
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly active and selective photochemical reduction of CO2 to CO using molecular-defined cyclopentadienone iron complexes†
Alonso
Rosas-Hernández
a,
Pamela G.
Alsabeh
a,
Enrico
Barsch
ab,
Hernrik
Junge
a,
Ralf
Ludwig
ab and
Matthias
Beller
*a
aLeibniz Institute for Catalysis at the University of Rostock, Albert Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: matthias.beller@catalysis.de
bInstitute of Chemistry, Department of Physical Chemistry, University of Rostock, Dr. Lorenz Weg 1, 18059 Rostock, Germany
First published on 8th June 2016
Abstract
Herein, we report highly active (cyclopentadienone)iron–tricarbonyl complexes for CO2 photoreduction using visible light with an Ir complex as photosensitizer and TEOA as electron/proton donor. Turnover numbers (TON) of ca. 600 (1 h) with initial turnover frequencies (TOF) up to 22.2 min−1 were observed. Operando FTIR measurements allowed for the proposal of a plausible mechanism for catalyst activation.
The development of molecular-defined catalysts that allow for the conversion of carbon dioxide as a ubiquitous C1-building block to valuable chemicals using sunlight is of broad and current interest.1 Over the past few decades following the seminal work of Lehn et al.,2 several transition metal complexes have been developed to drive the photochemical reduction of CO2 to primarily carbon monoxide and formic acid. Most of these catalytic systems are based on less abundant second and third row transition metals.3 For instance, metals such as Re,4 Ru,5 and Ir6 are regularly present within the most active, robust and selective catalysts known to date. Hence, from the viewpoint of basic science but also to develop economically viable solar-to-fuel technologies in the long run, there is a crucial need for new visible light-driven CO2 reduction catalysts, especially based on benign, nontoxic, and earth-abundant elements.
A series of metal complexes based on first row transition metals supported by multidentate macrocyclic ligands have been reported as efficient catalysts for the photoreduction of CO2 to CO using visible light.7 Specifically, Chang et al. showed that the photocatalytic system including a nickel N-heterocyclic carbene–isoquinoline complex and the photosensitizer (PS) Ir(ppy)3 (ppy = 2-phenylpyridine) drives the conversion of carbon dioxide to carbon monoxide with excellent turnover number, albeit using a highly diluted catalyst concentration.7e In addition, Bonin and Robert used an iron(0) porphyrin complex, previously employed in electrocatalytic CO2 reduction, and demonstrated that such a complex is also able to operate under photochemical conditions, obtaining mixtures of CO and H2 with a selectivity of 75% and a TON of 30 for carbon monoxide after 10 h of irradiation.7d Furthermore, when this iron complex is coupled with Ir(ppy)3 the overall performance of the photocatalytic reaction is increased, since CO is obtained with a selectivity of 93% and a TON of 140 after 55 h of irradiation; in addition, with the goal of replacing the Ir-based PS, a cheap organic sensitizer was used instead, resulting in a less active photocatalytic system.7c Chan et al. then developed a Co(II) complex supported by the tetradentate tripodal ligand TPA (tris(2-pyridylmethyl)amine) and tested it as catalyst together with Ir(ppy)3, obtaining a CO selectivity of 85% and TONs of 953 after 70 h of irradiation.7b Most recently, Lau and Robert reported catalytic activity, in the presence of Ir(ppy)3, with either a Co(II) or an Fe(II) complex supported by a pentadentate macrocyclic ligand. Interestingly in this case, carbon monoxide was formed with the cobalt complex (catalytic CO selectivity of ca. 97% and TON up to 270 after 22 h irradiation), while formic acid was obtained only with the iron-based catalyst (TON of ca. 5 after 20 h).7a Although there has been a growing interest in developing noble metal-free CO2-reduction photocatalysts, challenges remain. For instance, improvements in the turnover frequencies (TOF) and selectivities are highly desirable.
