
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic dehydrocoupling of amine-boranes and amines into diaminoboranes: isolation of a Pt(II), Shimoi-type, η1-BH complex†
Marta
Roselló-Merino
a,
Raquel J.
Rama
a,
Josefina
Díez
b and
Salvador
Conejero
*a
aInstituto de Investigaciones Químicas (IIQ), Departamento de Química Inorgánica, Centro de Innovación en Química Avanzada (ORFEO-CINCA), CSIC and Universidad de Sevilla, Avda. Américo Vespucio 49, 41092 Sevilla, Spain. E-mail: sconejero@iiq.csic.es; Tel: +34 954489563
bLaboratorio de Compuestos Organometálicos y Catálisis (Unidad asociada al CSIC), Departamento de Química Orgánica e Inorgánica, Universidad de Oviedo, C/Julián Clavería 8, 33006, Oviedo, Spain
First published on 3rd June 2016
Abstract
The platinum complex [Pt(ItBu′)(ItBu)][BArF] is a very efficient catalyst in the synthesis of diaminoboranes through dehydrocoupling of amine-boranes and amines. Shimoi-type, η1-BH complexes are key intermediates in the process.
In the last few years amine-boranes and related base-stabilised borane adducts have been shown to produce rich chemistry in which metal catalysed dehydrocoupling processes are involved.1 During some of these reactions the B–H bonds of the amine borane establish an initial interaction with the metal centre to form complexes exhibiting η1 or η2 coordination modes, undergoing thereafter subsequent reactivities.1a,2
Dehydrogenation of amine-boranes has been mainly focused on the production and release of dihydrogen and on the generation of amino-boranes that can undergo further dimerization, oligomerization or polymerization processes.1,2j,3 Less attention has been paid to the production of other boranes, through boron–boron coupling4 or the formation of diaminoboranes, (NR2)2BH.5 With regard to the latter process, only a couple of catalytic processes have been reported by Alcaraz and Sabo-Etienne, using ruthenium5a,c and by Hill,5b with calcium and magnesium based catalysts to produce the corresponding diaminoboranes. Catalyst loadings of 2.5% (Ru, Ca, Mg), together with long reaction times (Ru), and even heating (Ca, Mg) were required.
In this communication we wish to report a very efficient platinum(II) complex that promotes diaminoborane formation at a catalyst loading of 0.5% in a few minutes for most of the substrates tested. In addition, we have been able to characterise by means of X-ray diffraction studies the first Shimoi-type η1-BH complex of a Lewis base-borane adduct and platinum, which provides structural insights into a key intermediate in the dehydrogenation process of amine-boranes and amines.
Previously, we had reported that the [Pt(ItBu′)(ItBu)][BArF] complex (where ItBu stands for 1,3-di-tert-butylimidazolylidene and ItBu′ its cyclometalated form), 1,6 is able to promote the dehydrocoupling of dimethylamineborane (NMe2H·BH3) into cyclic [NMe2BH2]2 (Scheme 1).7 During NMR mechanistic studies, we observed that bis(dimethylamino)borane (NMe2)2BH is also formed in small amounts and its yield increases as the concentration of free NMe2H in the reaction media increases. Therefore, we analysed the ability of complex 1 to act as a catalyst for the formation of diaminoboranes.
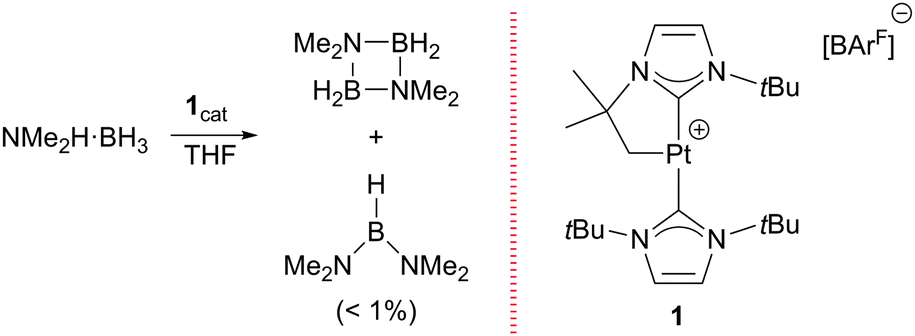 | ||
| Scheme 1 Dehydrocoupling of NMe2H·BH3 catalysed by complex 1. | ||
We have first studied the benchmark reaction of tert-butylamineborane (tBuNH2·BH3) and 1 equiv. of tert-butylamine (tBuNH2) in CD2Cl2, using a catalyst loading of 0.5% (Scheme 2). The reaction was monitored both by NMR spectroscopy and by measuring the increase of gas pressure in a closed system due to the generated H2 (see the ESI†). The 11B NMR spectrum revealed the formation of a single species in nearly quantitative yield after 11.5 min showing a doublet signal at δ 25.8 ppm (1JBH = 127 Hz). In addition, the 1H NMR spectrum exhibits a broad quartet signal centred at 4.12 ppm that sharpens upon 11B decoupling into a triplet (JHH = 8.2 Hz). These values are in agreement with the formation of diaminoborane (tBuNH)2BH,8s1 (Scheme 2). The calculated TON and TOF values for this process are 400 and 2087 h−1, the highest reported to the best of our knowledge (Table 1). At the very end of the reaction, the catalyst remained the cyclometalated species 1, but is slowly hydrogenated into complex [PtH(ItBu)2][BArF], 2. Other amine-boranes were tested under identical reaction conditions to form symmetrical diaminoboranes. Diethylamine-borane was also shown to be efficiently dehydrogenated in ca. 10 min in the presence of 1 equiv. of NEt2H and catalyst 1 (TON = 400, TOF = 2400 h−1). On the other hand, pyrrolidine-borane (CH2)4NH·BH3 required longer reaction times (2 h) to be converted into [(CH2)4N]2BH (TON = 380, TOF = 193 h−1). In the latter case, 5% of cyclic dimer [(CH2)4NBH2]2 is observed in the 11B NMR spectra. Bulkier amine-boranes, such as iPr2NH·BH3, do not undergo dehydrocoupling under similar reaction conditions, while heating at 60 °C for 12 h results in the formation of amino-borane NiPr2BH2, with no evidence of the corresponding diaminoborane.
