
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe drastic effect of cobalt and chromium catalysts in the borylation of arylzinc reagents†
Kimihiro
Komeyama
*,
Shinnosuke
Kiguchi
and
Ken
Takaki
Department of Applied Chemistry, Graduate School of Engineering, Hiroshima University, Higashi-Hiroshima 739-8527, Japan. E-mail: kkome@hiroshima-u.ac.jp; Fax: +81-82-424-5494; Tel: +81-82-424-7747
First published on 28th April 2016
Abstract
A new synthetic approach to arylboronic esters from arylzinc reagents with boryl electrophiles MeOB(OR)2 has been developed. Furthermore, this protocol could be applied to the cyclization/borylation of alkynylaryl iodides to afford cyclized vinylboronic esters.
Arylboronic esters are recognized as being indispensable building blocks in organic synthesis because of their stability and wide range applicability to C–X (X = C, N and O) bond-forming reactions in chemical, medicinal and materials science.1 Over the past 20 years, transition-metal-catalysed borylations of aryl (pseudo)halides with diborons, hydroboranes or metal–boryl reagents have been developed using Pd, Rh, Ni, Cu, Zn, Fe and Co complexes.2 These methods facilitate the concise synthesis of arylboronic esters bearing useful functional groups (route a, Scheme 1). More recently, the synthetic strategies used to produce arylboronic esters have shifted to favour the direct C–H borylation of arenes, which has been rapidly evolved by precious metal catalysts such as Rh and Ir (route b).3,4 Moreover, very recently, the direct borylations have been also accomplished by using abundant and inexpensive transition-metal catalysts.5 In contrast, transition-metal-free borylation protocols, such as electrophilic borylations of electron-rich arenes (route c),6 the Sandmeyer-type borylation (route d),7 and boryl substitution reactions with borylsilanes8 or borylzincates9 (route e), have been frequently reported, although there are still problems with these strategies, such as their narrow substrate scope, loss of the boron component and/or the scarcity of suitable boron sources.
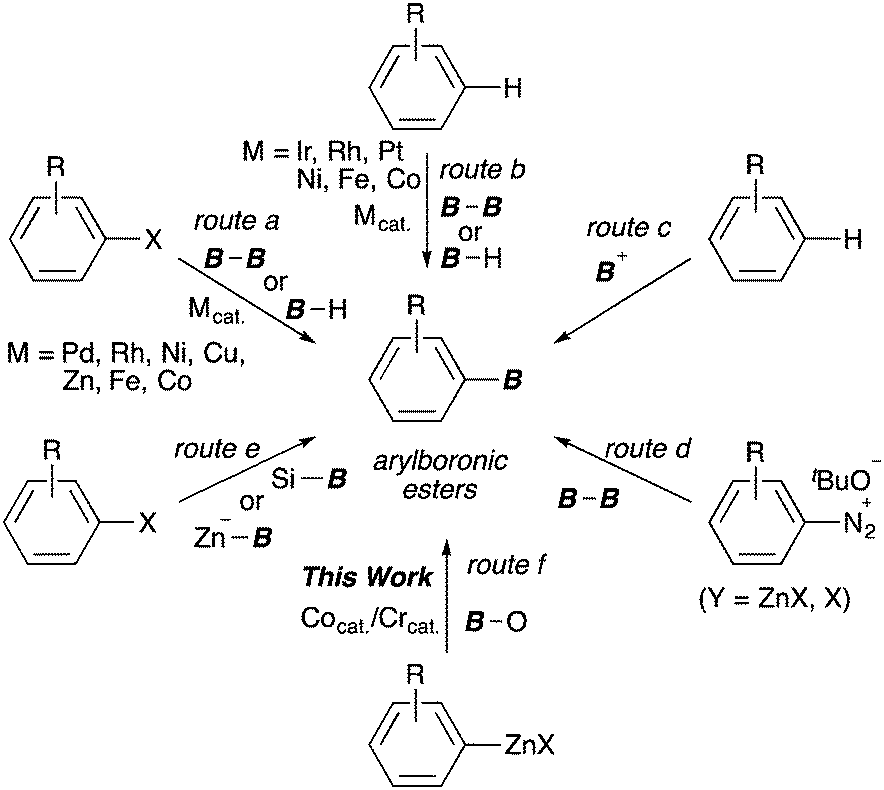 | ||
| Scheme 1 Synthetic methods for arylboronic esters. | ||
A classical but powerful method for the synthesis of arylboronic esters is the substitution of aryllithium or arylmagnesium reagents with trialkylboric esters.10 This method, however, suffers from poor functional group tolerance. Conversely, compared with the above organometallics, organozinc compounds are highly compatible with a broad range of polar functional groups due to the relatively weak ionic character of the C–Zn bond, which may undergo chemoselective transformation.11 However, the nucleophilicity of organozinc reagents is quite low; therefore, their reactions with organic electrophiles often require the use of transition-metal catalysts.12 Despite their high potential for the development of tractable organic transformations, the borylation of organozinc reagents using easy-to-handle boric esters has not been reported,13 except for highly electrophilic B-chlorocatecholborane and subporphyrins.14
Transmetalation is a highly effective route to drastically changing the reactivity of an organometallic species. For example, Takagi reported that highly nucleophilic arylchromium intermediates,15 afforded by the reaction of arylzinc reagents with chromium(III) salts, underwent addition to aldehydes under mild conditions. A similar strategy of changing the nucleophilicity of organometallics via transmetalation has been demonstrated in the Nozaki–Hiyama–Kishi reaction.16 Here we report the marked additive effect of low-valence cobalt and chromium catalysts in the borylation of arylzinc reagents with MeOB(OR)2 as the boryl electrophile in the presence of TMSCl (route f). Furthermore, this protocol could be applied to the direct synthesis of arylboronic esters from ubiquitous aryl halides under CoBr2, xantphos and CrCl3(thf)3 catalyst systems in the presence of Zn.
