
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyaromatic N-heterocyclic carbene ligands and π-stacking. Catalytic consequences
Eduardo
Peris
Institute of Advanced Materials (INAM), Universitat Jaume I, Av. Vicente Sos Baynat, s/n, 12071 Castellón, Spain. E-mail: eperis@uji.es
First published on 4th April 2016
Abstract
In the course of our most recent research, we demonstrated how homogeneous catalysts with polyaromatic functionalities possess properties that clearly differ from those shown by analogues lacking these polyaromatic systems. The differences arise from the ability of the polyaromatic groups to afford non-covalent interactions with aromatic molecules, which can either be substrates in a homogeneous catalysed reaction, or the same catalysts to afford self-assembled systems. This article summarizes all our efforts toward understanding the fundamental effects of π-stacking interactions in homogenous catalysis, particularly in those cases where catalysts bearing polyaromatic functionalities are used. The study reveals several important implications regarding the influence of ligand–ligand interactions, ligand–additive interactions, and ligand–substrate interactions, in the performance of the catalysts used. In particular, the electronic properties of ligands with fused polyconjugated systems, are modified if molecules with π-stacking abilities are added, via a ligand–additive interaction. Also, the kinetics of the reactions in which aromatic substrates and catalysts with polyaromatic ligands are used, are strongly influenced by the self-association of the catalysts and by the non-covalent interaction between the catalyst and the aromatic substrates. The nature and the magnitude of these supramolecular interactions were unveiled by using host–guest chemistry methods applied to organometallic catalysis. Finally, non-covalent interactions afford a very convenient approach for the immobilization of catalysts decorated with polyaromatic systems onto the surfaces of graphene derivatives, hence affording an easy yet extremely effective way to support catalysts and facilitate recycling. The results given have fundamental implications in the design of future catalysts containing rigid polyaromatic systems, and may inspire future researchers in the design of improved homogeneous catalysts, by taking into account that the activities of the metal complexes are strongly modified by supramolecular interactions.
 Eduardo Peris | Eduardo Peris graduated in Chemistry in 1988 in Valencia. He received his PhD Degree in Chemistry (1991) in the Universidad de Valencia. In 1994 he joined Robert Crabtree's group at Yale University, where he stayed for two years. In October 1995 he moved to the Universitat Jaume I (Castellón-Spain) where he is currently Professor of Inorganic Chemistry. In 2012 he was awarded the ‘Spanish Royal Society of Chemistry’ award in the field of Inorganic Chemistry. In September 2014, he became President of the Spanish Organometallic Chemistry Division (GEQO), from the Spanish Royal Society of Chemistry (RSEQ). |
1. Introduction
The chemical properties of a complex can be tuned by the set of ligands that are bound to the metal. Consequently, understanding the properties of ligands, may be used for the design of complexes for specific purposes. In the field of homogeneous catalysis many groups focus the objective of their research on the design of sophisticated ligands that go beyond their traditional role of providing well-defined stereoelectronic properties.1 This interest is based on the fact that the properties of a metal complex can be modified if a ligand carries an additional functional group that undergoes ligand-based reactivity as a result of an external stimulus. Typical functionalities that may influence the properties of the complex include, proton responsive functional groups, hydrogen bonding sites, redox-sensitive moieties and photoresponsive