
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of a stable radical by oxidation of a tetraorganoborate†
Holger
Braunschweig
*a,
Ivo
Krummenacher
a,
Lisa
Mailänder
a,
Leanne
Pentecost
b and
Alfredo
Vargas
b
aInstitut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bDepartment of Chemistry, School of Life Sciences, University of Sussex, Brighton BN1 9QJ, Sussex, UK
First published on 9th May 2016
Abstract
Herein, we describe the selective formation of a stable neutral spiroborate radical by one-electron oxidation of the corresponding tetraorganoborate salt Li[B(C4Ph4)2], formally containing a tetrahedral borate centre and a s-cis-butadiene radical cation as the spin-bearing site. Spectroscopic and computational methods have been used to determine the spin distribution and the chromism observed in the solid state.
The oxidation of tetraaryl, -alkenyl or -alkyl borates with the general formula [BR4]− usually leads to the formation of C–C coupled products by skeletal rearrangements.1,2 Oxidation thus represents a versatile method for the synthesis of disubstituted alkynes or biaryl compounds.3 As depicted in Scheme 1, such reactions are also a feature of spiroborates, which in the case of a spirocyclic borafluorene derivative (1) afford a bis(biaryl) compound.4,5 In general, these reactions can be initiated by a variety of oxidising agents or reaction conditions, such as photo- or electrochemically.6 In a few instances, [BR4]˙ radicals have been suggested as intermediates in the oxidative ligand coupling process, but never directly observed.7 Herein, we show with a spiroborate (2) that the oxidation can be directed to the exclusive formation of such a boranyl radical species.
 | ||
| Scheme 1 Two examples of oxidation reactions of organoborate salts resulting in C–C coupled products. | ||
Spiroborate 2 was synthesised as previously reported by our group by salt elimination of two equivalents of 1,2-dilithio-1,2,3,4-tetraphenylbutadiene with BF3·Et2O.8 Whereas the redox chemistry of boroles, five-membered boron-containing, antiaromatic and highly Lewis acidic heterocycles,9 has been explored extensively,10 the redox behaviour of the related spiroborate salt 2 had not been investigated so far. As judged by cyclic voltammetry in tetrahydrofuran solution, compound 2 undergoes an irreversible oxidation at −0.35 V vs. the [FeCp2]/[FeCp2]+ couple (see ESI†) in the potential window. We wondered if the oxidation step could be realised on a preparative scale and if the formed product could be isolated. To this end, a stoichiometric amount of the one-electron oxidation agent ferrocenium hexafluorophosphate was added to a solution of 2 in benzene at rt (Scheme 2). Addition of the Fe(III) salt led to an immediate colour change from yellow to dark green and the precipitation of a colourless solid. Upon completion, the 11B NMR resonance of the starting material (2, δB = −1.4 ppm) disappeared without the occurrence of a new signal. Similarly, the 1H NMR spectrum of the crude reaction mixture showed only the formation of ferrocene but no additional signals for the oxidation product. After the separation of Li[PF6] and ferrocene, compound 3 was isolated as green amorphous, or alternatively, red crystalline, solids (80% crystalline yield).