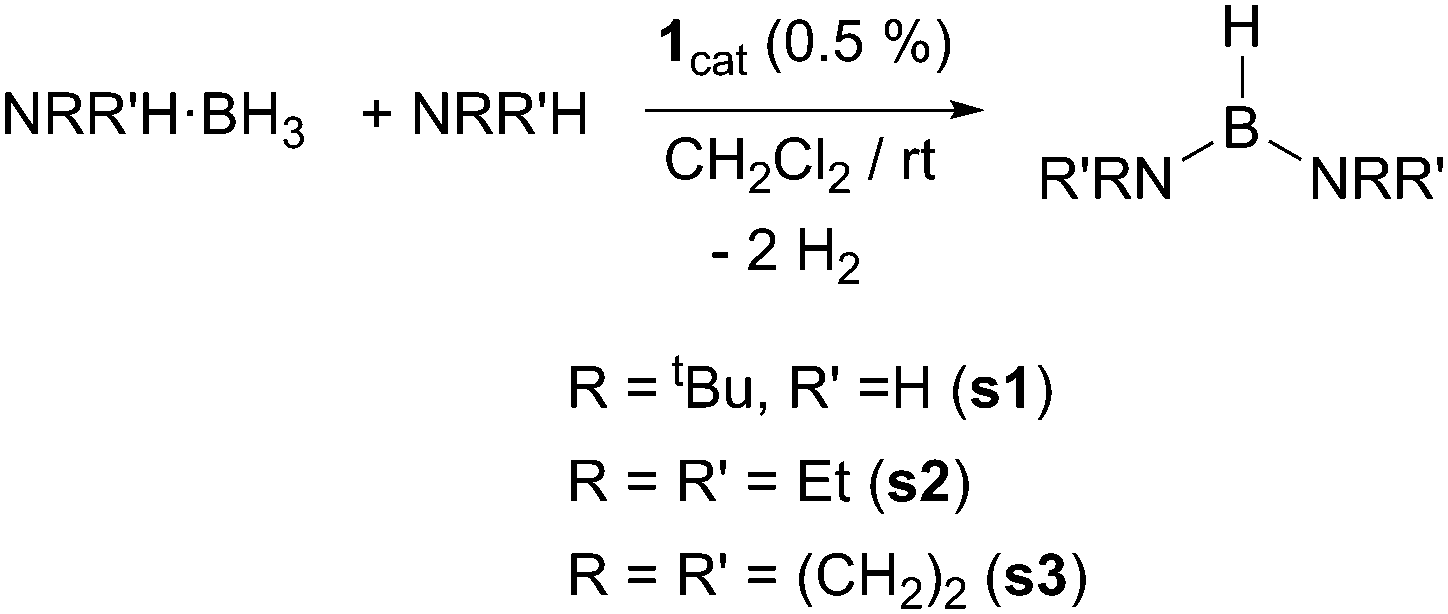 | ||
| Scheme 2 Catalytic synthesis of symmetric diaminoboranes. | ||
| Entrya | Amineborane | Amine | % Yieldb (isolated) | TON | TOF (h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: CH2Cl2, rt. b Yields determined by 11B NMR spectroscopy with respect to all diaminoboranes formed. c Cyclic dimers [NR2BH2]2 and other unidentified species constitute the remaining reaction products (see the ESI). d Isolated yield corresponds to all the possible products formed. | |||||
| 1 | t BuNH2·BH3 | t BuNH2 | >99 (99) | 400 | 2087 |
| 2 | NEt2H·BH3 | NEt2H | >99 (88) | 400 | 2400 |
| 3 | (CH2)4NH·BH3 | (CH2)4NH | 95c (81) | 380 | 193 |
| 4 | t BuNH2·BH3 | NEt2H | >99 (99) | 400 | 3692 |
| 5 | t BuNH2·BH3 | (CH2)4NH | 94 (68) | 376 | 376 |
| 6 | NEt2H·BH3 | (CH2)4NH | 93 | 372 | 248 |
| 7 | NMe2H·BH3 | t BuNH2 | 78c (71)d | 312 | 2023 |
| 8 | NMe2H·BH3 | NEt2H | 76c | 316 | 2257 |
Asymmetrical diaminoboranes are challenging substrates to be produced catalytically in a selective manner. Hill et al. have recently reported a method for the preparation of this type of substance using group 2 metal catalysts.5b Although in most of the cases the reaction proceeds with excellent selectivities, long reaction times (24–92 h) and mild heating are usually necessary. Complex 1 has shown very good catalytic behaviour in the formation of unsymmetrical diaminoboranes (Scheme 3). The reaction of tBuNH2·BH3 with NEt2H takes place very fast (6.5 min, TON = 400, TOF = 3692 h−1). According to NMR spectroscopy, borane (tBuNH)(NEt2)BH is formed almost exclusively (ca. 4% of (tBuNH)2BH is observed by 1H NMR). If the reaction is carried out in the inverse way, starting from NEt2H·BH3 and tBuNH2, the process is slower (ca. 20 min), but selectivities are comparable. The same behaviour is observed in the catalytic dehydrocoupling of (CH2)4NH·BH3 and tBuNH2. In this case, the reaction exhibits good selectivities towards the asymmetric diaminoborane (tBuNH)[(CH2)4N]BH (92%), whereas the only symmetric borane detected is [(CH2)4N]2BH (8%), with complete conversion after 2 h at rt. When the reaction is carried out the other way around, that is, starting from tBuNH2·BH3 and (CH2)4NH, the selectivities are only slightly different (85% of (tBuNH)[(CH2)4N]BH, 15% of [(CH2)4N]2BH), although the reaction proceeds in 1 h. The combination of