
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamic disulfide metathesis induced by ultrasound†
Urs F.
Fritze
and
Max
von Delius
*
Friedrich-Alexander University Erlangen-Nürnberg (FAU), Department of Chemistry and Pharmacy & Interdisciplinary Center of Molecular Materials (ICMM), Henkestrasse 42, 91054 Erlangen, Germany. E-mail: max.vondelius@fau.de
First published on 29th March 2016
Abstract
The reversible metathesis of disulfide bonds is generally induced by a combination of a reducing agent and base or by irradiation with ultraviolet light. Here we report that ultrasound irradiation is suitable for generating clean equilibrium mixtures of disulfides within one hour or one day, depending on the sonication source. Preliminary mechanistic investigations suggest that the solvent plays an active role in producing initiator radicals.
The dynamic chemistry of disulfide bonds plays a vital part in cysteine-containing proteins,1 and since 20002 it has emerged as one of the most powerful tools in the field of dynamic covalent/combinatorial chemistry (DCC).3
In nature, as well as in most man-made systems, disulfide exchange proceeds by the nucleophilic attack of a free thiolate at the disulfide bond, furnishing a new pair of disulfide and thiolate (Fig. 1a).4 When starting from air-stable disulfides, this type of reaction is somewhat limited in scope, because it requires long equilibration times and a combination of a reducing agent and base, which is not always compatible with the application one has in mind for a dynamic library. Progress towards alleviating these limitations was recently reported by several groups. Ramström and coworkers developed a Brønsted base-free, phosphine-catalyzed method for disulfide metathesis,5a while Belenguer, Friščić and Sanders described a mechanochemical approach that requires a base, but no reducing agent.5b,c The group of Pittelkow found that diselenides readily exchange at neutral pH, which in turn can be used to catalyze disulfide exchange.5d
 | ||
| Fig. 1 Disulfide metathesis: (a and b) conventional methods; (c) ultrasound-induced. | ||
An alternative approach for facilitating disulfide metathesis is based on the homolytic cleavage of the disulfide bond and a subsequent radical chain reaction (Fig. 1b). For disulfides, this process can be induced by irradiation with harsh ultraviolet (UV) light,6 but for diselenides Xu and coworkers have recently demonstrated that visible light is suitable for generating sufficient initiator radicals for an efficient exchange reaction.7a Interestingly, the authors were able to use this process for a spatially controlled “healing” of damaged areas in diselenide-based polymers.7b These studies illustrate that unprecedented methods for generating dynamic libraries ultimately translate into new applications of dynamic systems.
Here we report that the metathesis of two or more disulfides can be induced by simple sonication of a chloroform solution of the starting materials (Fig. 1c). While the use of a sonotrode leads to remarkably short equilibration times, it is also possible to carry out the reaction in an ultrasonic bath, a piece of equipment that is available in most chemical laboratories.
Researchers in the field of polymer mechanochemistry8 routinely make use of intense ultrasound for transmitting mechanical force onto functional groups (mechanophores). This process is not applicable to small molecules, however, because it is based on cavitational shear forces that only affect macromolecules (Mw > 30 kDa). Against this background, we were surprised when we discovered that small-molecule disulfides readily exchanged when subjected to ultrasound.
As shown in Fig. 2, irradiation of benzylic disulfides AA and BB for one hour with ultrasound (sonotrode) furnished mixed disulfide AB in a statistical mixture with AA and BB. We confirmed that the reaction outcome corresponds indeed to an equilibrium distribution by also using the established methods for the same reaction (Table 1, entries 6 and 7).