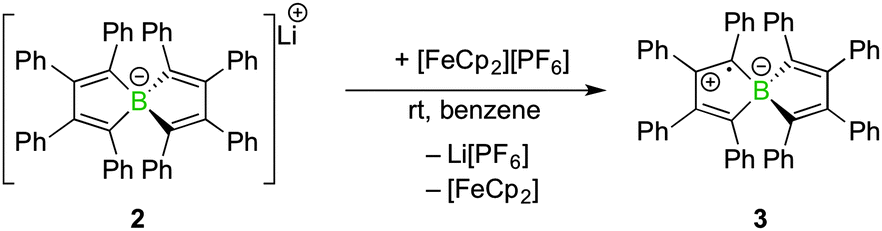 | ||
| Scheme 2 Oxidation of spiroborate salt 2 with [FeCp2][PF6] to radical 3 (only one possible resonance form is drawn). | ||
The absence of NMR signals suggested the radical character of 3, which was supported by EPR spectroscopy (vide infra). X-ray diffraction of red single crystals of 3 confirmed the formation of a charge-neutral species, consistent with a one-electron oxidation of spiroborate 2.8 The D2d symmetry is lifted in the oxidised product as can be seen by the changes of the geometric parameters around the spiro boron atom (cf. Table S1 in the ESI†). For instance, the two essentially planar BC5 rings in 3 deviate significantly from a perpendicular arrangement (79.0(1)°) and are displaced relative to each other from the twofold axis (Fig. 1, right). In addition, the one-electron oxidation leads to a shortening of the B–C distances of about 0.1 Å (3: ØB–C = 1.535 Å vs.2: ØB–C = 1.631 Å) and to a reduction in the bond length alternation of the bicyclic rings. The formal double bonds of the borate anion (C1–C2, C3–C4, C5–C6, and C7–C8) are elongated in the oxidised product 3, consistent with removal of an electron from the HOMO of the anion (2) which shows π-bonding interactions between these carbon atoms.
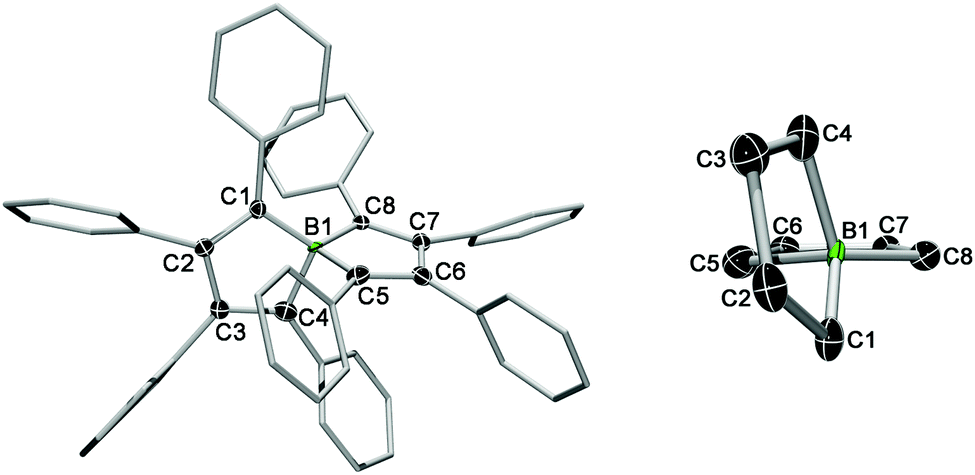 | ||
| Fig. 1 Left: Molecular structure of 3 in the solid state. Hydrogen atoms and the ellipsoids of the phenyl residues are omitted for clarity. The ellipsoids are set to 50% probability. For bond lengths and angles see Table S1 in the ESI.† Right: Side view showing the relative arrangement of the BC4 rings. | ||
The phenyl substituents around the spirocycle are arranged in a propeller-like manner as observed for 2. However, it is important to note that the phenyl ring at C1 is only tilted by 30.2° with respect to the fused ring plane, whereas the other aryl substituents have torsion angles between 43 and 69°. This might be due to effects of molecular packing or to some degree of spin delocalisation into the aryl π system.