We began by examining the borylation of the preformed 4-MeC6H4ZnI·LiCl17 with 2-methoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2a, MeOBpin) as model substrates in the presence of TMSCl (1.2 equiv.) at 60 °C for 16 h. These results are summarized in Table 1. As expected, the arylzinc compound was inactive and hardly participated in the borylation, wherein the arylzinc remained unchanged after the reaction (Table 1, entry 1).18 The addition of the CrCl3(thf)3 or CrCl2 catalyst (20 mol%) did not afford the arylboronic ester (entries 2 and 3). Conversely, we observed an unexpected effect arising from the combination of cobalt and chromium catalysts under reduction conditions (entries 4 and 5). Thus, a mixture of CoBr2/xantphos19 and CrCl3(thf)3 complexes in the presence of zinc powder (0.5–2.0 equiv.) effectively promoted the borylation, giving rise to 4-tolylboronic pinacol ester (3aa) in satisfactory yields (67–73%). Intriguingly, all the components, i.e. the cobalt, chromium and zinc reductants, are crucial for efficient borylation (entries 6–8). Finally, we found that low-valence cobalt and chromium complexes play important roles in the borylation reaction (entries 9 and 10). The role of the xantphos ligand is unclear,20 and it is not absolutely necessary to the borylation. There is no reaction in the absence of the ligand (entry 11); however, the presence of dppe afforded 3aa in 53% yield (entry 12).
|
|
|||
|---|---|---|---|
| Entry | Additives (mol%) | Zn (x equiv.) | Yieldb (%) |
| a All reactions were carried out in the presence of TMSCl (1.2 equiv.). b NMR yield. c The borylation was performed after reduction with 20 mol% zinc powder to form the low-valence cobalt complex. | |||
| 1 | — | — | Trace |
| 2 | CrCl3(thf)3 (20) | — | Trace |
| 3 | CrCl2 (20) | — | Trace |
| 4 | CoBr2/xantphos (10), CrCl3(thf)3 (20) | 2.0 | 73 |
| 5 | CoBr2/xantphos (10), CrCl3(thf)3 (20) | 0.5 | 67 |
| 6 | CoBr2/xantphos (10), CrCl3(thf)3 (20) | — | Trace |
| 7 | CrCl3(thf)3 (20) | 2.0 | Trace |
| 8 | CoBr2/xantphos (10) | 2.0 | 32 |
| 9 | CoBr/xantphos (10),c CrCl3(thf)3 (20) | — | 13 |