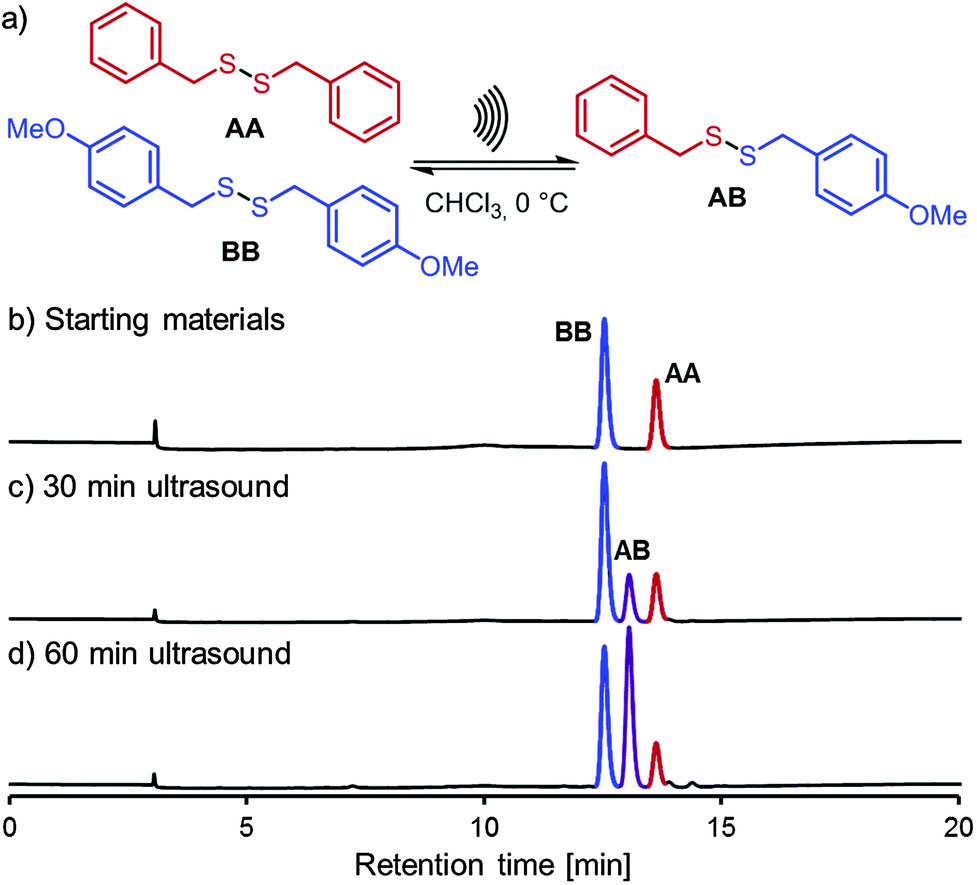 | ||
| Fig. 2 (a) Ultrasound-induced disulfide metathesis. Reaction conditions: AA (10 mM) and BB (10 mM), sonotrode irradiation (25% amplitude, pulse sequence 0.5 s on, 1.0 s off), CHCl3, 0 °C. (b) Analytical HPLC (70% → 100% gradient acetonitrile/water, 0.3 mL min−1, Ascentis amide column) of starting materials AA and BB. (c) Analytical HPLC after 30 minutes of sonication. (d) Analytical HPLC after 60 minutes of sonication. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Method | Conc. [mM] | Solvent | Timea | AB [%] |
| a Minimum equilibration times. b Reaction progress was monitored by analytical HPLC (area percent). c 13 mm titanium tip, pulsing at 20 kHz with 25% of maximal amplitude, 0.5 s pulse and 1.0 s break, 0 °C (ice bath). d Investigated solvents: acetone, tetrahydrofuran (THF), acetonitrile, dichloromethane, 1,1,2,2-tetrachloroethane, chloroform/water (emulsion). e AB not detected. f 1 h sonotrode irradiation, 44 h equilibration at RT. g Reaction conducted in a sealed glass tube, non-stop sonication, 27 °C. h DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene; DTT: 1,4-dithio-D-threitol. i 400 W Hg lamp. j 300 W, 170 °C, 11 bar. k DBPO: dibenzoyl peroxide. | |||||
| 1 | Sonotrodec | 10 | CHCl3 | 1 h | 48 |
| 2 | Sonotrodec | 10 | CHBr3 | 45 s | 50 |
| 3 | Sonotrodec | 10 | Otherd | 1 h | <5e |
| 4 | CHI3 additive (100%) | 10 | THF | 1 hf | 49 |
| 5 | Ultrasonic bath | 10 | CHCl3 | 24 h | 53 |
| 6 | 10% DBU/DTTh | 10 | CHCl3 | 6 d | 49 |
| 7 | UV lighti | 10 | CHCl3 | 3 h | 49 |
| 8 | 110 °C | 10 | CHCl3 | 5 h | —e |
| 9 | Microwave, 170 °Cj | 10 | CHCl3 | 1 h | <5 |
| 10 | 5% DBPO, 120 °Ck | 10 | CHCl3 | 5 h | 47 |

Intrigued by this initial finding, we proceeded to investigate a range of reaction parameters for the reaction of AA with BB. A screening of various organic solvents (Table 1, entry 3 and the ESI†) revealed that chloroform is uniquely suited for this reaction: even similar chlorinated solvents, such as dichloromethane or 1,1,2,2-tetrachloroethane, failed to produce significant quantities of product AB. Only bromoform (CHBr3) could be identified as a second excellent, albeit more toxic, solvent for the reaction (entry 2), providing a first indication that a radical mechanism might be at hand.