In this work, we aimed to explore the use of (cyclopentadienone)iron–tricarbonyl complexes as catalysts for the 2H+/2e− reduction of carbon dioxide (Fig. 1). These complexes are well-known catalysts for the hydrogenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O8 and CN bonds,9 and most recently for bicarbonates as well as CO2.10 An important feature of this class of complexes is their bifunctional nature, which translates to the presence of both, a proton-donor site (ligand) and a hydride-donor site (metal center). In addition, as previously observed in other energy-related photocatalytic reactions11 or in the case of the Fe-only hydrogenases,12 the implementation of a protic functional moiety in the second-coordination-sphere of the catalyst leads to an acceleration of the photocatalysis, as a result of intramolecular proton transfer pathways being kinetically favored.13 Thus, we envisioned to exploit this cooperative interaction between the iron center and its ligand to achieve a more efficient photochemical reduction of CO2.
O8 and CN bonds,9 and most recently for bicarbonates as well as CO2.10 An important feature of this class of complexes is their bifunctional nature, which translates to the presence of both, a proton-donor site (ligand) and a hydride-donor site (metal center). In addition, as previously observed in other energy-related photocatalytic reactions11 or in the case of the Fe-only hydrogenases,12 the implementation of a protic functional moiety in the second-coordination-sphere of the catalyst leads to an acceleration of the photocatalysis, as a result of intramolecular proton transfer pathways being kinetically favored.13 Thus, we envisioned to exploit this cooperative interaction between the iron center and its ligand to achieve a more efficient photochemical reduction of CO2.
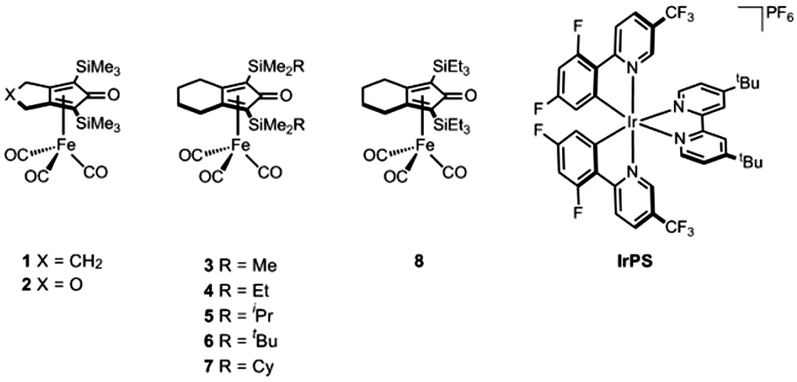 | ||
| Fig. 1 Molecular structure of the complexes used in the photocatalytic reduction of CO2. | ||
Accordingly, we started our investigation with [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) (IrPS) (where dF(CF3)ppy = cyclometalated 2-(2,4-difluorophenyl)-5-trifluoro-methylpyridine and dtbbpy = 4,4′-di-tert-butyl-2-2′-bipyridyl) as PS, due to its extended excited-state life-time (2300 ns) and strong reducing strength of the excited state (Eox* = 1.21 V).14 Recently, this iridium complex has been successfully applied in a wide range of photoredox catalytic processes.15 The photocatalytic reactions were carried out using NMP (N-methyl-2-pyrrolidone) as solvent and TEOA (triethanolamine) as the electron and proton donor, and irradiated using a Hg-lamp fitted with a built-in filter (400–700 nm) at 2.5 W of power output. It is worth mentioning that the active species when these types of complexes are used in hydrogenation reactions is an Fe(II) hydride species8b–d (vide infra). However, due to the sensitivity of these hydride complexes, we decided to use the air-stable Fe(0) tricarbonyl complexes depicted in Fig. 1 instead, expecting to form the catalytic active species in situ as achieved in previous reports.8a,9c,d
When complex 1 was used as catalyst, carbon monoxide was selectively formed in the head-space of the reaction vessel with a TON of 421 after 5 h of irradiation. Notably, the previously reported and structurally related iron(0) complex [Fe(CO)3(bpy)] as the catalyst produced CO with a poor selectivity of 43% over H2 production and a TON of 129.16 Thus, we can infer that the cyclopentadienone ligand in 1 plays a positive and important role in the overall performance of this photocatalytic system. Control experiments were carried out to establish that all components are essential to achieve this improved activity (Table S1, ESI†). For instance, when the reactions were performed in the absence of either complex 1 or IrPS, no catalytic activity was observed; the same behavior was detected when the experiments were carried out in the absence of TEOA or light irradiation. With the optimized conditions in hand, we investigated the catalytic activity of the other iron