| 10 | CoBr/xantphos (10),c CrCl2 (20) | — | 65 |
| 11 | CoBr (10)c | — | Trace |
| 12 | CoBr/dppe (10),c CrCl2 (20) | — | 53 |
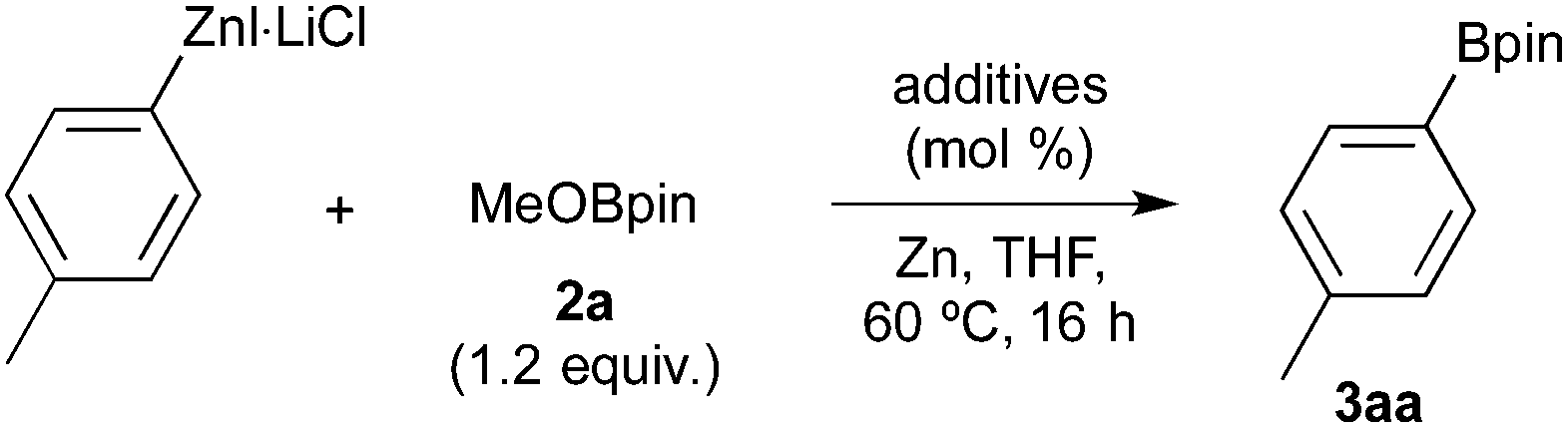
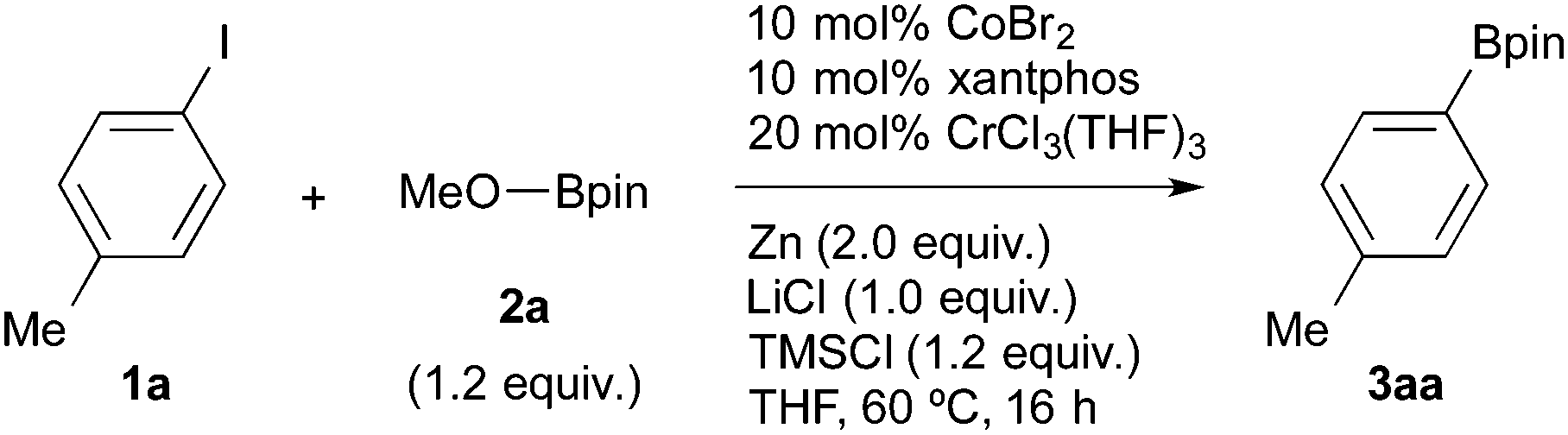
The cobalt/xantphos complex has been reported to be an equally effective catalyst for the transformation of aryl halides into arylzinc reagents under similar reduction conditions.21 Based on these reports and our previous work on cobalt-catalysed reactions,22 the present Co/Cr-catalysed borylation with arylzinc reagents was refined into a more simplified protocol starting from 4-tolyl iodide (Table 2, entry 1). Without TMSCl, the desired boronic ester 3aa was obtained in low yield (27%), even with a longer reaction time (entry 2). This result indicates robust Cr–O bond formation during the reaction, because the bond appears to be difficult to cleave in the catalytic cycle without the aid of TMSCl.23 Chromium complexes ligated by nitrogen ligands,24 such as 2,2′-bipyridyl (bpy) and 1,10-phenanthroline (1,10-phen), inhibit the borylation in THF solvent (entries 6 and 7). However, the borylation proceeded in acetonitrile with the replacement of xantphos with 1.10-phen, albeit with a slightly lower yield (entry 5).25
|
|
||
|---|---|---|
| Entry | Change from standard conditions | Yield/% of 3aaa |
| a NMR yield. The parenthesis value indicates an isolated yield. b Reaction time: 48 h. c MeCN solvent was used instead of THF. | ||
| 1 | — | 79 (75) |
| 2b | Without TMSCl | 27 |
| 3 | CrCl3(bpy) in place of CrCl3(THF)3 | 12 |
| 4 | CrCl3(1,10-phen) in place of CrCl3(THF)3 | 4 |
| 5c | 1,10-Phen in place of xantphos | 60 |
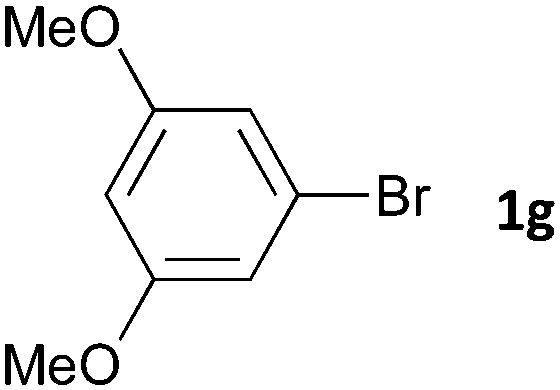
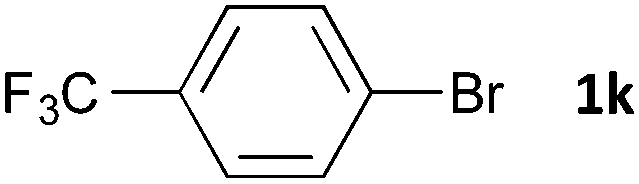
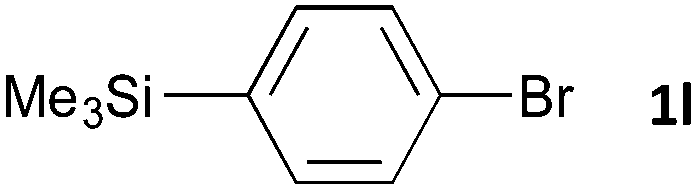
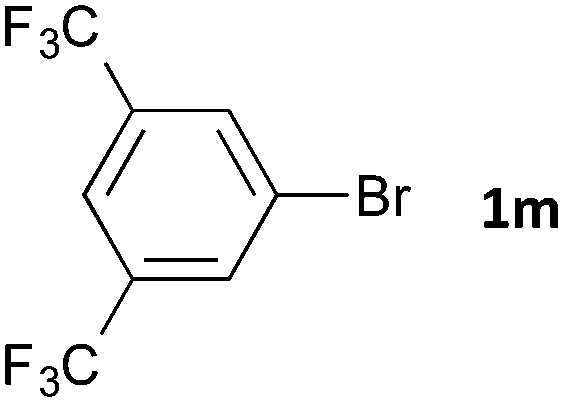
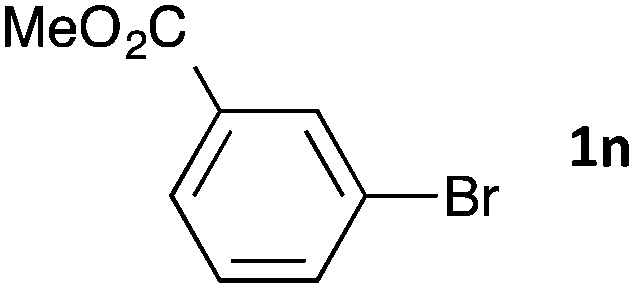
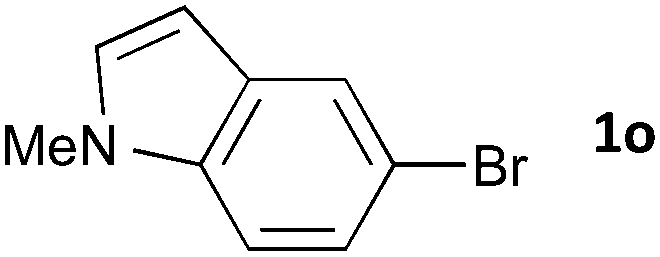
