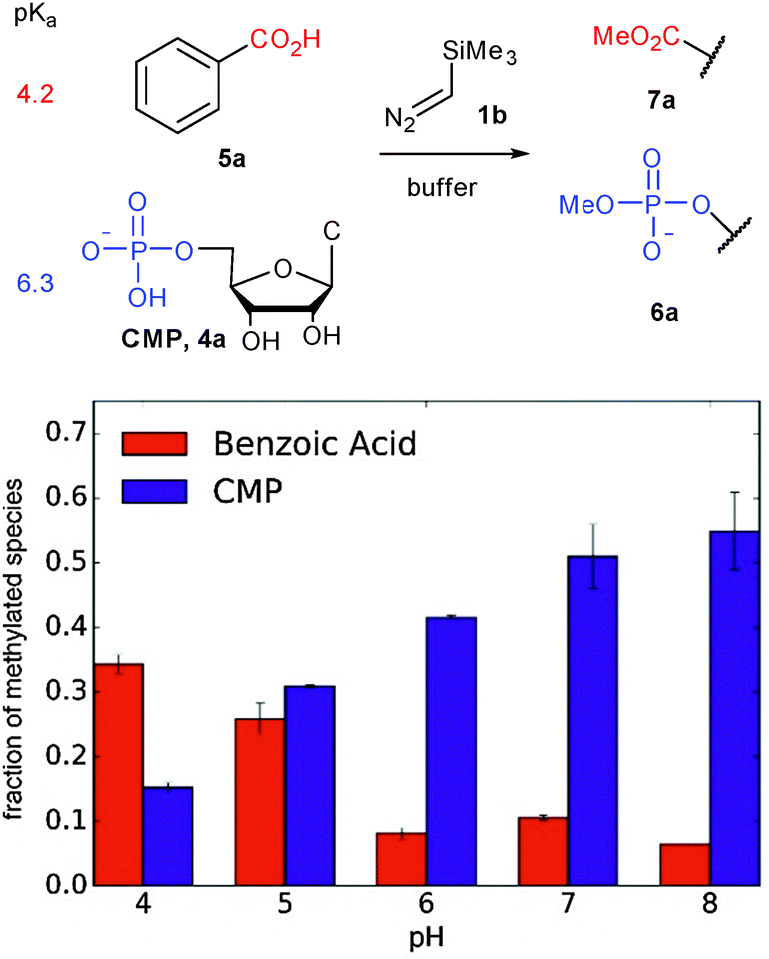 | ||
| Fig. 2 Competition experiments at varying pH support the role of pKa in reactions of Brønsted acids with trimethylsilyl diazomethane. | ||
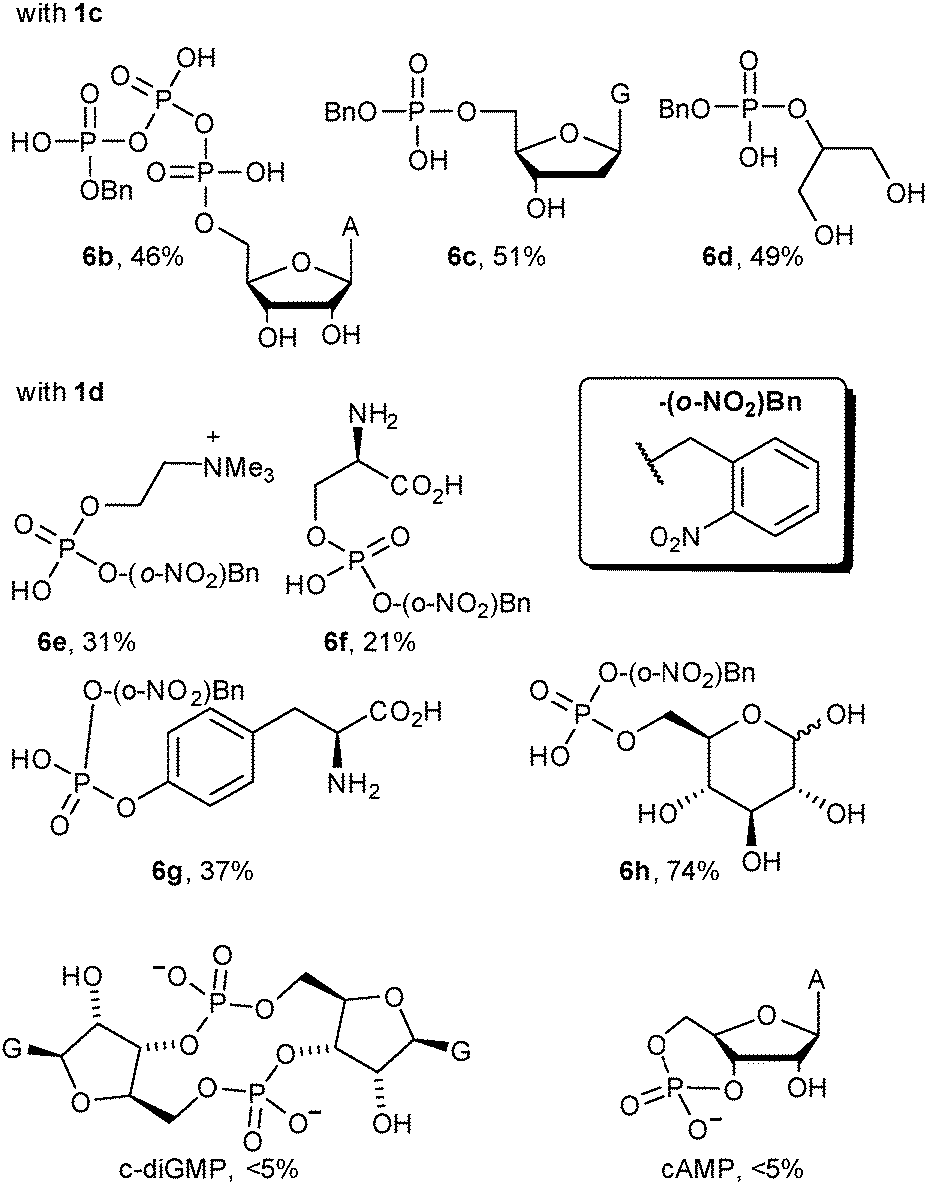
Although 1b was a convenient, highly basic, diazo compound for preliminary hypothesis testing, its extreme reactivity (comparable to diazomethane) would limit its practical application. The ideal diazo compound would have pKb that matches the pKa of the Brønsted acid. Unfortunately there has been no tabulation of pKas for diazo compounds. Raines13 selected the pKa of the parent alkyl group as an expedient for diazo acidity in their discussion of ester alkylation and found that diazo compounds derived from fluorene or α-aryl amides delivered optimal alkylation yields. The respective carbon acids of these have pKa values in the mid-twenties. We elected to choose a different series of structures based on practical considerations (diazo compounds 1c, 1d, and ethyl α-diazoacetate (not shown) were tested in a standard reaction with CMP), but our results are qualitatively in agreement with those of the Raines lab. In our case the most effective diazo compound was 1d, whose parent carbon acid has a pKa of approximately 25 in water.31 The more basic compound 1c also gave product, but it was prone to rapid degradation; the resonance stabilized compound ethyl α-diazoacetate gave undetectable levels of alkylation product.
The reaction is general. We selected a range of biologically relevant monophosphates and looked at their reaction with either diazo compounds 1c or 1d (see Table 1). The yields are not high (21–74% depending on the substrate) but the approach's simplicity and conciseness should override the practical disadvantage of moderate yields. In general we consider 1d the more valuable reagent because the ortho-nitrobenzyl (o-NO2-Bn) adduct is photolabile, offering opportunities for photo-uncaging applications (see the ESI† pages 15 & 41 for photo-uncaging examples with compounds 6g and 14).321d has the additional advantage that it is more stable than most alkyl diazo compounds since the ortho-nitro group provides resonance stabilization of the diazo anion.
