
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A thermoresponsive poly(ionic liquid) membrane enables concentration of proteins from aqueous media†
Yuki
Kohno
*abc,
Douglas L.
Gin
cd,
Richard D.
Noble
c and
Hiroyuki
Ohno
*ab
aDept. of Biotechnology, Tokyo University of Agriculture and Technology, Naka-cho, Koganei, Tokyo 184-8588, Japan. E-mail: ohnoh@cc.tuat.ac.jp; yuki_k@cc.tuat.ac.jp
bFunctional Ionic Liquid Laboratories, Graduate School of Engineering, Tokyo University of Agriculture and Technology, Naka-cho, Koganei, Tokyo 184-8588, Japan
cDept. of Chemical & Biological Engineering, University of Colorado, Boulder, CO 80309, USA
dDept. of Chemistry & Biochemistry, University of Colorado, Boulder, CO 80309, USA
First published on 16th May 2016
Abstract
A new type of poly(ionic liquid) membrane, which shows switchable hydrated states via lower critical solution temperature-type phase behaviour, enables concentration of some water-soluble proteins from aqueous media.
Affinity control between bio-polymers (e.g., proteins, enzymes, and DNA) and synthetic polymers plays a crucial role in the design of attractive bio-engineering processes including bio-separations,1 drug/gene deliveries,2 and catalytic processes.3 Stimuli-responsive polymers (i.e., “smart” polymer materials that alter their properties in response to external stimuli) have been exploited for many of these processes because of their excellent sensitivities toward temperature, pH, and concentration of specific molecules.4 Poly(N-isopropylacrylamide) and some other non-ionic polymers are known to show lower critical solution temperature (LCST)-type phase behaviour in aqueous media, in which homogeneously dissolved polymers reversibly become immiscible in water.4 Although such thermal responsiveness in these non-ionic polymers has long offered opportunities for use in bio-engineering processes, very little work has been carried out to design highly charged polyelectrolytes with the same type of LCST-type phase behaviour for the above-mentioned applications.
In the last several years, increasing attention has been focused on ionic liquids (ILs) and poly(ionic liquid)s (PILs) that show thermoresponsive phase transitions.5 Our intensive and systematic investigations have revealed that the total hydrophobicity of both cations and anions governs IL phase behaviour with water, and suitably-designed ILs with hydrophobicity between hydrophilic and hydrophobic ILs have been found to undergo highly temperature-sensitive and reversible LCST behaviour.6 Through the same design criteria, a few polymerisable ILs (i.e., ILs with polymerisable groups on one of the component ions) have been prepared and also found to show the LCST behaviour with water even after polymerisation.7 Such highly charged, highly thermoresponsive, and IL-derived polyelectrolyte materials should contribute to various bio-engineering processes after fabrication of various types of solid-state materials such as membranes, gels, and nanoparticles. The potential (expected) applications of these PIL-based solid-state materials have recently been summarised by us.5b Very recently, a handful of PIL-based materials have been proven to act as draw agents in forward osmosis systems for desalination applications.8 However, concrete and practical applications for thermoresponsive PILs are still undeveloped.
Herein, we report a new type of thermoresponsive PIL membrane (PILM) for protein concentration from aqueous media. There are a few papers on the thermoresponsive, non-polymerisable ILs to extract proteins from aqueous to IL phases via homogeneous liquid/liquid extraction techniques.9 However, to our knowledge, a thermoresponsive PIL material that is capable of controlling the partition of proteins via LCST behaviour is unprecedented, offering potential bio-engineering process applications especially for the separation and immobilisation of proteins without recognition of proteins through strong electrostatic interactions.
A series of PILMs was prepared from two IL monomers, tetra-n-butylphosphonium p-styrene sulfonate ([P4444][SS])7a and an analogous IL monomer comprising a cation with a longer alkyl chain ([P4448][SS]) (Fig. 1(a)). Free-standing, transparent, and flexible PILMs with a thickness of 150 μm were fabricated (Fig. 1(b)) by radical photo-cross-linking the two IL monomers in the presence of poly(ethylene glycol) (PEG) diacrylate (average number–average molecular weight (Mn) = 700, Fig. 1(a)) as a cross-linker and 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone as a radical photo-initiator. The ratio of the IL monomers/cross-linker/photo-initiator of these photo-cross-linking mixtures was 100/1/1 by mol. The detailed procedures for the fabrication of these PILMs are described in the ESI.† The completion of polymerisation was confirmed using infrared spectroscopy.