Having established the optimum conditions (Table 2, entry 1), we explored the substrate scope through the Co/Cr-catalysed borylation of various aryl bromides (Table 3). Electron-neutral (1b and 1c) and -rich aryl bromides (1d, 1e and 1f) are efficiently converted to the corresponding arylboronic acid pinacol esters 3 in 66–71% yields (entries 1–5). However, electron-deficient aryl halides with inductively and/or resonance withdrawing substituents were less reactive (entries 6, 8, 10, 12, 14 and 17). In particular, the strongly electron-deficient aryl bromides bearing CN (1j) and CF3 (1k, 1m) substituents were markedly less reactive towards trapping with 2a (entries 12, 14 and 17). Fortunately, a significant improvement was achieved by the replacement of 2a with 2-methoxy-5,5-dimethyl-1,3,2-dioxaborinane (MeOBnep, 2b), which is a less sterically hindered electrophile compared to 2a.26 This boronate preferentially afforded the desired boronic esters 3jb, 3kb and 3mb in good yields (entries 13, 15 and 18). We also explored the effect of the leaving group, such as the 2-methoxyethoxy substituent, on the boron 2, but such substituents did not significantly affect the reactivity.27 In the Co/Cr-catalysed borylation, trimethylsilyl aryls (1l, entry 16) as well as heteroaryl skeletons such as indole (1o, entry 21) and thiophene (1p, entry 22) also took part in the borylation. In contrast, the use of aryl chlorides leads to lower product yields.
|
|
||||
|---|---|---|---|---|
| Entry | Ar–Br 1 | Time/h | Product & yielda/% | |
| a Isolated yield. b 2 equivalents of the boryl electrophile were employed. c MeOB(nep) was used instead of MeOB(pin). | ||||
| 1 |
|
16 | 3ba | 71 |
| 2 |
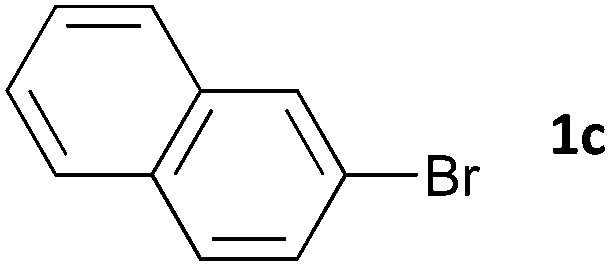
|
16 | 3ca | 66 |
| 3 |
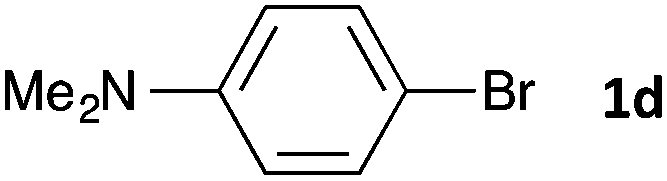
|
16 | 3da | 66 |
| 4 |
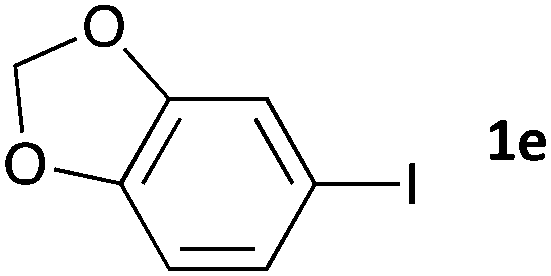
|
16 | 3ea | 67 |
| 5b |
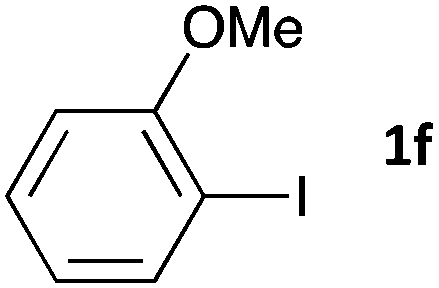
|
48 | 3fa | 68 |
| 6 |
|
16 | 3gb | 38 |
| 7c | 16 | 3ga | 66 | |
| 8 |
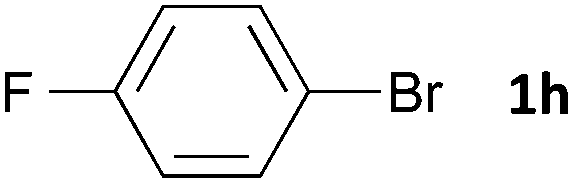
|
16 | 3ha | 62 |
| 9c | 16 | 3hb | 75 | |
| 10 |
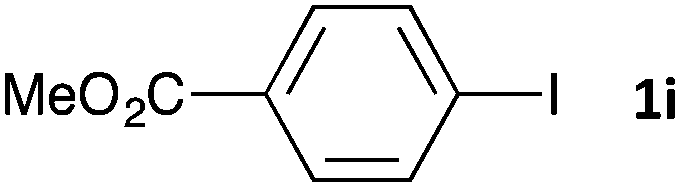
|
48 | 3ia | 68 |
| 11c | 48 | 3ib | 74 | |
| 12b |
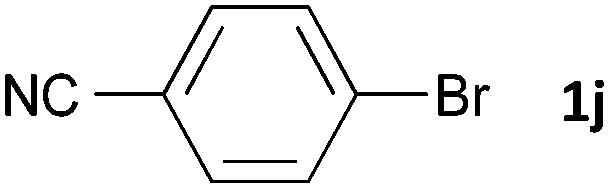
|
48 | 3ja | 38 |
| 13b,c | 48 | 3jb | 83 | |
| 14b |
|
48 | 3ka | 23 |
| 15b,c | 48 | 3kb | 83 | |