|
|
|---|
| a Conditions: Diazo compound as a stock solution was added to a 50 mM solution of the phosphate in 250 mM MES buffer. Reactions were stopped when diazo compound was completely consumed as determined by the disappearance of the orange colour. |
|
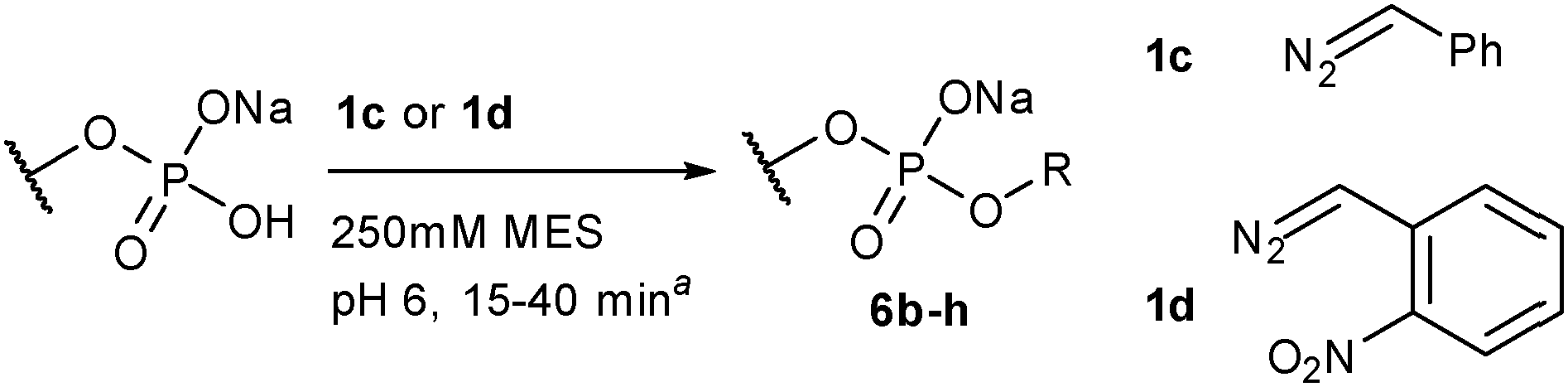
Modifications of phosphates within large biopolymers would be a valuable application of diazo alkylation. While this reaction has previously been employed in the alkylation of nucleoside monophosphates and nucleic acid oligomers, past work suffered from a lack of selectivity, giving both mono- and diester alkylation.16,18 The exception is the work of the Friedman lab, who have shown that terminal phosphates are strongly preferred over phosphate diesters in oligonucleotide alkylation.20,21 We propose that pH buffering in the vicinity of the Brønsted acid's pKa is the decisive difference for selectivity. This is supported by the poor reactivity of the phosphate diesters shown in the bottom two entries of Table 1 where less than 5% conversion is observed – identical conditions lead to clean alkylation of phosphate monoesters. There are cases where phosphate diester alkylation has been used in practice, but an enormous excess is always employed.19,25 With 30 equivalents of 1d we begin to see phosphate diester alkylation (data not shown), but under these forcing conditions substantial amounts of sulfonate alkylation of the MES buffer salt were also observed (see the ESI† page 9). The dramatic reactivity difference between mono- and diesters augured well for a selective 5′-phosphate alkylation in larger oligonucleotides.
Oligonucleotides bearing terminal phosphates are alkylated selectively (Fig. 3). A simple hexanucleotide (8, see trace a in Fig. 3) bearing a 5′-phosphate was treated with the diazo compound 1d in varying amounts at pH 7 (see traces b–g). Optimal conversion was observed at 20 equivalents of 1d with further increases leading to little improvement. The reactions are clean with most by-products derived from side-reactions of the diazo compound with itself or water (see trace j for control sample where 8 is not added to reaction) and not with the backbone or the bases of the oligonucleotide (see trace h for control experiment of 1d's reaction with dephosphorylated 8).
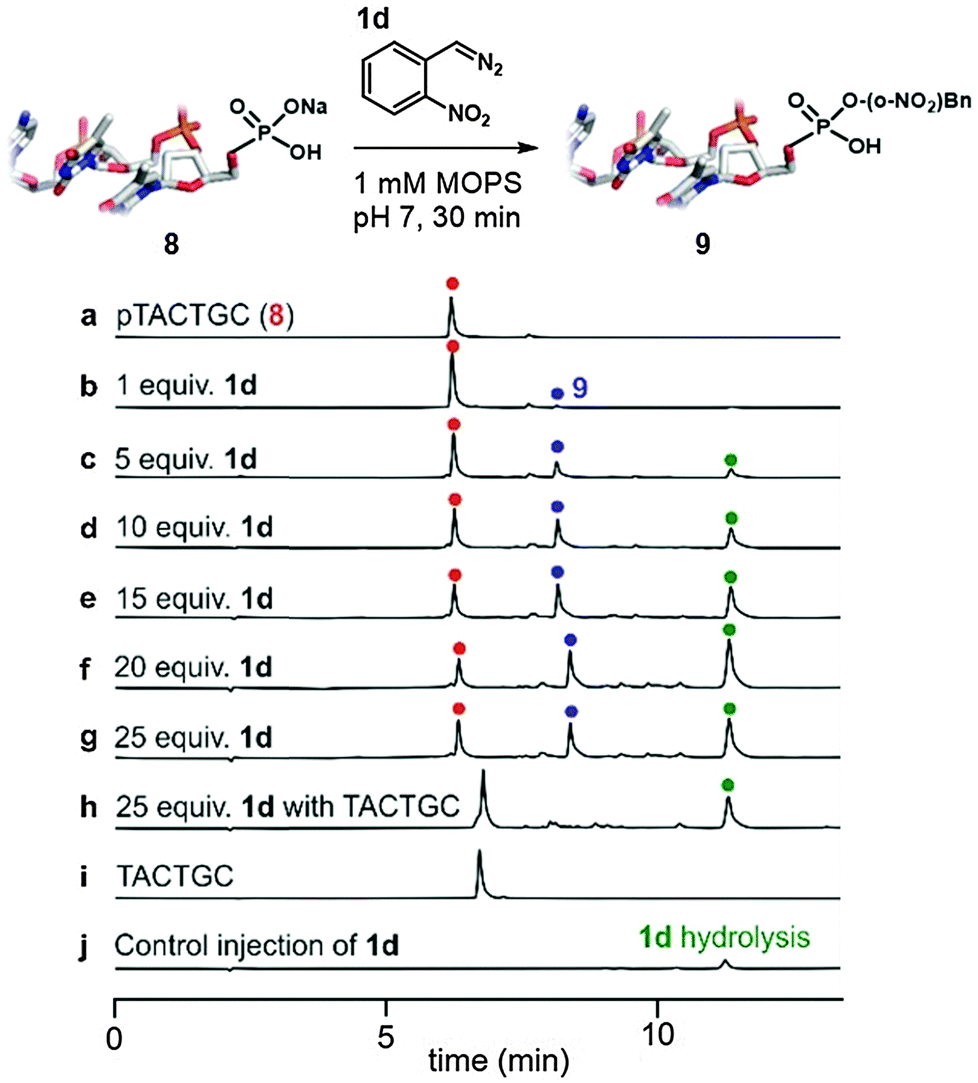 | ||
| Fig. 3 5′-Phosphates in DNA are alkylated cleanly by diazo compound 1d. Conditions: the appropriate amount of 1d stock solution was added to a 100 μM solution of the oligonucleotide and aliquots were analysed by HPLC at 30 minutes. | ||