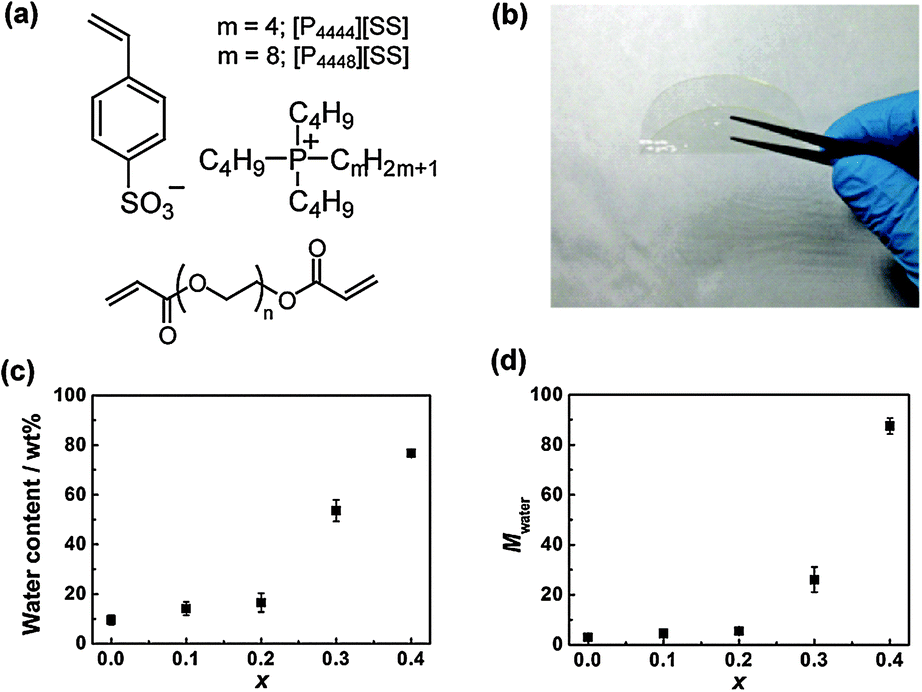 | ||
| Fig. 1 (a) Structure of IL monomers and a cross-linker; (b) Photograph of a cross-linked PILM used in this study; (c) water content of the poly([P4444][SS]x-co-[P4448][SS]1−x)-type PILMs (x corresponds to the molar fraction of [P4444][SS]) with different x values at ambient temperature (22 ± 1 °C); (d) the number of water molecules per ion pair (Mwater) of PILMs with different x values. | ||
To introduce LCST-type thermal responsiveness into non-polymerisable and polymerisable ILs, it is important to control the saturated water content of the IL phases after phase separation with water. Specifically, ILs having a number of water molecules per ion pair (Mwater) of 7 or more show LCST behaviour.6b,10 Furthermore, the Mwater value can be arbitrarily controlled by mixing multiple ILs with different hydrophobicities.6a Based on our previous investigation of thermoresponsive ILs, we anticipated that LCST behaviour is possible for PILMs by copolymerising suitable IL monomers with adequate hydrophobicity values (i.e., [P4444][SS] and [P4448][SS]). Fig. 1(c) shows the water content of the as-prepared PEG-diacrylate-cross-linked poly([P4444][SS]x-co-[P4448][SS]1−x)-type PILMs (x denotes the molar fraction of [P4444][SS] in the copolymer) after immersing them in pure water for 24 h at ambient temperature. The water content increased with the increasing molar fraction of the less hydrophobic [P4444][SS] monomer in the PIL, reaching a water content of 54 wt% when x = 0.3. This value corresponds to a Mwater value of 26 (Fig. 1(d)), which is above the threshold to show LCST behaviour.