pyrrolidine and diethylamine substrates indicates that the reaction is not selective yielding all possible reaction products (NEt2)2BH, [(CH2)4N]2BH and (NEt2)[(CH2)4N]BH in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5.4:3.1 ratio, respectively, independent of the two possible combinations utilised. A similar behaviour was observed in the reaction of NMe2H·BH3 with NEt2H, although we could not establish the exact ratio due to signal overlapping in the 1H NMR (see the ESI†). Nevertheless, good selectivities are produced in the reaction of NMe2H·BH3 and tBuNH2 (Scheme 3). Very interestingly, the reaction of tBuNH2·BH3 with iPr2NH generates diaminoborane (tBuNH)2BH (s1) and amino-borane iPr2N–BH2 in ca. 5 min at rt. Since 1 does not catalyse the dehydrocoupling of iPr2NH·BH3 at rt, iPr2N–BH2 is probably formed through the reorganization of amino-borane tBuNH–BH2 and iPr2NH.9
:5.4:3.1 ratio, respectively, independent of the two possible combinations utilised. A similar behaviour was observed in the reaction of NMe2H·BH3 with NEt2H, although we could not establish the exact ratio due to signal overlapping in the 1H NMR (see the ESI†). Nevertheless, good selectivities are produced in the reaction of NMe2H·BH3 and tBuNH2 (Scheme 3). Very interestingly, the reaction of tBuNH2·BH3 with iPr2NH generates diaminoborane (tBuNH)2BH (s1) and amino-borane iPr2N–BH2 in ca. 5 min at rt. Since 1 does not catalyse the dehydrocoupling of iPr2NH·BH3 at rt, iPr2N–BH2 is probably formed through the reorganization of amino-borane tBuNH–BH2 and iPr2NH.9
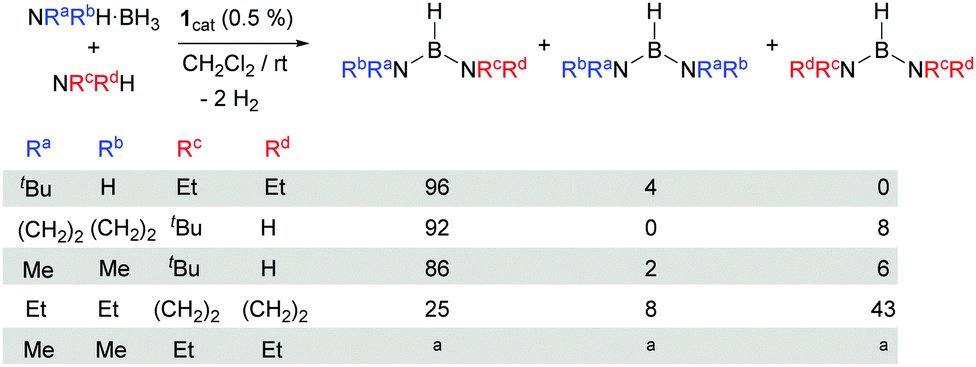 | ||
| Scheme 3 Catalytic synthesis of asymmetric diaminoboranes. aSignal overlapping in the 1H and 11B{1H} NMR spectra precluded establishing the exact amount of each diaminoborane (see the ESI†). | ||
With regard to the mechanism by which these transformations occur, it has been previously shown that dehydrogenation of amine-boranes NR2H·BH3 leading to the corresponding aminoboranes NR2BH2 is a key step.5 Previously, we have reported that complex 1 is able to dehydrogenate NMe2H·BH3 into NMe2BH2 (that then dimerises) through a distinct mechanism that involves a first step in which the amine-borane interacts with the platinum atom through the BH protons (complex 3a in Scheme 4) followed by nucleophilic addition of free amine present in solution to the activated boron atom.7 