systems, among others. Complementary to this type of research, many efforts have also been devoted to finding ways to profit from the weak reversible non-covalent interactions between pre-designed molecular building blocks for the development of more effective catalysts. Hence the term ‘supramolecular catalysis’ was coined in 2008,2 and since then a number of very important review articles have appeared.3Although the building of a supramolecular catalyst needs to incorporate a high degree of design, non-covalent interactions are less predictable, and for this reason the supramolecular effects that influence the catalytic performance of a catalyst are often recognized post-factum. The understanding of the nature and strength of the interaction between the catalyst and the substrate is one of the main challenges that may help to improve the basis of the design of supramolecular catalysts. Current studies in the field have focused primarily on molecular recognition phenomena, trying to determine the thermodynamic stabilization of the aggregates formed by the bonding between complexes and substrates.4 Non-covalent interactions play an important role in catalysis by lowering the energy of the transition states. These forces are responsible for the accelerations and selectivities induced by enzymes. In the last decade these interactions have been successfully exploited in organocatalysis with small organic molecules,4 but have been rarely studied in the area of organometallic catalysis.5
Supramolecular chemistry has often been considered as a bridge between homogeneous catalysis and biocatalysis.3h In biocatalysis, a term that is often used is ‘allosteric’, a word that derives from the Greek root ‘allo’, meaning ‘the other’. In an enzyme an alteration of the conformation of the catalyst indirectly modifies the reactivity of the catalytic active site, hence this indirect mode of action is the most widely accepted meaning of ‘allosteric’.6 In molecular catalysis, an event taking place at one structural part of the catalyst may cause an effect in the catalytic active site, much like the behavior of a telecommunications network in which receivers and transceivers communicate with each other remotely, and therefore the term allosteric may be equally appropriate, although is less commonly used. In an excellent review article by Raynal and co-workers, interactions that are typically observed in supramolecular catalysis are classified in three fundamental groups, and these are: (i) interactions between ligands, (ii) interactions between a ligand and an additive, and (iii) interactions between ligands and substrates. These are depicted in Scheme 1. In the ligand–ligand interaction case, two molecules of catalyst non-covalently interact forming a dimer, thus the catalytic activity of the dimer is different compared to the activity of the related monomer. In the ligand–additive interaction an additive is added to the vessel in which the catalytic reaction is taking place, so that an interaction between the additive and a ligand of the catalyst modifies the electronic and steric properties of the ligand, hence changing the catalytic properties of the complex. Finally, the substrate–ligand interaction involves non-covalent interactions of the substrate and the ligand, thus placing the substrate in a privileged position from which it can more easily interact with the metal. Other types of supramolecular effects in catalysis are based on the encapsulation of the substrate or the catalyst in pre-defined cages,3f,i spheres, baskets,3a clusters3b or metallo-organic frameworks (MOFs),3d and therefore are more related to host–guest chemistry, in which the ‘confinement effects’ create a different environment around the substrate than in the bulk solvent,3f hence the substrate has to adjust to the size and shape of the cavity.
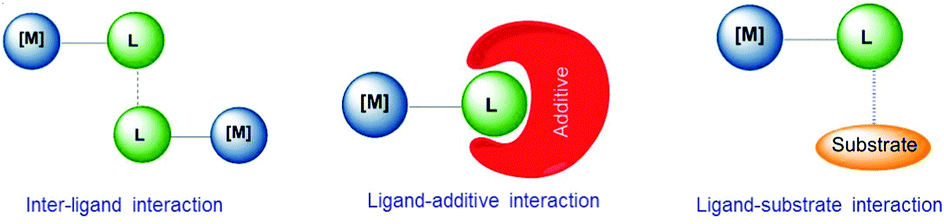 | ||