complexes (Table 1). As in the case of 1, all the iron complexes proved to selectively reduce CO2 over H+ since only carbon monoxide was found by calibrated GC in the head-space of the photocatalytic reactions together with traces of H2. In addition, analysis of the liquid phase by capillary electrophoresis resulted in the quantification of up to 40 TONs of HCOOH. We observed that the activity of the photocatalytic system is heavily influenced by the steric and electronic properties of the cyclopentadienone ligand. When we compare the activities of the complexes 3 and 7 (Table 1, entry 3 vs. entry 10), it is evident that increasing the steric hindrance of the substituents at the silicon atom has a negative impact on the photocatalytic activity, specifically, the TON is reduced from 429 to 319. Moreover, the activity of the catalytic system was drastically diminished when a heteroatom (oxygen) is present in the cyclic structure of the ligand (Table 1, entry 6). The same catalytic activity was observed regardless whether a 5- or 6-membered ring was fused to the cyclopentadienone moiety in the ligand (Table 1, entry 1 vs. entry 3). When the catalytic reactions were irradiated for longer times (18 h) an enhancement in the activity was not achieved, suggesting that the catalyst is no longer active after 5 h of reaction time (Table 1, entries 1 and 3 vs. entries 2 and 4). In fact, the conversion plot in Fig. S1 (ESI†) shows that the production of CO stops after 1 h of irradiation when complex 3 is used. Attempts to reactivate the catalytic system after 5 h of reaction time, by adding a second equivalent of either catalyst 3 or photosensitizer, were unsuccessful. In addition, when the catalyst concentration was lowered to 5 μM a TON up to 596 was observed (Table 1, entry 5). It should be noted that the catalytic activities observed in this work surpass in terms of TONs all previous reports using an iron-based CO2-reduction catalyst. Furthermore, the TOF value of 22.2 min−1 for catalyst 3 represents a high activity for a photocatalytic system containing a noble metal-free catalyst using >μM loadings of the catalyst.
|
|
|||
|---|---|---|---|
| Entry | Complex | CO (TON)b | CO (TOF, min−1)c |
a Reaction conditions: N-methyl-2-pyrrolidone and triethanolamine (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) 7.5 mL; catalyst 0.13 mM; photosensitizer IrPS 1.67 mM; Hg-lamp (light output 2.5 W) equipped with a 400–700 nm filter.
b Determined using calibrated GC; TON = n(CO)/n(catalyst).
c Determined after 15 min of irradiation.
d Reaction performed for 18 h.
e Reaction performed in the presence of 5 μM catalyst. :1, v/v) 7.5 mL; catalyst 0.13 mM; photosensitizer IrPS 1.67 mM; Hg-lamp (light output 2.5 W) equipped with a 400–700 nm filter.
b Determined using calibrated GC; TON = n(CO)/n(catalyst).
c Determined after 15 min of irradiation.
d Reaction performed for 18 h.
e Reaction performed in the presence of 5 μM catalyst.
|
|||
| 1 | 1 | 421 | 9.1 |
| 2d | 1 | 413 | n.d. |
| 3 | 3 | 429 | 10.9 |
| 4d | 3 | 413 | n.d. |
| 5e | 3 | 596 | 22.2 |
| 6 | 2 | 41 | 1.7 |
| 7 | 4 | 380 | 9.6 |
| 8 | 5 | 390 | 10.5 |
| 9 | 6 | 336 | 9.9 |
| 10 | 7 | 319 | 6.2 |
| 11 | 8 | 392 | 9.1 |

We then proceeded to investigate the mechanism of the photochemical CO2 reduction mediated by iron complex 3. First, the quantum yield (Φ) of the overall photocatalytic cycle was determined using an iron actinometer to measure the number of incident photons (see ESI† for details). The quantum yield was calculated using monochromatic light (440 nm) at different light intensities after 5 h of irradiation. An excellent value of 68% was determined when an intensity of 2.4 × 10−7 einstein s−1 was used (Table S2, ESI†). However, if we consider the possibility of a dark electron transfer from a TEOA carbon-based neutral radical17 to complex 3, only one photon is then required to reduce one molecule of CO2 and therefore, the value of Φ would be 34%. In any case, these values represent excellent quantum efficiencies for CO2 photoreduction using noble metal-free catalysts.