According to DFT calculations the most stable coordination mode of the BH3 moiety is η1-BH (Shimoi type complex) but, unfortunately, no experimental evidence for this type of coordination could be provided. Here we further investigate these adducts by carrying out stoichiometric reactions between complex 1 and amine-boranes at 0 °C in CD2Cl2. tBuNH2·BH3 reacts with 1 leading to a new species that has been postulated as the η1-BH derivative 3b (Scheme 4). The BH3 group resonates as a broad signal centred at 0.12 ppm that sharpens upon 11B broadband decoupling. This signal is shifted upfield from the free tBuNH2·BH3 (δ 1.36). In addition, JPtH of the CH2–Pt fragment has a value of 90 Hz, that is, 30 Hz smaller than in complex 1 (120 Hz), consistent with the coordination of a ligand trans to the CH2 moiety.10 This value is smaller than that observed in dimethylamine-borane 3a (103 Hz) suggesting that the interaction with the platinum atom is stronger. The 11B NMR spectrum shows a signal at −18.2 ppm that is shifted with respect to free amine-borane (−21.1 ppm). All these data agree well with the formulation of 3b as depicted in Scheme 4.11 Unfortunately, the instability of 3a and 3b due to their propensity to undergo dehydrocoupling precluded their isolation as pure compounds and further characterization. To avoid this problem, tertiary amine-borane NMe3·BH3 was used. However, no interaction with 1 was observed, probably due to steric constrains, thus allowing the establishment of a correlation between the bulkiness of the amine-borane and its interaction with 1 (stronger interaction: tBuNH2·BH3 > NMe2H·BH3 > NMe3·BH3). Consequently, the less hindered Lewis base stabilised borane C6H5N·BH3 was a judicious choice. This borane reacts with 1 to yield 3c (Scheme 5) quantitatively by NMR spectroscopy. The main feature of 3c is the broad NMR signal centred at 1.25 ppm attributed to the BH3 protons (2.55 ppm in free C6H5N·BH3). The 11B{1H} spectrum exhibits a signal at −8.9 ppm, nearly 3 ppm down-field shifted with respect to C6H5N·BH3. The apparent coupling constant 1JB,H is ca. 85 Hz, that is, 12 Hz smaller than in free C6H5N·BH3, and compares well with previously described Shimoi-type complexes.11,12 The coupling constant of the CH2–Pt protons with 195Pt of 93 Hz is similar to that of complex 3b. A definite proof of the nature of this compound came