| 16c |
|
24 | 3lb | 92 |
| 17 | 48 | 3ma | Trace | |
| 18b,c |
|
48 | 3mb | 84 |
| 19b | 48 | 3na | 65 | |
| 20b,c |
|
48 | 3nb | 66 |
| 21 |
|
48 | 3oa | 72 |
| 22c |
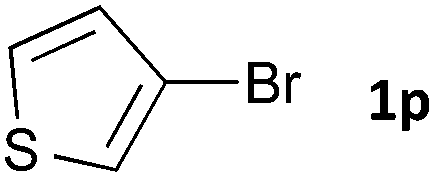
|
48 | 3pb | 80 |
The borylation reaction presumably involves the generation of aryl cobalt and aryl chromium intermediates. In contrast, the migratory insertion of an alkyne into both aryl–metal complexes has been established.25b,28 Based on these findings and our previous results,22 we assumed that if these aryl–metal complexes are active in alkyne-insertion reactions to form a vinyl-metal species, and the products undergo a substitution reaction with boryl electrophiles under identical conditions to those presented above, a useful three-component reaction (carboboration) could be developed. The carboboration of alkynes is a very important process for the regio- and stereoselective synthesis of multisubstituted olefins.29 Similar catalytic reactions for alkynes13,30 have been reported; however, all examples employed carbon electrophiles. The transformation using boryl electrophiles has never been demonstrated, except in cases employing Grignard reagents.31 Our initial attempts at the addition/borylation of 4-octyne with 4-tolyl bromide 1a and MeOBpin 2a under identical conditions failed, wherein the major product is arylboronic ester 3aa. This may be because the reaction of the in situ generated aryls-metal species with the boryl electrophile is fast compared to the intermolecular reaction with the alkyne. Based on these experimental results, we next attempted the cyclisation/borylation of alkynyl aryl iodides 4 (Scheme 2). Having refined the conditions,32 the desired cyclisation/borylation was accomplished, affording the cyclised vinylboronic esters 5a, 5b and 5c in 50, 41 and 43% yields, respectively (Scheme 2). The stereochemistry of the products was determined by NOE measurements, which clearly indicated syn-carboboration. In addition, an alkynyl aryl iodide tethered with nitrogen also reacted to afford the corresponding boronic ester 5d in 26% yield.