The resulting poly([P4444][SS]0.3-co-[P4448]0.7)-type PILM 1 indeed exhibits LCST behaviour as predicted. Fig. S1 in the ESI† shows photographs of PILM 1 in pure water at different temperatures. At 5 °C, PILM 1 absorbed a relatively large amount of water. Upon increasing the temperature up to 22 °C, PILM 1 became opaque and eventually transparent again at 30 °C while shrinking. As shown in Fig. 2, the water content of PILM 1 decreased exponentially with a slight increase in the temperature at around 25 °C. For instance, the water content decreased from 54 to 14 wt% upon increasing the membrane temperature from 22 to 30 °C. This behaviour clearly indicates that PILM 1 is able to control the hydrated state via LCST behaviour. We have already reported thermoresponsive PIL gels showing reversible water uptake/release via LCST behaviour.11 These previously reported PIL gels absorbed a large amount of water (saturated water content >99 wt%) and desorbed water at higher temperatures (e.g., 50 °C), with a large amount of water still remaining in the gel.11 Very distinct from these reported PIL gels, PILM 1 is able to control the water content in a very narrow temperature range around physiological temperatures. This feature will be particularly beneficial when it comes to the affinity control of various molecules dissolved in water via an LCST response. A potential advantage of this technique is the use of indirect interactions between sufficiently hydrated polymer membranes and target molecules. Direct interactions such as electrostatic interaction and hydrogen bonding usually affect the conformation and even a higher-order structures of proteins.3a
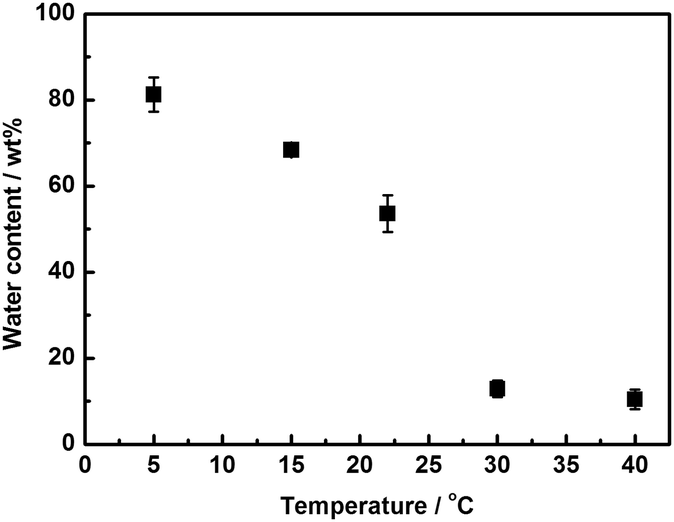 | ||
| Fig. 2 Temperature-dependent water content of PILM 1 (x value = 0.3). | ||
In addition to the PILM/pure water systems, it is also of great importance to analyse the phase behaviour of these PILMs with aqueous buffer solutions for protein engineering. We subsequently investigated the temperature-dependent phase behaviour of PILM 1 after soaking it in 0.01 and 0.1 mol L−1 aq. potassium phosphate buffer (PKB, pH = 7.0). As shown in Fig. S2 (ESI†), the absolute water content of PILM 1 decreased upon increasing the concentration of K2HPO4/KH2PO4 salts. This behaviour can be comprehended as the “salting-out” effect of the salts that results in the dehydration of PILM 1. In addition, water content of the PILM 1 also decreased upon increasing the temperature, clearly indicating that PILM 1 retains LCST-type thermal responsiveness in PKB solution. On the other hand, the decrease in the water content via the LCST response was smaller than that in pure water systems. Since this behaviour was seen in aqueous solutions with both salt concentrations tested, the existence of K2HPO4/KH2PO4 salts tends to make the LCST behaviour of PILM 1 rather sluggish.
A few reports have already been reported that analyse the phase behaviour of PILs in aq. salt solutions.7b,12 According to these previous investigations, phase transition temperatures of the PIL materials were significantly affected by the existence of the inorganic salts, but the salt effects (i.e., “salting-in” and “salting-out” effects) varied widely with both the salt species and the structure of PILs. We are currently performing systematic studies to clarify salt effects on the phase behaviour of PIL materials, including PILMs, in combination with different IL monomers and salt species.
The extraction efficiency values (EE) of some water-soluble, heme-proteins (e.g., cytochrome c from equine heart (Cyt. c), myoglobin from equine heart (Mb), and horseradish peroxidase (HRP)) in PILM 1 were then measured by soaking the membrane sample in PKB containing the corresponding proteins. A detailed procedure for the evaluation of EE was mentioned in the ESI.†Table 1 summarises the EE values of the proteins at 5, 22, and 30 °C for PILM 1. The observed EE value varies widely depending on both the protein species and the temperature. The highest EE was obtained for Cyt. c, and this value increased with the increasing temperature to reach 99.7% at 30 °C. Similarly, the EE of Mb increased for higher temperatures. On the other hand, the EE of HRP remained negligible at any temperature tested.