from the solid-state structure obtained by X-ray diffraction studies. Colourless crystals suitable for this analysis were obtained by slow diffusion of a concentrated solution of 3c in CH2Cl2 into pentane at 0 °C. Fig. 1 depicts an ORTEP-type view of the cation of complex 3c. The complex is four-coordinated, with two N-heterocyclic carbene units in the expected trans arrangement (C(1)–Pt(1)–C(12): 168.38(19)°) one of which is cyclometalated. The fourth coordination site is occupied by the C6H5N·BH3 ligand in which one of the hydride atoms of the BH3 unit bridges the platinum nucleus. The Pt(1)–H(1B) and H(1B)–B(1) bond distances are 1.96(5) and 1.03(2) Å, respectively. The Pt(1)–H(1B)–B(1) angle of 147.48(485)° together with the long Pt(1)⋯B(1) bond distance (2.8436(5) Å) indicates a negligible interaction between platinum and boron, and therefore this complex is best described as a η1-BH or Shimoi-type complex.12,13
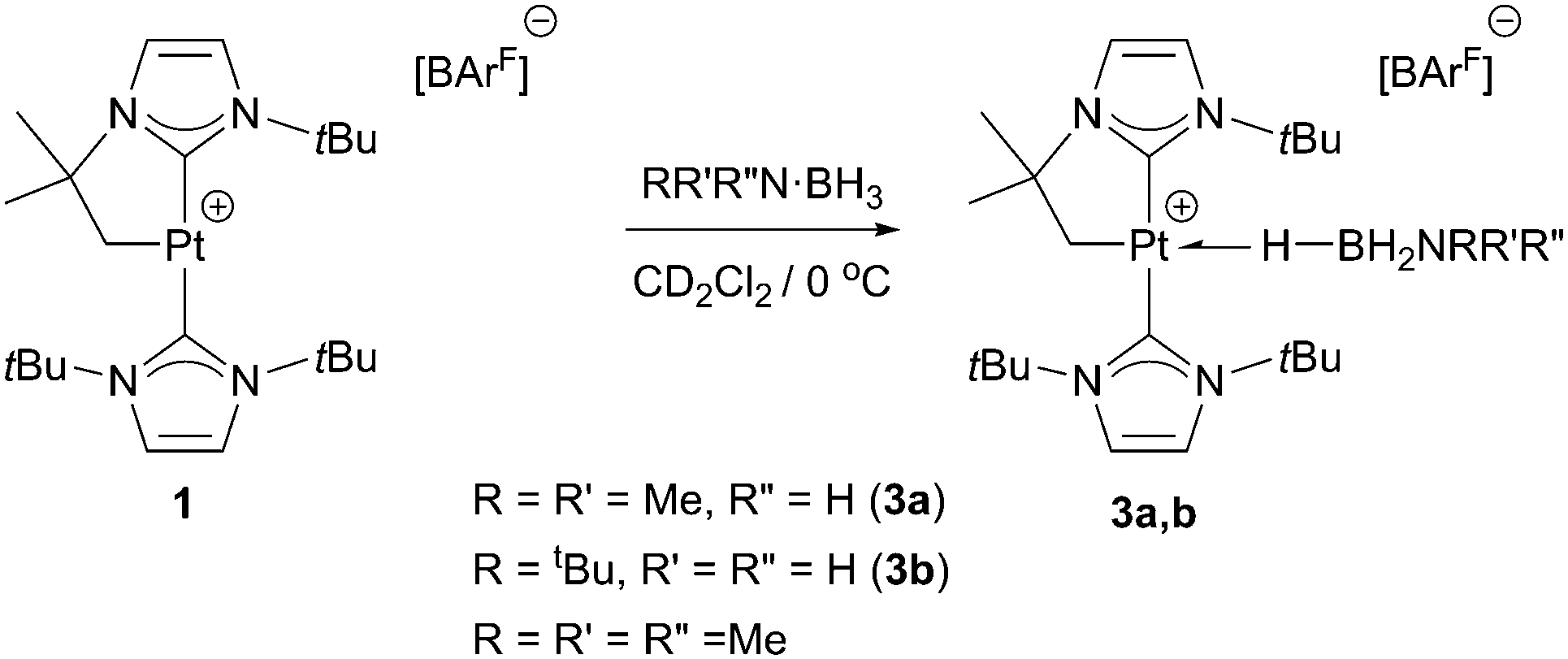 | ||
| Scheme 4 Formation of complexes 3a,b. | ||
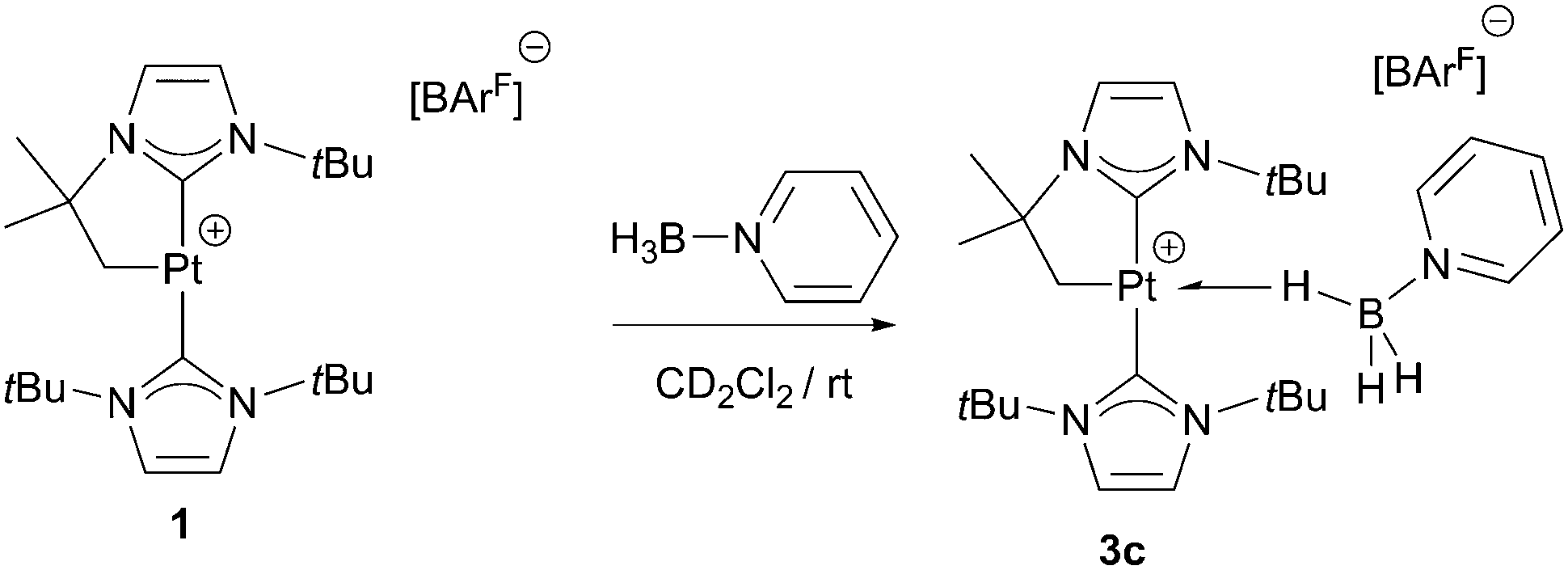 | ||
| Scheme 5 Synthesis of complex 3c. | ||
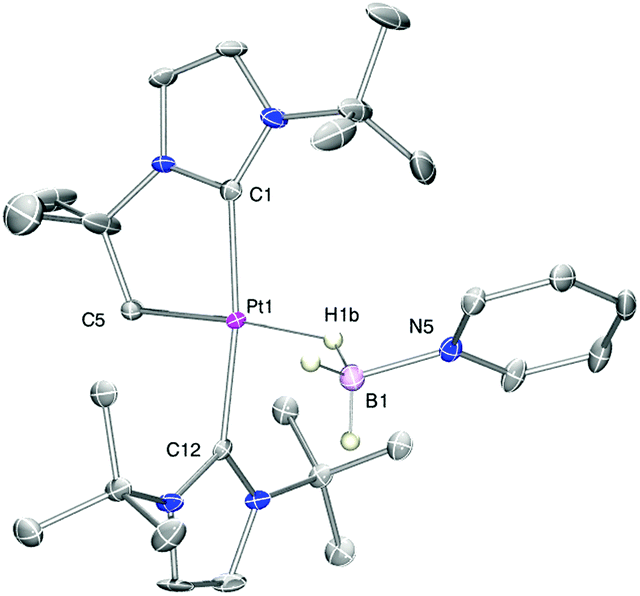 | ||
| Fig. 1 ORTEP-type view of the cation of complex 3c. Hydrogen atoms, except those of the BH3 unit, have been omitted for clarity (ellipsoids are drawn at 30% probability). Selected bonds (Å) and angles (°): Pt1–H1b 1.96(5), Pt1–C5 2.045(5), B1–H1b 1.03(2); Pt1–H1b–B1 147.48(485), C1–Pt1–C12 168.38(19). | ||
Variable temperature NMR spectroscopy was used to study the dynamic behaviour due to exchange between bridging and terminal BH protons. The resonance for the BH3 protons collapses into the baseline at ca. 223 K. Upon cooling to 203 K, a new signal of relative integral 2 appears at 2.47 ppm, in the expected region for terminal BH protons. Nevertheless, at this temperature, the signal corresponding to the Pt–H–B proton is not discernible.14 Further cooling to 183 K provided evidence for a very broad resonance at ca. −4 ppm (approx. integral value of 1), with the terminal BH protons resonating at 2.44 ppm. With respect to the 11B NMR spectra, a very broad signal centred at δ −12.2 is detected at this temperature.