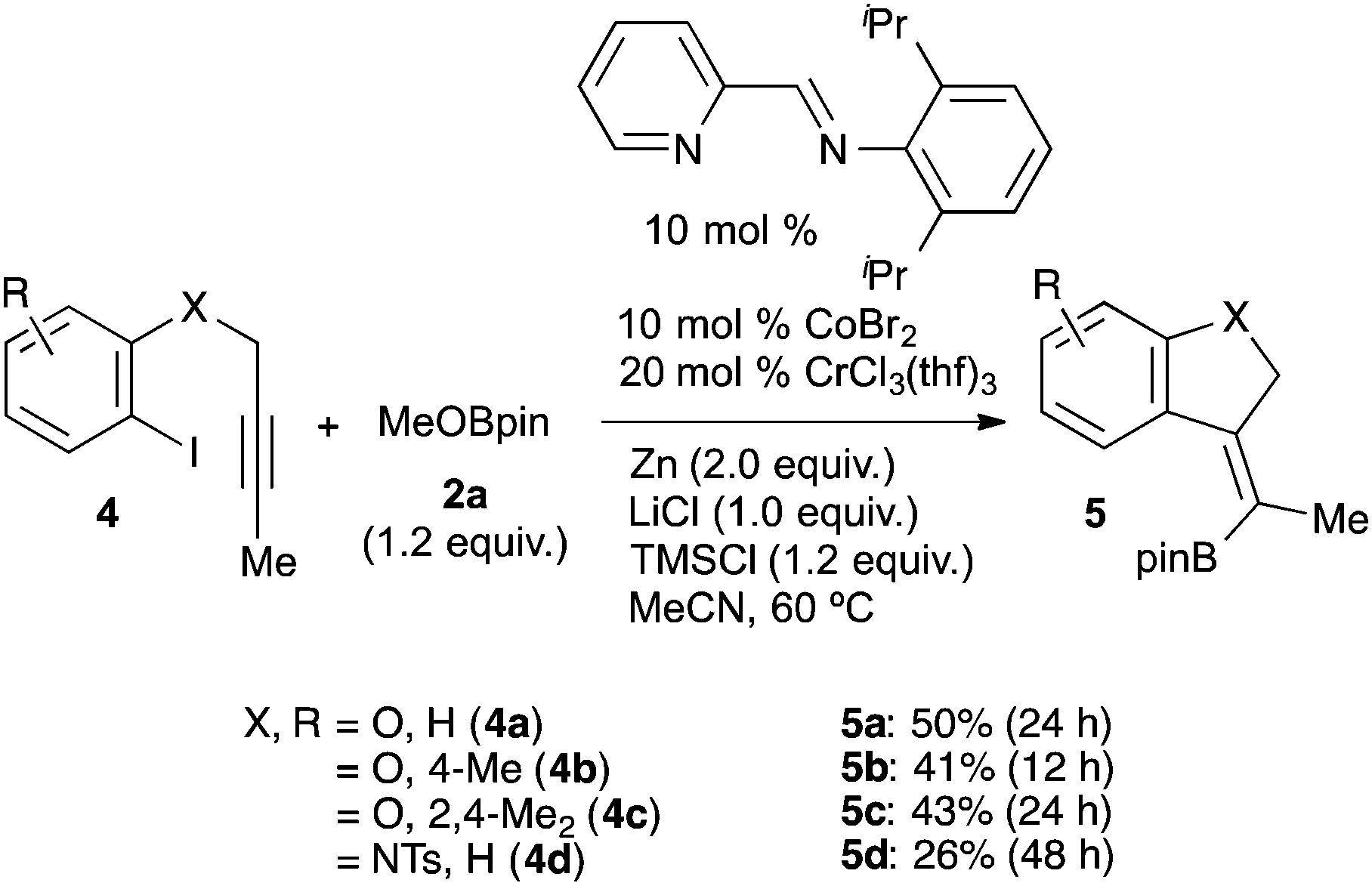 | ||
| Scheme 2 Substrate scope for the Co/Cr-catalysed cyclisation/borylation. | ||
Although the actual role of the cobalt catalyst is still unclear, the arylchromium(II) species seems to trigger the borylation step. Thus, the reaction of 4-MeC6H4CrCl2 (prepared via the reaction of the 4-MeC6H4Li with CrCl3(thf)3)33 with MeOBpin afforded 3aa in only 3% GC yield.34 In contrast, the reaction of the 4-MeC6H4CrCl provided 3aa in 67% yield.
In conclusion, we have reported a drastic additive effect for increasing the reactivity of arylzinc reagents, enabling the borylation of unreactive arylzinc compounds to afford various arylboronic esters. In addition, we found that this protocol could be applied to a more practical route to borylation starting from ubiquitous aryl halides. The easy-to-operate procedure avoids the preparation of air- and moisture-sensitive arylzinc reagents, making it more practical for arylboronic ester synthesis. Furthermore, cyclisation/borylation of arylalkynyl iodides was accomplished by the modified catalytic system to give cyclized vinylboronic esters, in which carboboration proceeded in an exclusively syn-addition manner. Control experiments revealed the important roles of low-valence cobalt and chromium in the borylation. This suggests that a key intermediate is an aryl chromium(II) species, although more data are required in order to understand the mechanistic details of the reaction. Further studies on the mechanism and synthetic applications of the Co/Cr catalyst system are currently underway in our laboratory.
This work was partially supported by a Grant-Aid for Scientific Research (KAKENHI) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (No. 15K05502).
Notes and references
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine, Wiley-VCH, Weinheim, 2005 Search PubMed.