Operando FTIR measurements were then carried out to establish the nature of the catalytic intermediates formed in solution during the course of the reaction. When the iron complex 3 was placed in the solvent mixture NMP:TEOA (5:1, v/v) under argon atmosphere, three bands at 2052, 1999 and 1980 cm−1 were observed corresponding to the carbonyl ligands present in the complex (Fig. 2a). When this solution was irradiated under visible light, a first order decarbonylation process was observed with a constant rate of 0.23 min−1 (Fig. S3 and S4, ESI†). On the other hand, when a solution of complex 3 and IrPS in the same solvent mixture and under Ar atmosphere was irradiated, a new species formed with signals at 1995 and 1933 cm−1 (Fig. 2b). Based on previous literature data,18 this species can be unambiguously assigned to the hydride Fe(II) species 9 (Fig. 3). This suggests that complex 3, after the loss of a CO ligand, is reduced in situ to form the catalytic intermediate 9 by 2 protons donated by TEOA and 2 electrons transferred from either the excited or reduced state of IrPS. Finally, by continuously monitoring a photocatalytic CO2 reduction reaction mediated by complex 3, both the iron precursor and the hydride 9 were observed in solution along with carbon monoxide dissolved in the reaction mixture (Fig. 2c).
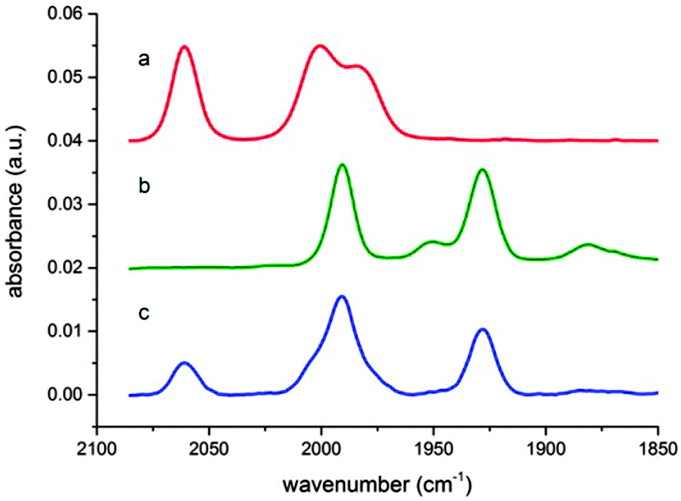 | ||
| Fig. 2 Operando FTIR experiments. (a) Spectrum of complex 3 in NMP/TEOA (5:1) under Ar atmosphere. (b) Spectrum of a solution containing 3 and IrPS under Ar atmosphere and visible light irradiation. (c) Spectrum of a solution containing 3 and IrPS under CO2 atmosphere and irradiation. | ||
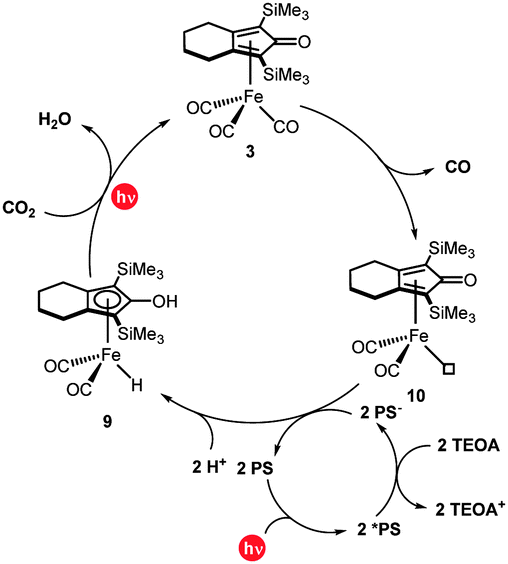 | ||
| Fig. 3 Plausible mechanism for the photochemical reduction of carbon dioxide using the cyclopentadienone iron complex. | ||
The quenching rate of the IrPS excited state by the catalyst 3 and TEOA was then measured using Stern–Volmer analysis (see ESI† for details). As observed in Fig. S5 and S6 (ESI†), both 3 and TEOA effectively quenched the emission intensity of IrPS excited state. The measured quenching rate constants for 3 and TEOA are 9.0 × 108 M−1 s−1 and 1.7 × 107 M−1 s−1, respectively. Although 3 follows a Stern–Volmer behavior, the cyclic voltammetry for this compound showed an irreversible reduction peak at −1.44 V vs. SCE (Fig. S7, ESI†), which, when compared to the redox potential of the couple [*IrPS/IrPS+] (−0.89 V vs. SCE) indicates that an electron transfer from the excited state of the photosensitizer to the catalyst is not feasible from a thermodynamic consideration. Thus, the possibility of an oxidative quenching mechanism is excluded and the reductive quenching of IrPS by TEOA is more favorable in our photocatalytic system (Fig. S8, ESI†).