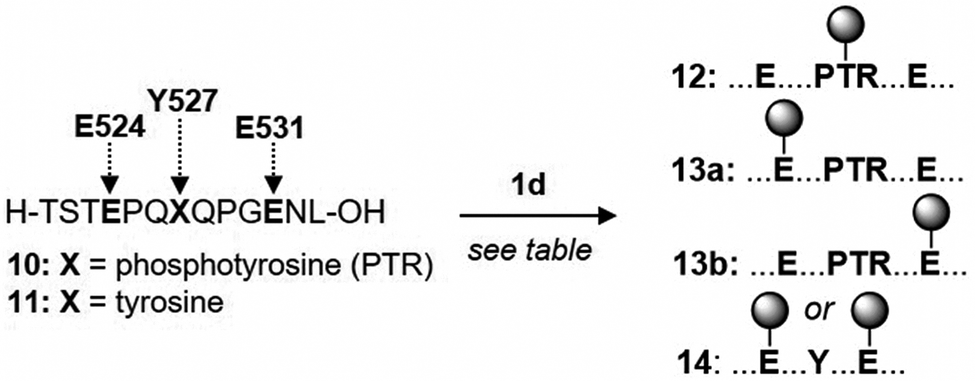
Protein phosphorylation is a widespread post-translational modification that regulates numerous functions.33 The ability to selectively modify phosphates in a native protein would be a powerful tool for studying phosphorylation networks.34 Alkylation to create products 6f and 6g in Table 1 already suggested that a selective phosphate alkylation in a peptide or protein might be feasible. Although whole proteins with a precisely defined phosphorylation state are difficult to obtain, many phosphorylated model peptides are commercially available. Phosphorylation of Y527 in the C-terminal region of c-Src causes this domain to fold over on its SH2 domain, holding the protein in an inactive state.35 A 13-mer phosphorylated peptide of the c-Src C-terminal domain (residues 521–533, see 10 in Table 2 header) has therefore become an important model peptide in studying Src signalling. If the commercially available TFA salt of 10 is dissolved in unbuffered water (final pH 4.3) and treated directly with diazo, alkylation of glutamic acid 531 (E531) is the major product (see entry 2). Consistent with the pKas controlling selectivity, performing the same reaction in pH 7 MOPS buffer switches the alkylation preference to Y527 (cf. entries 2 & 3). Although there are three carboxylates available for alkylation (C-terminus, E524 and E531), phosphate alkylation is the major product along with a small amount of carboxylate alkylation (13a & 13b, sites of alkylation are determined by MS/MS sequencing, see the ESI† for pages 39–41 for details). In addition, while 2 equivalents of 1d leads to 43% conversion of peptide 10, dephosphorylated peptide 11 is not detectably alkylated under the same conditions (see ESI† page 32). Entries 3–7 investigate the effect of adding more diazo compound and the ideal seems to be 3 equivalents; further addition of 1d increases consumption of peptide 10, but also leads to substantial amounts of double alkylation with little change in the yield of 12 (see number in parenthesis in the conversion column of Table 2). A second study on a model 5-mer phosphorylated peptide is given in the ESI† on pages 43–46 and supports the generality of the observations in Table 2. The results in Table 2 again indicate how pH can be exploited to control selectivity in Brønsted acid alkylation, even in the presence of competing acids.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | pHa | Equiv. 1d | Major product | % conv.b | Y527: (E524 + E531)c |