| Protein | EEa (%) at 5 °C | EE (%) at 22 °C | EE (%) at 30 °C |
|---|---|---|---|
| a EE was calculated using the following equation; EE (%) = (Absbefore × Vbefore − Absafter × Vafter)/(Absbefore × Vbefore) × 100, where Absbefore, Absafter, Vbefore, and Vafter respectively denote absorbance and volume of the aqueous phase before and after the protein extraction experiments. The reported values are the averages of at least 3 independent sample measurements with std. dev. error bars. | |||
| Cytochrome c | 93 ± 1 | 97.9 ± 0.2 | 99.7 ± 0.3 |
| Myoglobin | 38 ± 6 | 64 ± 4 | 83.4 ± 0.7 |
| Peroxidase | 5 ± 3 | 1 ± 2 | 2 ± 3 |
Fig. 3 displays photographs of PILM 1 after protein concentration experiments at 22 °C. Typical colours of Cyt. c and Mb were seen in the PILM 1 samples, whereas no colour was apparently seen in the membrane in contact with HRP. UV-visible spectroscopy on each membrane was then carried out. As for Cyt. c, a strong Soret band and a broad Q band were seen at 408 nm and around 530 nm, respectively, even in PILM 1 (see Fig. S3, ESI†), which is consistent with those in aq. PKB solution. The UV-visible spectrum of Mb confirmed the presence of the Soret band at 413 nm, as well as the Qα and Qβ bands at 564 and 536 nm, respectively (see Fig. S4, ESI†). This shift in the spectrum compared to the aqueous Mb solution (the Soret band at 408 nm and the Qα band at 620 nm) has already been reported in Mb-surfactant systems, which retain the O2 binding ability of Mb.13 On the other hand, no absorption derived from HRP was observed in PILM 1 (see Fig. S5, ESI†). This clearly shows that HRP remains in the aqueous phase, not in the PILM 1 film. These results strongly suggest that PILM 1 enables selective, stable, and thermoresponsive concentration of proteins from buffered aqueous solution into PILM 1.
 | ||
| Fig. 3 Photographs of PILM 1 after immersion in 0.1 mol L−1 aq. PKB solution with the proteins Cyt. c (left), Mb (middle), and HRP (right) at 22 °C for 24 h. | ||
In terms of the factors for concentration of proteins from an aqueous medium to the PILM 1 domain, we previously reported partition coefficients of various proteins between the aqueous phase and the IL phase using the liquid-state IL/water mixtures showing LCST behaviour.9a Among the proteins analysed, a significant difference in the partition coefficient was found in proteins in spite of the almost same isoelectric points (e.g., Mb and HRP).9a This behaviour could not be explained by simple electrostatic interaction between ILs and proteins. As a result of our investigations to comprehend the mechanism of this selectivity, both the “salting-out” and “salting-in” profiles of proteins depending on the IL concentration were concluded to mainly dominate the selectivity.9b Namely, in the case of Cyt. c, the salting-out profile was followed by the salting-in profile upon increasing the concentration of IL. The salting-in profile in a highly concentrated IL phase (it should be better to call “hydrated IL”14) enhances the solubility of Cyt. c9b while only the salting-out profile was found in HRP at higher IL concentrations; this resulted in a negligible solubility of HRP in the IL phase.9b Given the knowledge from these liquid-state IL studies, excellent selectivity in EE for Cyt. c and HRP should result from the salting-out and salting-in profiles of the proteins in hydrated PILM 1. We are currently undertaking further studies to fully comprehend these phenomena. These studies include a detailed analysis of the hydrated states of PILMs having different component ions, and the correlation between the hydrated states and concentration ability of many proteins with different properties (e.g., molecular weight, isoelectric point, and hydrophobicity).
A variety of thermoresponsive, non-ionic polymer materials have been utilised in the literature for affinity control of various water-soluble proteins.15 In many cases, the proteins were released from the polymer phase to the aqueous phase above the LCST.15c Also, high selectivity of proteins in these polymers was rarely observed, and copolymerisation of charged monomers was often employed to improve the selectivity.16 Apart from previous polymer systems, the EE values in PILM 1 increased with slight increase in temperature. In addition, excellent selectivity was found for specific proteins. Our new protein concentration process using PILM 1 has some obvious benefits compared to existing polymer materials when considering selectivity based on the salting-out and salting-in profiles of proteins, as well as the unique combination of properties derived from ILs. Our ongoing work with these PILMs includes: (i) detailed analysis and control of the hydrated state of the PILMs under multiple stimuli, (ii) establishment of membrane separation systems for proteins and other bio(macro)molecules, and (iii) activity analysis of proteins/enzymes in the PILMs.
In conclusion, a chemically cross-linked, sufficiently hydrated poly([P4444][SS]0.3-co-[P4448][SS]0.7)-type PILM 1 shows hydration/dehydration transition behaviour upon a slight temperature change. This behaviour enables selective concentration of proteins without a significant loss of their higher-order structures because of no direct interaction between proteins and the membrane.
Financial support of this work was provided by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (KAKENHI, No. 26248049). Y. K. thanks the Japan Society for the Promotion of Sciences for financial support in the form of a Postdoctoral Fellowship for Research Abroad.