For comparison purposes, we have analysed the interaction of C6H5N·BH3 with the hydrogenated form of complex 1, the hydride derivative [PtH(ItBu)2][BArF], 2.15 No evidence of interaction between 2 and C6H5N·BH3 is observed at 298 K. At this temperature, the BH3 protons resonate at δ 2.49, whereas the platinum-hydride signal appears at δ −25.47 exhibiting a JPt,H of 2550 Hz, only marginally different from that of complex 2 in the absence of C6H5N·BH3 (JPt,H = 2564 Hz).15 However, the 1H NMR spectra at temperatures below 233 K indicate the presence of a new species, 4 (Scheme 6), coexisting with complex 2, in a 0.3 to 1 ratio (4:2). This compound is characterised by a signal in the hydride region at −21.85 ppm showing a reduced coupling constant with 195Pt (JPt,H = 1920 Hz) and another broad signal at 0.28 ppm (3H relative integral) that sharpens upon 11B decoupling. As the temperature decreases, the concentration of 4 increases at the expense of 2, reaching a maximum at 188 K (4:2 ratio, ca. 4:1). The 11B{1H} NMR spectra at all temperatures exhibit a single very broad signal at −12.3 ppm that does not show splitting upon coupling to 1H below 208 K. Interestingly, even at 188 K the BH3 group still shows fast exchange between terminal and bridging hydrogens. The different behaviour of compounds 1 and 2 toward the interaction with C6H5N·BH3 can be easily rationalised in terms of the different steric protection that the cyclometalated ItBu ligand exerts on the platinum atom compared to its non-cyclometalated form. When the ItBu ligand is cyclometalated the tBu group is tilted16 away from the metal centre favouring the interaction with amine-boranes.
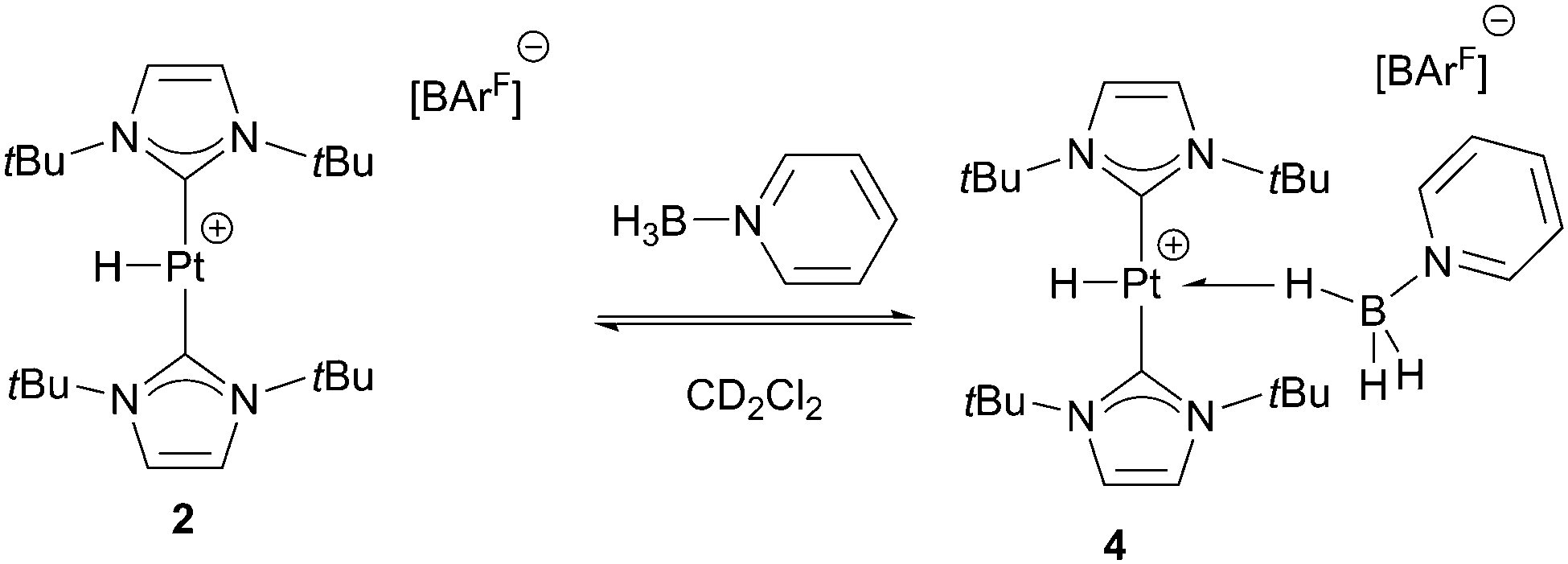 | ||
| Scheme 6 Equilibrium between complexes 2 and 4 in the presence of C6H5N·BH3. | ||
Once the amine-borane binds the metal in a Shimoi-type fashion, dehydrogenation leading to amino-boranes, NR2–BH2, takes place through a mechanism that involves the intermediacy of boronium cations (NHR2)2BH2+ and the neutral platinum hydride [PtH(ItBu′)(ItBu)], 5.7 It must be noted that we have not observed an interaction between complex 1 and amino-borane NMe2BH2 (either using cyclic dimer [NMe2BH2]2 as a precursor17 or during monitoring the dehydrogenation reaction of 1 and NMe2H·BH3 at 0 °C).7 At this point, we do not have further information on the mechanism by which the amino-borane is converted into diaminoboranes and the role of complex 1 in the process.