Based on the aforementioned mechanistic studies, we proposed a plausible reaction mechanism for the photocatalytic CO2 reduction mediated by 3 (Fig. 3). In the first step, irradiation results in the loss of a carbonyl ligand from the initial iron complex to form 10. Simultaneously, IrPS is excited to *IrPS and then quenched by TEOA to give IrPS−. Subsequently, 2 equivalents of IrPS− and 2H+ react with 10 and the hydride Fe(II) complex 9 is formed. This is a key reaction intermediate since it is directly responsible for the activation of carbon dioxide and its reduction to carbon monoxide. In fact, when a solution of complex 9 in NMP is exposed to 1 atm of CO2 under visible-light irradiation, the transformation to complex 3 is observed using 13C NMR spectroscopy (Fig. S9, ESI†). Accordingly, CO2 reacts under irradiating conditions with complex 9, resulting in the formation of an intermediate containing an Fe–COOH moiety, which, through a dehydration reaction using the –OH group of the ligand as a local proton source, regenerates the original complex 3.
In conclusion, we have disclosed for the first time the use of cyclopentadienone iron complexes as highly selective catalysts in the photochemical reduction of CO2 to carbon monoxide. In particular, complexes 1 and 3 showed excellent activities (initial TOF up to 22.2 min−1), which represents the highest value for any non-noble metal-based molecular homogeneous catalyst in this photoredox reaction. Based on results from operando FTIR measurements, it was possible to determine the nature of the catalytic intermediates formed in solution and to propose a plausible reaction mechanism. Presumably, the role of the second-coordination-sphere of the catalyst is crucial to understand the high activity of the system through the enhancement of the rate of the dehydration step, which usually is considered the rate-limiting step in this photocatalytic reaction.19
This work has been supported by the State of Mecklenburg-Vorpommen, the European Union (European Social Funds, ESF) within the project “CADIAC” and the BMBF. We thank Dr Robert Francke for his support in the electrochemical measurements. We also thank C. Fischer, S. Bucholz, S. Schareina, N. Rudat, Dr W. Baumann and DI A. Koch (all LIKAT) for their analytical and technical support. P. G. A. and A. R.-H. acknowledge financial support from the Alexander von Humboldt Foundation and the DAAD, respectively.
Notes and references
- (a) A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621 CrossRef CAS PubMed; (b) Y. Amao, ChemCatChem, 2011, 3, 458 CrossRef CAS; (c) E. B. Cole and A. B. Bocarsly, in Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, 2010, p. 291 Search PubMed.
- (a) J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990 CrossRef CAS; (b) J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536 RSC.
- (a) S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Chem. Soc. Rev., 2014, 43, 7501 RSC; (b) C. D. Windle and R. N. Perutz, Coord. Chem. Rev., 2012, 256, 2562 CrossRef CAS; (c) T. Yui, Y. Tamaki, K. Sekizawa and O. Ishitani, Top. Curr. Chem., 2011, 303, 151 CrossRef CAS PubMed; (d) A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983 CrossRef CAS PubMed.