‡ The use of an entirely different solvent (tetrahydrofuran), an important aspect regarding possible applications of this method, turned out to be possible when a stoichiometric amount of additive iodoform (CHI3) was added to the reaction mixture (entry 4).
Because sonotrodes are not widely available in chemical laboratories, we tested whether the reaction would also take place in an ultrasonic bath. We were pleased to find that a clean equilibrium mixture could be obtained after irradiating a sealed flask in a temperature-controlled ultrasonic bath for 24 hours (entry 5). To make sure that this result was not due to the make-up of one particular piece of equipment, we tested three different ultrasonic baths from two manufacturers and obtained identical reaction outcomes in all three cases (see the ESI†).
In addition to the conventional protocols for disulfide metathesis (Table 1, entries 6 and 7), we conducted three control experiments (entries 8–10), designed to provide qualitative evidence for either a thermal (“hot spots”) or a radical-initiated reaction mechanism. Heating the reaction mixture at 110 °C in a sealed tube or at 170 °C under microwave irradiation led to only negligible formation of product AB, whereas the use of radical initiator DBPO at 120 °C proved to be effective, pointing towards a radical mechanism.
Studies on the scope of the described ultrasound-induced disulfide exchange are summarized in Table 2. Both benzylic and aliphatic compounds are suitable substrates (entries 1, 3 and 4), whereas aromatic disulfides react only sluggishly (entry 2). This reactivity trend is complementary to the classic base-induced method, in which aromatic thiols/disulfides are most commonly used.3 Our preliminary studies confirm that ether and alcohol functions are tolerated (substrates BB and EE), whereas a Boc-protected cystine substrate (substrate FF, entry 5) led to the formation of several side products, indicating that this protocol is not suitable for peptide substrates. Finally, a ternary mixture of substrates AA, BB and EE was found to lead to clean exchange (entry 6).