- Recent reviews for the transition metal-catalyzed borylation of aryl (pseudo)halides: (a) W. K. Chow, O. Y. Yuen, P. Y. Choy, C. M. So, C. P. Lau, W. T. Wong and F. Y. Kwong, RSC Adv., 2013, 3, 12518 RSC; (b) M. Murata, Heterocycles, 2012, 85, 1795 CrossRef CAS . With Pd: ; (c) J. Campos and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 14159 CrossRef CAS PubMed . With Rh: ; (d) M. Tobisu, H. Kinuta, Y. Kita, E. Rémond and N. Chatani, J. Am. Chem. Soc., 2011, 134, 115 CrossRef PubMed; (e) H. Kinuta, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2015, 137, 1593 CrossRef CAS PubMed . With Ni: ; (f) M. Tobisu, K. Nakamura and N. Chatani, J. Am. Chem. Soc., 2014, 136, 5587 CrossRef CAS PubMed; (g) X.-W. Liu, J. Echavarren, C. Zarate and R. Martin, J. Am. Chem. Soc., 2015, 137, 12470 CrossRef CAS PubMed; (h) T. Niwa, H. Ochiai, Y. Watanabe and T. Hosoya, J. Am. Chem. Soc., 2015, 137, 14313 CrossRef CAS PubMed; (i) J. Zhou, M. W. Kuntze-Fechner, R. Bertermann, U. S. D. Paul, J. H. J. Berthel, A. Friedrich, Z. Du, T. B. Marder and U. Radius, J. Am. Chem. Soc., 2016, 138, 5250 CrossRef CAS PubMed . With Cu: C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350 Search PubMed . With Zn: ; (j) S. Bose and T. B. Marder, Org. Lett., 2014, 16, 4562 CrossRef CAS PubMed; (k) S. K. Bose, A. Deißenberger, A. Eichhorn, P. G. Steel, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2015, 54, 11843 CrossRef CAS PubMed . With Fe: ; (l) R. B. Bedford, P. B. Brenner, E. Carter, T. Gallagher, D. M. Murphy and D. R. Pye, Organometallics, 2014, 33, 5940 CrossRef CAS; (m) F. Labre, Y. Gimbert, P. Bannwarth, S. Olivero, E. Duñach and P. Y. Chavant, Org. Lett., 2014, 16, 2366 CrossRef CAS PubMed . With Co: ; (n) W. Yao, H. Fang, S. Peng, H. Wen, L. Zhang, A. Hu and Z. Huang, Organometallics, 2016 DOI:10.1021/acs.organomet.6b00161.
- Review for the Rh or Ir catalysed aromatic C–H borylation: (a) T. Ishiyama and N. Miyaura, J. Organomet. Chem., 2003, 680, 3 CrossRef CAS; (b) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (c) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC; (d) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229 RSC.
- (a) S. M. Preshlock, D. L. Plattner, P. E. Maligres, S. W. Krska, R. E. Maleczka and M. R. Smith, Angew. Chem., Int. Ed., 2013, 52, 12915 CrossRef CAS PubMed; (b) S. Kawamorita, T. Miyazaki, H. Ohmiya, T. Iwai and M. Sawamura, J. Am. Chem. Soc., 2011, 133, 19310 CrossRef CAS PubMed; (c) Y. Saito, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 5193 CrossRef CAS PubMed; (d) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712 CrossRef CAS PubMed; (e) With Pt: T. Furukawa, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2015, 137, 12211 CrossRef CAS PubMed.
- With Ni: (a) T. Furukawa, M. Tobisu and N. Chatani, Chem. Commun., 2015, 51, 6508 RSC; (b) H. Zhang, S. Hagihara and K. Itami, Chem. Lett., 2015, 44, 779 CrossRef CAS . With Fe: ; (c) T. Hatanaka, Y. Ohki and K. Tatsumi, Chem. – Asian J., 2010, 5, 1657 CrossRef CAS PubMed; (d) G. Yan, Y. Jiang, C. Kuang, S. Wang, H. Liu, Y. Zhangb and J. Wang, Chem. Commun., 2010, 46, 3170 RSC; (e) T. Dombray, C. G. Werncke, S. Jiang, M. Grellier, L. Vendier, S. Bontemps, J.-B. Sortais, S. Sabo-Etienne and C. Darcel, J. Am. Chem. Soc., 2015, 137, 4062 CrossRef CAS PubMed . With Fe/Cu: ; (f) T. J. Mazzacano and N. P. Mankad, J. Am. Chem. Soc., 2013, 135, 17258 CrossRef CAS PubMed . With Co: ; (g) J. V. Obligacion, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 4133 CrossRef CAS PubMed.
- A. Del Grosso, M. D Helm, S. A. Solomon, D. Caras-Quintero and M. J. Ingleson, Chem. Commun., 2011, 47, 12459 RSC.
- F. Mo, Y. Jiang, D. Qiu, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 1846 CrossRef CAS PubMed.
- E. Yamamoto, K. Izumi, Y. Horita and H. Ito, J. Am. Chem. Soc., 2012, 134, 19997 CrossRef CAS PubMed.
- Y. Nagashima, R. Takita, K. Yoshida, K. Hirano and M. Uchiyama, J. Am. Chem. Soc., 2013, 135, 18730 CrossRef CAS PubMed.
- (a) H. C. Brown and T. E. Cole, Organometallics, 1983, 2, 1316 CrossRef CAS; (b) J. W. Clary, T. J. Rettenmaier, R. Snelling, W. Bryks, J. Banwell, W. T. Wipke and B. Singaram, J. Org. Chem., 2011, 76, 9602 CrossRef CAS PubMed.
- P. Knochel and R. D. Singer, Chem. Rev., 1993, 93, 2117 CrossRef CAS.
- P. Knochel, M. A. Schade, S. Bernhardt, G. Manolikakes, A. Metzger, F. M. Piller, C. J. Rohbogner and M. Mosrin, Beilstein J. Org. Chem., 2011, 7, 1261 CrossRef CAS PubMed.