- (a) G. Sahara and O. Ishitani, Inorg. Chem., 2015, 54, 5096 CrossRef CAS PubMed; (b) A. Nakada, K. Koike, T. Nakashima, T. Morimoto and O. Ishitani, Inorg. Chem., 2015, 54, 1800 CrossRef CAS PubMed; (c) A. Nakada, K. Koike, K. Maeda and O. Ishitani, Green Chem., 2016, 18, 139 RSC; (d) E. Kato, H. Takeda, K. Koike, K. Ohkubo and O. Ishitani, Chem. Sci., 2015, 6, 3003 RSC; (e) C. Liu, K. D. Dubois, M. E. Louis, A. S. Vorushilov and G. Li, ACS Catal., 2013, 3, 655 CrossRef CAS; (f) H. Takeda, K. Koike, T. Morimoto, H. Inumaru and O. Ishitani, in Adv. Inorg. Chem., ed. E. Rudi van and S. Grażyna, Academic Press, 2011, vol. 63, p. 137 Search PubMed; (g) J. Schneider, K. Q. Vuong, J. A. Calladine, X.-Z. Sun, A. C. Whitwood, M. W. George and R. N. Perutz, Inorg. Chem., 2011, 50, 11877 CrossRef CAS PubMed; (h) J. Ettedgui, Y. Diskin-Posner, L. Weiner and R. Neumann, J. Am. Chem. Soc., 2011, 133, 188 CrossRef CAS PubMed; (i) H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023 CrossRef CAS PubMed; (j) H. Tsubaki, A. Sekine, Y. Ohashi, K. Koike, H. Takeda and O. Ishitani, J. Am. Chem. Soc., 2005, 127, 15544 CrossRef CAS PubMed; (k) B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326 CrossRef CAS PubMed; (l) H. Hori, F. P. A. Johnson, K. Koike, K. Takeuchi, T. Ibusuki and O. Ishitani, J. Chem. Soc., Dalton Trans., 1997, 1019 RSC.
- (a) A. Rosas-Hernández, H. Junge and M. Beller, ChemCatChem, 2015, 7, 3316 CrossRef; (b) Y. Kuramochi, J. Itabashi, K. Fukaya, A. Enomoto, M. Yoshida and H. Ishida, Chem. Sci., 2015, 6, 3063 RSC; (c) Y. Kuramochi, K. Fukaya, M. Yoshida and H. Ishida, Chem. – Eur. J., 2015, 21, 10049 CrossRef CAS PubMed; (d) Y. Kuramochi, M. Kamiya and H. Ishida, Inorg. Chem., 2014, 53, 3326 CrossRef CAS PubMed; (e) Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673 CrossRef CAS PubMed.
- (a) R. O. Reithmeier, S. Meister, A. Siebel and B. Rieger, Dalton Trans., 2015, 44, 6466 RSC; (b) R. O. Reithmeier, S. Meister, B. Rieger, A. Siebel, M. Tschurl, U. Heiz and E. Herdtweck, Dalton Trans., 2014, 43, 13259 RSC; (c) S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988 CrossRef CAS PubMed.
- (a) L. Chen, Z. Guo, X.-G. Wie, C. Gallenkamp, J. Bonin, E. Anxolabéhère-Mallart, K.-C. Lau, T.-C. Lau and M. Robert, J. Am. Chem. Soc., 2015, 137, 10918 CrossRef CAS PubMed; (b) S. L.-F. Chan, T. L. Lam, C. Yang, S.-C. Yan and N. M. Cheng, Chem. Commun., 2015, 51, 7799 RSC; (c) J. Bonin, M. Robert and M. Routier, J. Am. Chem. Soc., 2014, 136, 16768 CrossRef CAS PubMed; (d) J. Bonin, M. Chaussemier, M. Robert and M. Routier, ChemCatChem, 2014, 6, 3200 CrossRef CAS; (e) V. S. Thoi, N. Kornienko, C. G. Margarit, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 14413 CrossRef CAS PubMed; (f) T. Dhanasekaran, J. Grodkowski, P. Neta, P. Hambright and E. Fujita, J. Phys. Chem. A, 1999, 103, 7742 CrossRef CAS; (g) J. Grodkowski, D. Behar, P. Neta and P. Hambright, J. Phys. Chem. A, 1997, 101, 248 CrossRef CAS.