The continuous-wave ESR spectrum of 3 in benzene solution shows that the unpaired spin density is delocalised into the aryl substituents (Fig. 2). The best-fit parameters for the second-derivative ESR signal (giso = 2.003) relate to small proton hyperfine couplings (in the range of 0.05–0.15 mT) of four equivalent phenyl groups, indicating simultaneous delocalisation of the unpaired spin density over both π planes. The ESR results thus point to a spiroconjugated organic radical with a structure of higher symmetry (D2d) than observed in the solid state.11
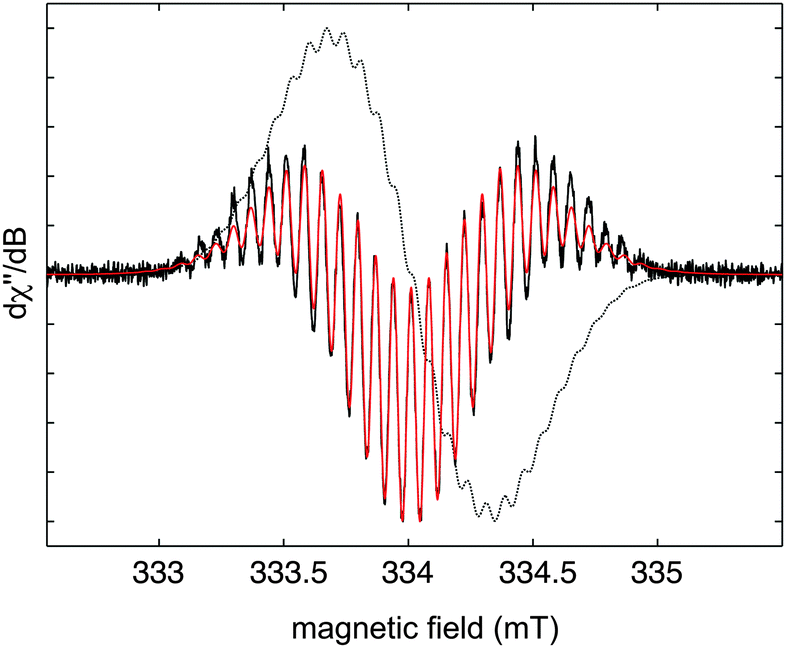 | ||
| Fig. 2 X-band first (grey dotted line) and second (solid black line) derivative ESR spectrum of 3 at room temperature in benzene solution. A simulation of the second derivative spectrum is shown in red. Instrumental parameters: ν = 9.379 GHz, modulation frequency = 100 kHz, modulation amplitude = 0.1 G, microwave power = 0.25 mW, conversion time = 40 ms, 4 scans. | ||
The UV-vis spectrum of 3 differs strongly from that of the borate salt 2, which shows a lowest absorption maximum at 365 nm (ε = 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 490 L mol−1 cm−1).8 In addition to bands in the same wavelength region (λ(ε) = 329 nm (24894 L mol−1 cm−1), λ(ε) = 352 nm (22919 L mol−1 cm−1)), radical 3 exhibits additional transitions at lower energies (λ(ε) = 463 nm (12967 L mol−1 cm−1), λ(ε) = 572 nm (2442 L mol−1 cm−1), λ(ε) = 604 nm (2307 L mol−1 cm−1), λ(ε) = ∼700.0 nm (∼750 L mol−1 cm−1)), which are extended into the near infrared range. The bands between 500–900 nm are significantly weaker in intensity than those between 300–500 nm and likely can be attributed to intervalence charge transfer transitions. The absorptions from 550 nm to the near IR range are consistent with other organic monoradicals.12–14
490 L mol−1 cm−1).8 In addition to bands in the same wavelength region (λ(ε) = 329 nm (24894 L mol−1 cm−1), λ(ε) = 352 nm (22919 L mol−1 cm−1)), radical 3 exhibits additional transitions at lower energies (λ(ε) = 463 nm (12967 L mol−1 cm−1), λ(ε) = 572 nm (2442 L mol−1 cm−1), λ(ε) = 604 nm (2307 L mol−1 cm−1), λ(ε) = ∼700.0 nm (∼750 L mol−1 cm−1)), which are extended into the near infrared range. The bands between 500–900 nm are significantly weaker in intensity than those between 300–500 nm and likely can be attributed to intervalence charge transfer transitions. The absorptions from 550 nm to the near IR range are consistent with other organic monoradicals.12–14
Another interesting aspect of this radical species is that its colour is strongly dependent on its physical state. The radical (3) is dark green in solution and in its amorphous solid state, whereas it is red in the crystalline state. Presumably, the locked conformation in the crystal structure limits the amount of π conjugation in the molecule, thus leading to a shift of the maximum absorption to shorter wavelengths.