| a Run in 5 mM MOPS buffer at pH 7, pH 4.3 experiment is run in unbuffered water and the pH is measured after the reaction. b First number is conversion of starting peptide; number in parenthesis refers to amount of alkylated phosphate. c Ratios are determined by HPLC integration. d A small side-product obscures phosphate alkylation hence we can only report a lower limit (see the ESI, Fig. S9–S13 for details). | ||||||
| 1 | 11 | 7 | 2 | 14 | <5 | n.a. |
| 2 | 10 | 4.3 | 20 | 13 | 40 | ≤1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9d :9d |
| 3 | 10 | 7 | 2 | 12 | 43 (35) | 13:1 |
| 4 | 10 | 7 | 3 | 12 | 52 (42) | 15:1 |
| 5 | 10 | 7 | 5 | 12 | 68 (44) | 11:1 |
| 6 | 10 | 7 | 10 | 12 | 75 (47) | 11:1 |
| 7 | 10 | 7 | 15 | 12 | 95 (40) | 11:1 |
The accepted mechanism of O-alkylation by diazo compounds predicts that the pH of the medium could be used to steer O-alkylation preference among Brønsted acids. We have validated this prediction and shown how this property can be used to selectively target phosphates in a mixture of other Brønsted acids including carboxylic acids and sulfonic acids.
In its present form the method is most useful for creating photo-caged variants of peptides, nucleic acids, and phosphate bearing metabolites. An exciting future application, however, would be in phosphoproteomics34,36 and we are currently working to apply the method in this area.
Notes and references
- F. H. Westheimer, Science, 1987, 235, 1173–1178 CAS.
- S. C. L. Kamerlin, P. K. Sharma, R. B. Prasad and A. Warshel, Q. Rev. Biophys., 2013, 46, 1–132 CrossRef CAS PubMed.
- P. Cohen, Nat. Cell Biol., 2002, 4, E127–E130 CrossRef CAS PubMed.
- J. V. Olsen, B. Blagoev, F. Gnad, B. Macek, C. Kumar, P. Mortensen and M. Mann, Cell, 2006, 127, 635–648 CrossRef CAS PubMed.
- T. J. Yeatman, Nat. Rev. Cancer, 2004, 4, 470–480 CrossRef CAS PubMed.
- S. J. Parsons and J. T. Parsons, Oncogene, 2004, 23, 7906–7909 CrossRef CAS PubMed.
- L. C. Kim, L. Song and E. B. Haura, Nat. Rev. Clin. Oncol., 2009, 6, 587–595 CrossRef CAS PubMed.
- M. R. J. Vallée, P. Majkut, D. Krause, M. Gerrits and C. P. R. Hackenberger, Chem. – Eur. J., 2015, 21, 970–974 CrossRef PubMed.
- S. Capolicchio, D. T. Thakor, A. Linden and H. J. Jessen, Angew. Chem., Int. Ed., 2013, 52, 6912–6916 CrossRef CAS PubMed.
- S. Capolicchio, H. Wang, D. T. Thakor, S. B. Shears and H. J. Jessen, Angew. Chem., Int. Ed., 2014, 53, 9508–9511 CrossRef CAS PubMed.