- (a) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170 CrossRef CAS PubMed; (b) X. Lu, Y. Zhang, P. Yun, M. Zhang and T. Li, Org. Biomol. Chem., 2013, 11, 5264 RSC; (c) G. Bauer and K. A. Kirchner, Angew. Chem., Int. Ed., 2011, 50, 5798 CrossRef CAS PubMed; (d) C. P. Casey and H. Guan, J. Am. Chem. Soc., 2007, 129, 5816 CrossRef CAS PubMed.
- (a) S. Zhou, S. Fleischer, H. Jiao, K. Junge and M. Beller, Adv. Synth. Catal., 2014, 356, 3451 CrossRef CAS; (b) T. Yan, B. L. Feringa and K. Barta, Nat. Commun., 2014, 5, 5602 CrossRef CAS PubMed; (c) S. Moulin, H. Dentel, A. Pagnoux-Ozherelyeva, S. Gaillard, A. Poater, L. Cavallo, J.-F. Lohier and J.-L. Renaud, Chem. – Eur. J., 2013, 19, 17881 CrossRef CAS PubMed; (d) A. Pagnoux-Ozherelyeva, N. Pannetier, M. D. Mbaye, S. Gaillard and J.-L. Renaud, Angew. Chem., Int. Ed., 2012, 51, 4976 CrossRef CAS PubMed; (e) S. Zhou, S. Fleischer, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5120 CrossRef CAS PubMed.
- F. Zhu, L. Zhu-Ge, G. Yang and S. Zhou, ChemSusChem, 2015, 8, 609 CrossRef CAS PubMed.
- M. Rakowski DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62 RSC.
- (a) C. Greco, M. Bruschi, L. De Gioia and U. Ryde, Inorg. Chem., 2007, 46, 5911 CrossRef CAS PubMed; (b) Y. Nicolet, A. L. de Lacey, X. Vernède, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596 CrossRef CAS PubMed; (c) H.-J. Fan and M. B. Hall, J. Am. Chem. Soc., 2001, 123, 3828 CrossRef CAS PubMed.
- (a) I. Siewert, Chem. – Eur. J., 2015, 21, 15078 CrossRef CAS PubMed; (b) O. S. Wenger, Acc. Chem. Res., 2013, 46, 1517 CrossRef CAS PubMed.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef CAS.
- (a) A. Noble, S. J. McCarver and D. W. C. MacMillan, J. Am. Chem. Soc., 2015, 137, 624 CrossRef CAS PubMed; (b) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433 CrossRef CAS PubMed; (c) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437 CrossRef CAS PubMed; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- P. G. Alsabeh, A. Rosas-Hernández, E. Barsch, H. Junge, R. Ludwig and M. Beller, Catal. Sci. Technol., 2016, 6, 3623 CAS.
- (a) C. Kutal, A. J. Corbin and G. Ferraudi, Organometallics, 1987, 6, 553 CrossRef CAS; (b) C. Kutal, M. A. Weber, G. Ferraudi and D. Geiger, Organometallics, 1985, 4, 2161 CrossRef CAS.
- H.-J. Knölker, E. Baum, H. Goesmann and R. Klauss, Angew. Chem., Int. Ed., 1999, 38, 2064 CrossRef.
- (a) C. Riplinger and E. A. Carter, ACS Catal., 2015, 5, 900 CrossRef CAS; (b) Y. C. Lam, R. J. Nielsen, H. B. Gray and W. A. Goddard, ACS Catal., 2015, 5, 2521 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, quantum yield calculations and emission-quenching experiments procedure. See DOI: 10.1039/c6cc01671e |
| This journal is © The Royal Society of Chemistry 2016 |