Notes and references
- (a) C. L. Cooper, P. L. Dubin, A. B. Kayitmazer and S. Turksen, Curr. Opin. Colloid Interface Sci., 2005, 10, 52 CrossRef CAS; (b) R. Ghosh, J. Chromatogr. A, 2002, 952, 13 CrossRef CAS PubMed.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113 CrossRef CAS PubMed.
- (a) R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289 CrossRef CAS; (b) B. Krajewska, Enzyme Microb. Technol., 2004, 35, 126 CrossRef CAS.
- (a) A. Kikuchi and T. Okano, Prog. Polym. Sci., 2002, 27, 1165 CrossRef CAS; (b) D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655 CrossRef CAS PubMed; (c) T. Okano, N. Yamada, M. Okuhara, H. Sakai and Y. Sakurai, Biomaterials, 1995, 16, 297 CrossRef CAS PubMed.
- (a) Y. Kohno and H. Ohno, Chem. Commun., 2012, 48, 7119 RSC; (b) Y. Kohno, S. Saita, Y. Men, J. Yuan and H. Ohno, Polym. Chem., 2015, 6, 2163 RSC.
- (a) Y. Kohno, H. Arai, S. Saita and H. Ohno, Aust. J. Chem., 2011, 64, 1560 CrossRef CAS; (b) Y. Kohno and H. Ohno, Phys. Chem. Chem. Phys., 2012, 14, 5063 RSC.
- (a) Y. Kohno and H. Ohno, Aust. J. Chem., 2012, 65, 91 CrossRef CAS; (b) Y. Kohno, Y. Deguchi and H. Ohno, Chem. Commun., 2012, 48, 11883 RSC.
- (a) Y. Cai, R. Wang, W. B. Krantz, A. G. Fane and X. M. Hu, RSC Adv., 2015, 5, 97143 RSC; (b) X. Fan, H. Liu, Y. Gao, Z. Zou, V. S. J. Craig, G. Zhang and G. Liu, Adv. Mater. DOI:10.1002/adma.201600205.
- (a) Y. Kohno, S. Saita, K. Murata, N. Nakamura and H. Ohno, Polym. Chem., 2011, 2, 862 RSC; (b) Y. Kohno, N. Nakamura and H. Ohno, Aust. J. Chem., 2012, 65, 1548 CrossRef CAS; (c) H. Passos, A. Luís, J. A. P. Coutinho and M. G. Freire, Sci. Rep., 2016, 6, 20276 CrossRef CAS PubMed.
- H. Ohno, K. Fujita and Y. Kohno, Phys. Chem. Chem. Phys., 2015, 17, 14454 RSC.
- Y. Deguchi, Y. Kohno and H. Ohno, Chem. Commun., 2015, 51, 9287 RSC.
- (a) Y. Men, X.-H. Li, M. Antonietti and J. Yuan, Polym. Chem., 2012, 3, 871 RSC; (b) Y. Men, H. Schlaad, A. Voelkel and J. Yuan, Polym. Chem., 2014, 5, 3719 RSC; (c) A. Okafuji, Y. Kohno and H. Ohno, Macromol. Rapid Commun. DOI:10.1002/marc.201500752.
- A. W. Perriman, A. P. S. Brogan, H. Cölfen, N. Tsoureas, G. R. Owen and S. Mann, Nat. Chem., 2010, 2, 622 CrossRef CAS PubMed.
- (a) K. Fujita, D. R. MacFarlane and M. Forsyth, Chem. Commun., 2005, 4804 RSC; (b) K. Fujita, D. R. MacFarlane, M. Forsyth, M. Yoshizawa-Fujita, K. Murata, N. Nakamura and H. Ohno, Biomacromolecules, 2007, 8, 2080 CrossRef CAS PubMed; (c) N. Byrne, L.-M. Wang, J.-P. Belieres and C. A. Angell, Chem. Commun., 2007, 2714 RSC.
- (a) L. E. Bromberg and E. S. Ron, Adv. Drug Delivery Rev., 1998, 31, 197 CrossRef CAS; (b) K. Zhang and X. Y. Wu, Biomaterials, 2004, 25, 5281 CrossRef CAS PubMed; (c) J.-Y. Wu, S.-Q. Liu, P. W.-S. Heng and Y.-Y. Yang, J. Controlled Release, 2005, 102, 361 CrossRef CAS PubMed.
- Z. M. O. Rzaev, S. Dinçer and E. Pişkin, Prog. Polym. Sci., 2007, 32, 534 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedure to fabricate and characterise PILMs. See DOI: 10.1039/c6cc02703b |
| This journal is © The Royal Society of Chemistry 2016 |