Additional computational studies within the Kohn–Sham density functional theory (DFT) were undertaken. These calculations support the experimental results with respect to the generation of the radical species. Calculated structures of 2 and 3 were in reasonable agreement with those obtained experimentally (see ESI† for Computational details).
Energy minimisation calculations on 3 yielded structures in the gas phase that are distinct from the solvated structures. Furthermore, excited state calculations on the resulting geometries revealed different absorption profiles. These results strongly suggest environment-dependent geometries which in turn dictate the observed absorption profile – explaining the experimentally observed differing colours of the species depending on the physical state. Indeed, depending on the structure, e.g. the solvated structure, lower molecular orbitals are involved in the excitation process. Such observations lead to the idea of environment (e.g. solvent)-stabilised charge distribution within the system, which is further supported by computational results as shown by molecular electrostatic potentials (MEP) and natural charges. Further discussions on the theoretical results are given in the ESI.† Moreover, these calculations allowed identification of the experimentally observed transitions: HOMO−2 to LUMO (spin-down) at around 15500 cm−1 (645 nm), and HOMO to LUMO (spin-up) at around 21000 cm−1 (476 nm).
Another geometry-dependent feature of 3 is that of the spin-density distribution, which unsurprisingly is dictated by the structure. Calculations show that the spin population in the radical is essentially localised on one borole moiety in both the gas and solution phase; the dominant spin density is found on the two carbon atoms adjacent to the boron atom. Imposing D2 symmetry distributes the spin density evenly on all four carbon atoms adjacent to the boron atom that extends into the phenyl substituents. Furthermore, in this symmetrical configuration the molecular orbitals (MOs) are delocalised across both borole moieties (see Fig. 3). Both the spin density and MO profiles are well in line with experimental results (vide supra). The total electronic energy of the symmetrised structure is only slightly (0.6 kcal mol−1) higher than that of the non-symmetrised structure. This could be ascribed in part to interaction of the two perpendicular π planes connected by the boron centre, via so-called spiroconjugation.15
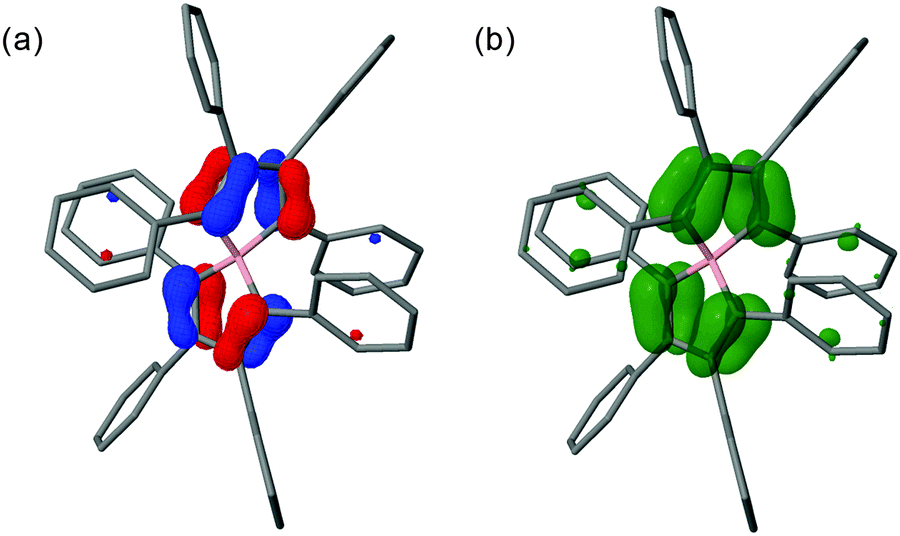 | ||
| Fig. 3 MO62X/6-311G* calculated (a) singly-occupied molecular orbital (SOMO), and (b) spin-density distribution for 3 with imposed D2 symmetry. | ||
The difference in reactivity of 2 compared to its benzannelated analogue 1, the latter undergoing coupling of the organic substituents upon oxidation,4,5 most likely arises from the stoichiometric use of the outer-sphere oxidant ferrocenium, which can accept precisely one electron from the borate. Isolation of the stable spiro radical (3) suggests that a radical mechanism might be operative in the oxidative ligand coupling of other organoborates, as previously proposed.4,5,7 The possible