|
|
|||
|---|---|---|---|
| Entry | Substratesa | Reaction outcomeb | Equilibration time |
| a Reaction conditions: equimolar quantities of the substrates (10 mM in CHCl3), Suslick cell under an Ar atmosphere, sonotrode irradiation as specified in Table 1. b All reactions were monitored by HPLC(MS) or NMR spectroscopy (see the ESI). c Values in parentheses correspond to reactions under “ultrasonic bath” conditions. | |||
| 1 | AA + BB | Clean exchange (HPLC) | 1 h (24 h)c |
| 2 | BB + CC | Sluggish reactivity (HPLC) | 1 h |
| 3 | BB + DD | Clean exchange (NMR) | 3.5 h (22 h)c |
| 4 | BB + EE | Clean exchange (NMR) | 1 h (17 h)c |
| 5 | BB + FF | Side product formation (HPLC) | 3 h |
| 6 | AA + BB + EE | Clean exchange (HPLC), formation of 6 compounds | 1.5 h |
To gain a deeper mechanistic understanding, we monitored the reaction between AA and BB under different reaction conditions (Fig. 3). The kinetic profiles shown in Fig. 3a provide two insights: (i) the formation of product AB follows a sigmoidal curve, indicating the presence of an induction period. (ii) The reaction rate is the highest for the most dilute reaction.§ Both these observations can be explained by a radical chain reaction whose initiator radicals are provided via sonolysis of the solvent. This rationale is strongly supported by a study of Riesz that described the sonochemical generation and subsequent trapping of ˙CHCl2 radicals derived from chloroform.9
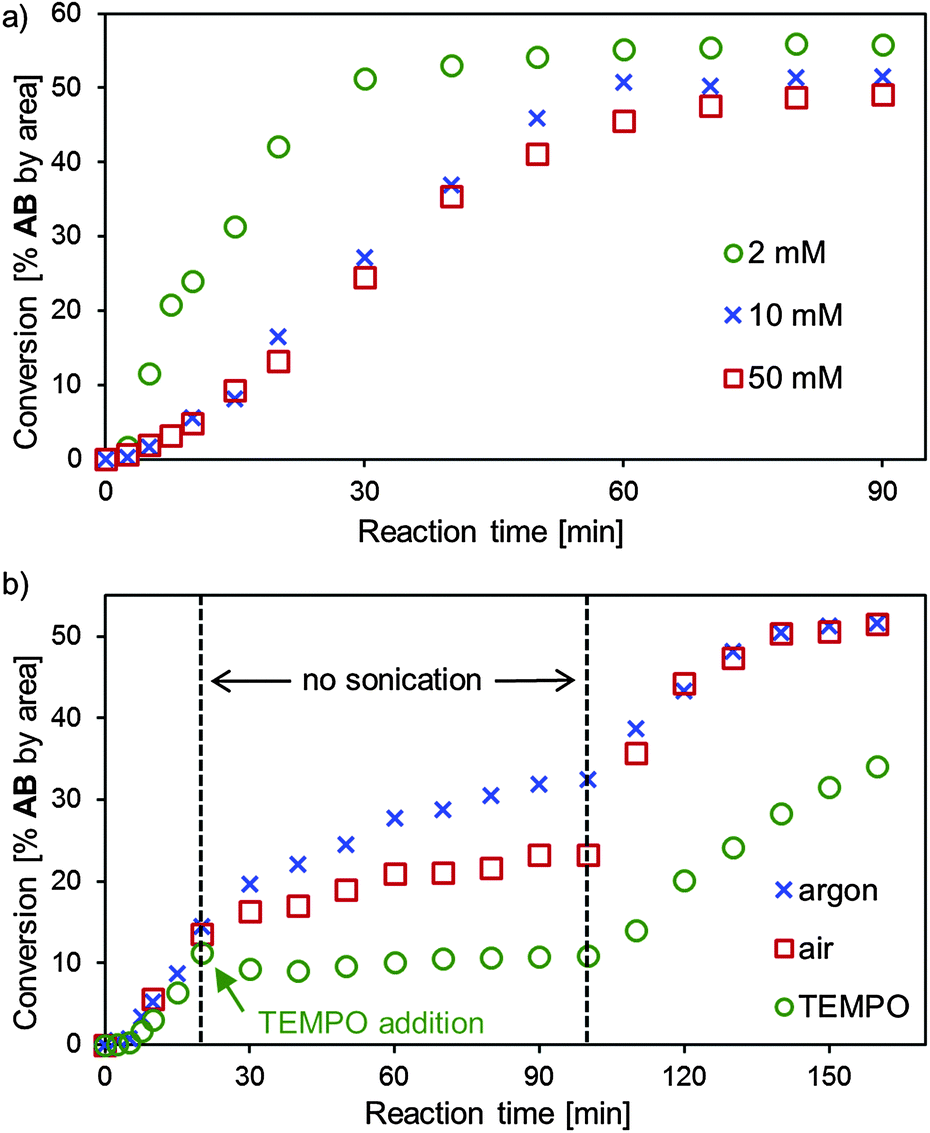 | ||
| Fig. 3 HPLC monitoring of the metathesis of disulfides AA and BB under sonotrode conditions in chloroform. (a) Influence of starting material concentration (2 mM, 10 mM, 50 mM). (b) Influence of switching off the ultrasound source (period: 20–100 min) in argon, air or in argon with addition of 40 mM of radical scavenger TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxyl) after 20 min. | ||
Further evidence for this mechanistic scenario is provided by the data shown in Fig. 3b. As one would expect for a radical chain reaction that has proceeded beyond the initial induction period, switching the ultrasound source off after 20 minutes does not stop the reaction. Whether the reaction is carried out in argon or air does not significantly affect the rate while ultrasound irradiation generates sufficient radicals, but during the “resting period” (Fig. 3b, 20–100 minutes) dioxygen supposedly leads to quenching of persistent radicals, thus slowing down the reaction. Addition of stoichiometric radical scavenger TEMPO after 20 minutes inhibits the reaction,¶ both in the presence and absence of ultrasound irradiation, providing further support for a radical mechanism.10