- I. Pavlovic, D. T. Thakor, L. Bigler, M. S. Wilson, D. Laha, G. Schaaf, A. Saiardi and H. J. Jessen, Angew. Chem., Int. Ed., 2015, 54, 9622–9626 CrossRef CAS PubMed.
- H.-H. Chou and R. T. Raines, J. Am. Chem. Soc., 2013, 135, 14936–14939 CrossRef CAS PubMed.
- N. A. McGrath, K. A. Andersen, A. K. F. Davis, J. E. Lomax and R. T. Raines, Chem. Sci., 2015, 6, 752–755 RSC.
- K. A. Mix and R. T. Raines, Org. Lett., 2015, 17, 2358–2361 CrossRef CAS PubMed.
- N. Nimura, T. Kinoshita, T. Yoshida, A. Uetake and C. Nakai, Anal. Chem., 1988, 60, 2067–2070 CrossRef CAS PubMed.
- A. Laayoun, M. Kotera, I. Sothier, E. Trévisiol, E. Bernal-Méndez, C. Bourget, L. Menou, J. Lhomme and A. Troesch, Bioconjugate Chem., 2003, 14, 1298–1306 CrossRef PubMed.
- M. Kotera, M.-L. Dheu, A. Milet, J. Lhomme and A. Laayoun, Bioorg. Med. Chem. Lett., 2005, 15, 705–708 CrossRef CAS PubMed.
- C. Bourget, E. Trévisiol, I. Bridon, M. Kotera, J. Lhomme and A. Laayoun, Bioorg. Med. Chem., 2005, 13, 1453–1461 CrossRef CAS PubMed.
- J. Engels, Bioorg. Chem., 1979, 8, 9–16 CrossRef CAS.
- S. Shah, P. K. Jain, A. Kala, D. Karunakaran and S. H. Friedman, Nucleic Acids Res., 2009, 37, 4508–4517 CrossRef CAS PubMed.
- P. K. Jain, S. Shah and S. H. Friedman, J. Am. Chem. Soc., 2011, 133, 440–446 CrossRef CAS PubMed.
- D. Brown, D. Magrath and A. R. Todd, J. Chem. Soc., 1955, 4396–4401 RSC.
- J. W. Walker, G. P. Reid, J. A. McCray and D. R. Trentham, J. Am. Chem. Soc., 1988, 110, 7170–7177 CrossRef CAS.
- J. W. Walker, J. Feeney and D. R. Trentham, Biochemistry, 1989, 28, 3272–3280 CrossRef CAS PubMed.
- H. Ando, T. Furuta, R. Y. Tsien and H. Okamoto, Nat. Genet., 2001, 28, 317–325 CrossRef CAS PubMed.
- K. Tishinov, K. Schmidt, D. Häussinger and D. G. Gillingham, Angew. Chem., Int. Ed., 2012, 51, 12000–12004 CrossRef CAS PubMed.
- K. Tishinov, N. Fei and D. Gillingham, Chem. Sci., 2013, 4, 4401–4406 RSC.
- N. Fei, D. Haussinger, S. Blumli, B.-J. Laventie, L. D. Bizzini, K. Zimmermann, U. Jenal and D. Gillingham, Chem. Commun., 2014, 50, 8499–8502 RSC.
- J. F. McGarrity and T. Smyth, J. Am. Chem. Soc., 1980, 102, 7303–7308 CrossRef CAS.
- E. Kühnel, D. D. P. Laffan, G. C. Lloyd-Jones, T. Martínez del Campo, I. R. Shepperson and J. L. Slaughter, Angew. Chem., Int. Ed., 2007, 46, 7075–7078 CrossRef PubMed.
- J. Lelievre, P. G. Farrell and F. Terrier, J. Chem. Soc., Perkin Trans. 2, 1986, 333–336 RSC.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2012, 113, 119–191 CrossRef PubMed.
- K. Schmelzle and F. M. White, Curr. Opin. Biotechnol., 2006, 17, 406–414 CrossRef CAS PubMed.
- K. Engholm-Keller and M. R. Larsen, Proteomics, 2013, 13, 910–931 CrossRef CAS PubMed.
- J. M. Summy and G. E. Gallick, Cancer Metastasis Rev., 2003, 22, 337–358 CrossRef CAS PubMed.
- B. Macek, M. Mann and J. V. Olsen, Annu. Rev. Pharmacol. Toxicol., 2009, 49, 199–221 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed protocols, assays, and characterization. See DOI: 10.1039/c6cc03561b |
| This journal is © The Royal Society of Chemistry 2016 |