In summary, the coordinatively unsaturated Pt(II) complex 1 is a very efficient catalyst for the synthesis of diaminoboranes achieving TON and TOF values of 400 and 3692 h−1, respectively, the highest reported to date. The process takes place through the initial coordination of the BH protons of amine-boranes to the platinum centre in an end-on mode (Shimoi type) that was demonstrated crystallographically in the pyridine·BH3 adduct 3c. Ongoing efforts are geared towards unveiling the mechanism by which amino-boranes are transformed into diaminoboranes.
Financial support (FEDER contribution) from the MINECO (Projects CTQ2013-45011-P and CTQ2014-51912-REDC) and the Junta de Andalucía (Project FQM-2126) is gratefully acknowledged.
Notes and references
- (a) H. C. Johnson, T. N. Hooper and A. S. Weller, Top. Organomet. Chem., 2015, 49, 153 CrossRef CAS; (b) E. M. Leitao, T. Jurka and I. Manners, Nat. Chem., 2013, 5, 817 CrossRef CAS PubMed; (c) N. E. Stubbs, A. P. M. Robertson, E. M. Leitao and I. Manners, J. Organomet. Chem., 2013, 730, 84 CrossRef CAS; (d) A. S. John, K. I. Goldberg and D. M. Heinekey, Top. Organomet. Chem., 2013, 40, 271 CrossRef.
- (a) H. C. Johnson and A. S. Weller, Angew. Chem., Int. Ed., 2015, 54, 10173 CrossRef CAS PubMed; (b) M. A. Esteruelas, A. M. López, M. Mora and E. Oñate, ACS Catal., 2015, 5, 187 CrossRef CAS; (c) A. G. Algarra, L. J. Sewell, H. C. Johnson, S. A. Macgregor and A. S. Weller, Dalton Trans., 2014, 43, 11118 RSC; (d) S. Muhammad, S. Moncho, E. N. Brothers and A. A. Bengali, Chem. Commun., 2014, 50, 5874 RSC; (e) L. J. Sewell, G. C. Lloyd-Jones and A. S. Weller, J. Am. Chem. Soc., 2012, 134, 3598 CrossRef CAS PubMed; (f) C. Y. Tang, N. Phillips, M. J. Kelly and S. Aldridge, Chem. Commun., 2012, 48, 11999 RSC; (g) R. Kumar and B. R. Jagirdar, Inorg. Chem., 2013, 22, 28 CrossRef PubMed; (h) H. C. Johnson, A. P. M. Robertson, A. B. Chaplin, L. J. Sewell, A. L. Thompson, M. F. Haddow, I. Manners and A. S. Weller, J. Am. Chem. Soc., 2011, 133, 11076 CrossRef CAS PubMed; (i) R. Dallanegra, A. P. M. Robertson, A. B. Chaplin, I. Manners and A. S. Weller, Chem. Commun., 2011, 47, 3763 RSC; (j) G. Alcaraz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2010, 49, 7170 CrossRef CAS PubMed; (k) C. Y. Tang, A. L. Thompson and S. Aldridge, J. Am. Chem. Soc., 2010, 132, 10578 CrossRef CAS PubMed; (l) T. M. Douglas, A. B. Chaplin, A. S. Weller, X. Yang and M. B. Hall, J. Am. Chem. Soc., 2009, 131, 15440 CrossRef CAS PubMed.