In conclusion, we have discovered a sonochemical method for generating dynamic mixtures of disulfides. Although this reaction is somewhat limited regarding solvents and substrates, we believe that it could be a practical alternative to the existing methods, particularly in scenarios where short equilibration times, base-free reaction conditions or a new vector for dynamic covalent orthogonality11 are needed.
This work was supported by the Deutsche Forschungs-gemeinschaft (Emmy-Noether grant DE1830/2-1) and the Evangelisches Studienwerk Villigst (PhD fellowship to U. F. F.). We thank Dr Konstantin Amsharov for providing access to a photoreactor.
Notes and references
-
(a) C. B. Anfinsen, Science, 1973, 181, 223 CAS
; (b) P. Dopieralski, J. Ribas-Arino, P. Anjukandi, M. Krupicka, J. Kiss and D. Marx, Nat. Chem., 2013, 5, 685 CrossRef CAS PubMed
- S. Otto, R. L. E. Furlan and J. K. M. Sanders, J. Am. Chem. Soc., 2000, 122, 12063 CrossRef CAS
-
(a) P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652 CrossRef CAS PubMed
- R. Singh and G. M. Whitesides, J. Am. Chem. Soc., 1990, 112, 1190 CrossRef CAS
-
(a) R. Caraballo, M. Rahm, P. Vongvilai, T. Brinck and O. Ramström, Chem. Commun., 2008, 6603 RSC
- F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587 CrossRef PubMed
-
(a) S. Ji, W. Cao, Y. Yu and H. Xu, Angew. Chem., Int. Ed., 2014, 53, 6781 CrossRef CAS PubMed
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181 CrossRef CAS PubMed
- V. Misik and P. Riesz, J. Phys. Chem., 1994, 98, 1634 CrossRef CAS
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2016, 55, 58 CrossRef CAS PubMed
-
(a) A. Wilson, G. Gasparini and S. Matile, Chem. Soc. Rev., 2014, 43, 1948 RSC
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental procedures and spectroscopic data. See DOI: 10.1039/c6cc02034h |
| ‡ In CHBr3, significant formation of side products was observed after extended reaction times (e.g. one hour), indicating that the reaction should be terminated after equilibrium is reached. In contrast to CHI3, CHBr3 was not a suitable additive in solvent THF (see the ESI,† page S30). |
| § We found that the reaction kinetics vary to some extent between different batches of solvent. However, the sigmoidal shape of the curves and the general trends remain unaffected. |
| ¶ We were unable to identify possible adducts of ˙CHCl2 radicals to the starting materials, which could be due to the fact that only small quantities of such compounds are formed. |
| This journal is © The Royal Society of Chemistry 2016 |