resonance forms of the zwitterionic species 3 all consist of a borate anion and a s-cis-1,3-butadiene radical cation bearing the unpaired spin density (see Scheme 1). The structure thus draws immediate comparison to simple butadiene radical cations which have attracted considerable theoretical and experimental attention,16 but have eluded characterisation by X-ray crystallography thus far.17 Moreover, radical 3 resembles the structures of the neutral radicals reported by Haddon and coworkers,18 in which two phenalenyl units are attached to a tetrahedral boron atom. In this family of compounds, however, the neutral spiro radical is based on N,N- or N,O-containing six-membered heterocyclic systems and is thus not susceptible to a competing oxidative ligand coupling reaction. Closely related to 3 are also the neutral borocyclic radicals of the general formula [(C6F5)2B(O2C14H8)]˙ and [(C6F5)2B(O2C16H8)]˙ in which the boron atom has two oxygen substituents.19
In summary, we have shown that by chemical one-electron oxidation of an organoborate, a neutral stable radical of the form [BR4]˙ can be generated. The stability and persistence of the radical species enabled its characterisation by X-ray crystallography, in addition to spectroscopic techniques (ESR and UV-vis). Assisted by DFT and TD-DFT calculations, the striking colour change between the crystalline and the solution/amorphous state could be attributed to structural changes of the spiro compound. Further investigations regarding the intriguing electronic and optical properties of the compound, as well as its reactivity, are underway.
We thank the Deutsche Forschungsgemeinschaft (H. B.), the University of Sussex and the EPSRC (A. V.) for support of this work.
Notes and references
- For references to early work, see: (a) H. W. Spier, Biochem. Z., 1952, 322, 467–470 CAS; (b) D. H. Geske, J. Phys. Chem., 1959, 63, 1062–1070 CrossRef CAS; (c) D. H. Geske, J. Phys. Chem., 1962, 66, 1743–1744 CrossRef CAS; (d) A. N. Nesmeyanov, V. A. Sazonova, G. S. Liberman and L. I. Emelyanova, Izv. Akad. Nauk SSSR, Otd. Khim. Nauk, 1955, 48 CAS.
- For different outcomes of organoborate oxidations see, for example: (a) S. T. Murphy, C. Zou, J. B. Miers, R. M. Ballew, S. D. Dlott and G. B. Schuster, J. Phys. Chem., 1993, 97, 13152–13157 CrossRef CAS; (b) A. Y. Polykarpov, S. Hassoon and D. C. Neckers, Macromolecules, 1996, 29, 8274 CrossRef CAS.
- (a) A. Suzuki, N. Miyaura, S. Abiko, M. Itoh, H. C. Brown, J. A. Sinclair and M. M. Midland, J. Am. Chem. Soc., 1973, 95, 3080–3081 CrossRef CAS; (b) M. Naruse, K. Utimoto and H. Nozaki, Tetrahedron, 1974, 30, 2159–2163 CrossRef CAS; (c) K. Yamada, N. Miyaura, M. Itoh and A. Suzuki, Tetrahedron Lett., 1975, 16, 1961–1964 CrossRef; (d) A. Suzuki, N. Miyaura, S. Abiko, M. Itoh, M. M. Midland, J. A. Sinclair and H. C. Brown, J. Org. Chem., 1986, 51, 4507–4511 CrossRef CAS; (e) A. Pelter and R. A. Drake, Tetrahedron Lett., 1988, 29, 4181–4184 CrossRef CAS.
- J. J. Eisch, J. H. Shah and M. P. Boleslawski, J. Organomet. Chem., 1994, 464, 11–21 CrossRef CAS.
- J. J. Eisch and R. J. Wilcsek, J. Organomet. Chem., 1974, 71, C21–C24 CrossRef CAS.