- (a) A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079 CrossRef CAS PubMed; (b) N. C. Smythe and J. C. Gordon, Eur. J. Inorg. Chem., 2010, 509 CrossRef CAS; (c) C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279 RSC.
- H. C. Johnson, C. L. McMullin, S. D. Pike, S. A. Macgregor and A. S. Weller, Angew. Chem., Int. Ed., 2013, 52, 9776 CrossRef CAS PubMed.
- (a) C. J. Wallis, G. Alcaraz, A. S. Petit, A. I. Poblador-Bahamonde, E. Clot, C. Bijani, L. Vendier and S. Sabo-Etienne, Chem. – Eur. J., 2015, 21, 13080 CrossRef CAS PubMed; (b) P. Bellham, M. S. Hill, G. Kociok-Köhn and D. J. Liptrot, Chem. Commun., 2013, 49, 1960 RSC; (c) C. J. Wallis, H. Dyer, L. Vendier, G. Alcaraz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2012, 51, 3646 CrossRef CAS PubMed; (d) It has been reported that aromatic amine-boranes can produce diaminoboranes without a catalyst: H. Helten, A. P. M. Robertson, A. Staubitz, J. R. Vance, M. F. Haddow and I. Manners, Chem. – Eur. J., 2012, 18, 4665 CrossRef CAS PubMed.
- O. Rivada-Wheelaghan, B. Donnadieu, C. Maya and S. Conejero, Chem. – Eur. J., 2010, 16, 10323 CrossRef CAS PubMed.
- M. Roselló-Merino, J. López-Serrano and S. Conejero, J. Am. Chem. Soc., 2013, 135, 10910 CrossRef PubMed.
- (a) P. Bellham, M. S. Hill and G. Kociok-Köhn, Organometallics, 2014, 33, 5716 CrossRef CAS; (b) Y. Kawano, M. Uruichi, M. Shimoi, S. Taki, T. Kawaguchi, T. Kakizawa and H. Ogino, J. Am. Chem. Soc., 2009, 131, 14946 CrossRef CAS PubMed.
- A. P. M. Robertson, E. M. Leitao and I. Manners, J. Am. Chem. Soc., 2011, 133, 19322 CrossRef CAS PubMed.
- M. A. Ortuño, S. Conejero and A. Lledós, Beilstein J. Org. Chem., 2013, 9, 1352 CrossRef PubMed.
- G. Alcaraz and S. Sabo-Etienne, Coord. Chem. Rev., 2008, 252, 2395 CrossRef CAS.
- (a) R. Dallanegra, A. B. Chaplin, J. Tsim and A. S. Weller, Chem. Commun., 2010, 46, 3092 RSC; (b) M. Shimoi, S. Nagai, M. Ichikawa, Y. Kawano, K. Katoh, M. Uruichi and H. Ogino, J. Am. Chem. Soc., 1999, 121, 11704 CrossRef CAS.
- (a) A. E. W. Ledger, C. E. Ellul, M. F. Mahon, J. M. J. Williams and M. K. Whittlesey, Chem. – Eur. J., 2011, 17, 8704 CrossRef CAS PubMed; (b) R. Dallanegra, A. B. Chaplin and A. S. Weller, Angew. Chem., Int. Ed., 2009, 48, 6875 CrossRef CAS PubMed; (c) Y. Kawano, K. Yamaguchi, S. Miyake, T. Kakizawa and M. Shimoi, Chem. – Eur. J., 2007, 13, 6920 CrossRef CAS PubMed; (d) Y. Kawano, M. Hashiva and M. Shimoi, Organometallics, 2006, 25, 4420 CrossRef CAS; (e) N. Merle, G. Koicok-Köhn, M. F. Mahon, C. G. Frost, G. D. Ruggerio, A. S. Weller and M. C. Willis, Dalton Trans., 2004, 3883 RSC; (f) T. Yasue, Y. Kawano and M. Shimoi, Angew. Chem., Int. Ed., 2003, 42, 1727 CrossRef CAS PubMed.
- A similar behavior has been observed in a ruthenium complex. See ref. 13a.
- (a) M. A. Ortuño, P. Vidossich, S. Conejero and A. Lledós, Angew. Chem., Int. Ed., 2014, 53, 14158 CrossRef PubMed; (b) O. Rivada-Wheelaghan, M. Roselló-Merino, M. A. Ortuño, P. Vidossich, E. Gutiérrez-Puebla, A. Lledós and S. Conejero, Inorg. Chem., 2014, 53, 4257 CrossRef CAS PubMed.
- J.-N. Luy, S. A. Hauser, A. B. Chaplin and R. Tonner, Organometallics, 2015, 34, 5099 CrossRef CAS.
- G. Bénac-Lestrille, U. Helmstedt, L. Vendier, G. Alcaraz, E. Clot and S. Sabo-Etienne, Inorg. Chem., 2011, 50, 11039 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, H2 evolution graphics and X-ray crystallographic data. CCDC 1468913. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc02720b |
| This journal is © The Royal Society of Chemistry 2016 |