- N. Nakagawa, T. Hatakeyama and M. Nakamura, Chem. – Eur. J., 2015, 21, 4257 CrossRef CAS PubMed.
- (a) S. Claudel, C. Gosmini, J. M. Paris and J. Périchon, Chem. Commun., 2007, 3667 RSC; (b) R. Kotani, K. Yoshida, E. Tsurumaki and A. Osuka, Chem. – Eur. J., 2016, 22, 3320 CrossRef CAS PubMed.
- (a) Y. Ogawa, M. Mori, A. Saiga and K. Takagi, Chem. Lett., 1996, 1069 CrossRef CAS; (b) Y. Ogawa, A. Saiga, M. Mori, T. Shibata and K. Takagi, J. Org. Chem., 2000, 65, 1031 CrossRef CAS PubMed.
- (a) K. Takai, K. Nitta and O. Fujimura, J. Org. Chem., 1989, 54, 4732 CrossRef CAS; (b) K. Takai, N. Matsukawa, A. Takahashi and T. Fujii, Angew. Chem., Int. Ed., 1998, 37, 152 CrossRef CAS.
- A. Krasovskiy, V. Malakhov, A. Gavryushin and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 6040 CrossRef CAS PubMed.
- After the reaction, treatment of the reaction mixture with I2 afforded 4-iodotoluene in nearly quantitative yield.
- M. M. P. Grutters, C. Müller and D. Vogt, J. Am. Chem. Soc., 2006, 128, 7414 CrossRef CAS PubMed.
- M.-N. Birkholz née Gensow, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099 RSC.
- M.-Y. Jin and N. Yoshikai, J. Org. Chem., 2011, 76, 1972 CrossRef CAS PubMed.
- (a) K. Komeama, T. Kashihara and K. Takaki, Tetrahedron Lett., 2013, 54, 5659 CrossRef; (b) K. Komeyama, Y. Okamoto and K. Takaki, Angew. Chem., Int. Ed., 2014, 53, 11325 CrossRef CAS PubMed; (c) R. Asakura, H. Fukuoka and K. Takaki, Tetrahedron Lett., 2015, 56, 1735 CrossRef.
- F. Alois and N. Shi, J. Am. Chem. Soc., 1996, 118, 12349 CrossRef.
- K. Namba and Y. Kishi, J. Am. Chem. Soc., 2005, 127, 15382 CrossRef CAS PubMed.
- (a) H. Fillon, C. Gosmini and J. Périchon, J. Am. Chem. Soc., 2003, 125, 3867 CrossRef CAS PubMed; (b) M. Corpet and C. Gosmini, Chem. Commun., 2012, 48, 11561 RSC.
- See ref. 13 and references therein.
- See Table S1 in the ESI†.
- With Co catalyst: (a) K. Murakami, H. Yorimitsu and K. Oshima, Org. Lett., 2009, 11, 2373 CrossRef CAS PubMed; (b) B.-H. Tan, J. Dong and N. Yoshikai, Angew. Chem., Int. Ed., 2012, 51, 9610 CrossRef CAS PubMed . With Cr catalyst: ; (c) K.-I. Kanno, Y. Liu, A. Iesato, K. Nakajima and T. Takahashi, Org. Lett., 2005, 7, 5453 CrossRef CAS PubMed.
- A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698 CrossRef CAS PubMed.
- With Pd catalyst: (a) M. Suginome, A. Yamamoto and M. Murakami, J. Am. Chem. Soc., 2003, 125, 6358 CrossRef CAS PubMed; (b) M. Daini and M. Suginome, Chem. Commun., 2008, 5224 RSC; (c) M. Daini, A. Yamamoto and M. Suginome, J. Am. Chem. Soc., 2008, 130, 2918 CrossRef CAS PubMed . With Ni catalyst: ; (d) A. Yamamoto and M. Suginome, J. Am. Chem. Soc., 2005, 127, 15706 CrossRef CAS PubMed; (e) M. Suginome, M. Shirakura and A. Yamamoto, J. Am. Chem. Soc., 2006, 128, 14438 CrossRef CAS PubMed . With Cu catalyst: ; (f) R. Alfaro, A. Parra, J. Aleman, J. L. G. Ruano and M. Tortosa, J. Am. Chem. Soc., 2012, 134, 15165 CrossRef CAS PubMed; (g) L. Zhang, J. Cheng, B. Carry and Z. Hou, J. Am. Chem. Soc., 2012, 134, 14314 CrossRef CAS PubMed; (h) H. Yoshida, I. Kageyuki and K. Takaki, Org. Lett., 2013, 15, 952 CrossRef CAS PubMed; (i) K. Kubota, H. Iwamoto, E. Yamamoto and H. Ito, Org. Lett., 2015, 17, 620 CrossRef CAS PubMed.
- G.-H. Fang, Z.-J. Yan and M.-Z. Deng, Synthesis, 2006, 1148 CAS.
- See Table S2 in the ESI†.
- J. J. Daly, R. P. A. Sneeden and H. H. Zeiss, J. Am. Chem. Soc., 1966, 88, 4287 CrossRef CAS.
- See Scheme S1 in the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental results, experimental procedures and characterisation of the products. See DOI: 10.1039/c6cc03086f |
| This journal is © The Royal Society of Chemistry 2016 |