- See, for example: (a) J. L. R. Williams, J. C. Doty, P. J. Grisdale, R. Searle, T. H. Regan, G. P. Happ and D. P. Maier, J. Am. Chem. Soc., 1967, 89, 5153–5157 CrossRef CAS; (b) E. E. Bancroft, H. N. Blount and E. G. Janzen, J. Am. Chem. Soc., 1979, 101, 3692 CrossRef CAS; (c) J. J. Eisch, M. P. Boleslawski and K. Tamao, J. Org. Chem., 1989, 54, 1627–1634 CrossRef CAS; (d) S. Boyatzis, J. D. Wilkey and G. B. Schuster, J. Org. Chem., 1990, 55, 4537–4544 CrossRef CAS; (e) Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414 CrossRef CAS; (f) G. Duret, R. Quinlan, P. Bisseret and N. Blanchard, Chem. Sci., 2015, 6, 5366 RSC.
- See, for example: (a) P. Abley and J. Halpern, J. Chem. Soc. D, 1971, 1238 RSC; (b) T. Ishikawa, S. Nonaka, A. Ogawa and T. Hirao, Chem. Commun., 1998, 1209 RSC.
- H. Braunschweig, C. Hörl, F. Hupp, K. Radacki and J. Wahler, Organometallics, 2012, 31, 8463–8466 CrossRef CAS.
- For general reviews, see: (a) H. Braunschweig, I. Krummenacher and J. Wahler, Adv. Organomet. Chem., 2013, 61, 1–53 CrossRef CAS; (b) H. Braunschweig and T. Kupfer, Chem. Commun., 2011, 47, 10903–10914 RSC; (c) A. Steffen, R. M. Ward, W. D. Jones and T. B. Marder, Coord. Chem. Rev., 2010, 254, 1950–1976 CrossRef CAS.
- For a general review, see: H. Braunschweig and I. Krummenacher, in Organic Redox Systems: Synthesis, Properties and Applications, ed. T. Nishinaga, John Wiley & Sons, Inc., 2016, pp. 503–519 Search PubMed.
- For an ESR study on a related spirobifluorene compound, see: F. Gerson, B. Kowert and B. M. Peake, J. Am. Chem. Soc., 1974, 96, 118–120 CrossRef CAS.
- A. Heckmann, C. Lambert, M. Goebel and R. Wortmann, Angew. Chem., Int. Ed., 2004, 43, 5851–5856 CrossRef CAS PubMed.
- L. Fajarí, R. Papoular, M. Reig, E. Brillas, J. L. Jorda, O. Vallcorba, J. Rius, D. Velasco and L. Juliá, J. Org. Chem., 2014, 79, 1771–1777 CrossRef PubMed.
- Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., 2015, 127, 7197–7201 CrossRef.
- For reviews, see: (a) H. Dürr and R. Gleiter, Angew. Chem., Int. Ed. Engl., 1978, 17, 559–569 CrossRef; (b) R. Gleiter and G. Haberhauer, Aromaticity and Other Conjugation Effects, Wiley-VCH, Weinheim, 2012 Search PubMed.
- For butadiene radical cations see, for example: (a) C. S. Q. Lew, J. R. Brisson and L. J. Johnston, J. Org. Chem., 1997, 62, 4047 CrossRef CAS; (b) J. Oxgaard and O. Wiest, J. Phys. Chem. A, 2001, 105, 8236 CrossRef CAS.
- A Cambridge Structural Database (CSD) search with the ConQuest program resulted in no match (accessed February 2, 2016).
- S. K. Mandal, M. E. Itkis, X. Chi, S. Samanta, D. Lidsky, R. W. Reed, R. T. Oakley, F. S. Tham and R. C. Haddon, J. Am. Chem. Soc., 2005, 127, 8185–8196 CrossRef CAS PubMed.
- L. E. Longobardi, L. Liu, S. Grimme and D. W. Stephan, J. Am. Chem. Soc., 2016, 138, 2500–2503 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional crystallographic, computational and electrochemical data. CCDC 1469228. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc02916g |
| This journal is © The Royal Society of Chemistry 2016 |