| Scheme 1 Types of supramolecular effects in homogeneous catalysis. | ||
The aim of this perspective article is to present our recent research in the design of NHC-based supramolecular catalysts. Our approach to this specific area of homogeneous catalysis began progressively by studying the catalytic behavior of a series of metal complexes with NHC ligands bearing extended polyaromatic systems. During the last few years, our research group focused the attention on the preparation of ditopic and tritopic N-heterocyclic carbenes (NHCs) connected by π-spacers, aiming to: (i) find clear examples that allowed us the study of the catalytic cooperativity between the metals comprised in the bi- or tri-metallic complexes, and (ii) prepare heterometallic catalysts in order to facilitate tandem reactions for which each of the metals catalyses a mechanistically distinct reaction, so that the number of single reactions that we could sequentially combine gave us access to highly sophisticated catalytic transformations.7 In the course of our research, we obtained a series of di- and tri-NHCs connected by spacers with extended polyaromatic systems (Scheme 2), which pursued to facilitate the electronic communications between the metals.8 However, we found that, despite the use of these extended π-conjugated linkers, the communication between the metals was negligible in most of the cases.9 Among these ligands, undoubtedly the most widely used one has been the benzobisimidazolylidene (B) first described by Bielawski and co-workers in 2005.10 All other ligands depicted in Scheme 2 were obtained by our research group or in collaborative works. By using the Janus-type bis-NHCs present in A–E, we could modulate the metal-to-metal separation from 6 Å, when we used the simpler triazol-di-ylidene ligand in A,8a to 22.5 Å for the nanosized di-carbene in E.8d Parallel to the development of these facially-opposed di-NHCs, we also prepared two threefold-symmetry tri-NHCs (F and G).11
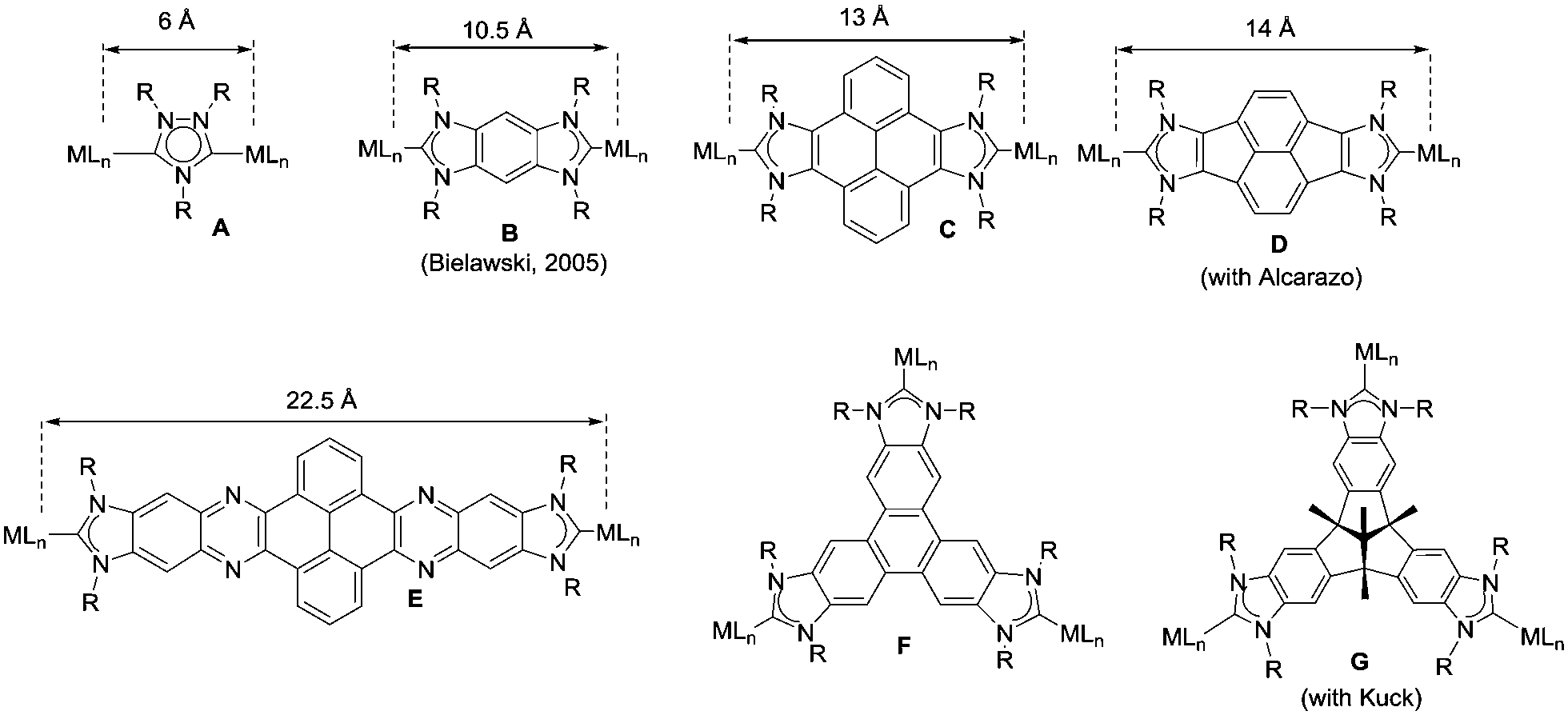 | ||
| Scheme 2 | ||
We found that a common feature of these ligands is that their metal complexes derived often display better catalytic performances than their related monometallic analogues. As will be described in the following sections, rather than attributing these benefits to the multimetallic nature of these complexes, we hypothesized that these catalytic benefits were due to supramolecular effects, because we thought that the extended polyaromatic linkers in our poly-NHC ligands, should be very prone to forming aggregates by π–π-stacking interactions with the substrates used in the catalytic reactions. The following sections of this article will try to disclose a series of arguments devoted to prove why π-stacking interactions should be taken into account both in homogeneous and heterogeneous catalysis, when catalysts with extended polyaromatic systems are used.
2. Catalytic benefits provided by polymetallic complexes with π-extended ligands
As mentioned above, in the course of our research we designed a series of dimetallic and trimetallic complexes with di- and tri-NHC ligands connected by rigid polyaromatic ligands (Scheme 2). In order to relate the effect of the polymetallic nature of a compound with its catalytic performance, it is very important to have a monometallic analogue compound with which a good comparative pattern can be established. We first studied the activities of a series of palladium and gold complexes using the di-NHC ligand present in D (pyracene-linked bisimidazolylidene, pyrabim) and the tri-NHC ligand in F (triphenylene-linked-trisimidazolylidene) and, in order to see if the polymetallic nature of the complexes provided any type of benefit, we compared the activity of these complexes with the activities provided by the palladium and gold complexes coordinated to an acetanaphtene-supported-NHC and a benzoimidazolylidene, respectively, as shown in Scheme 3.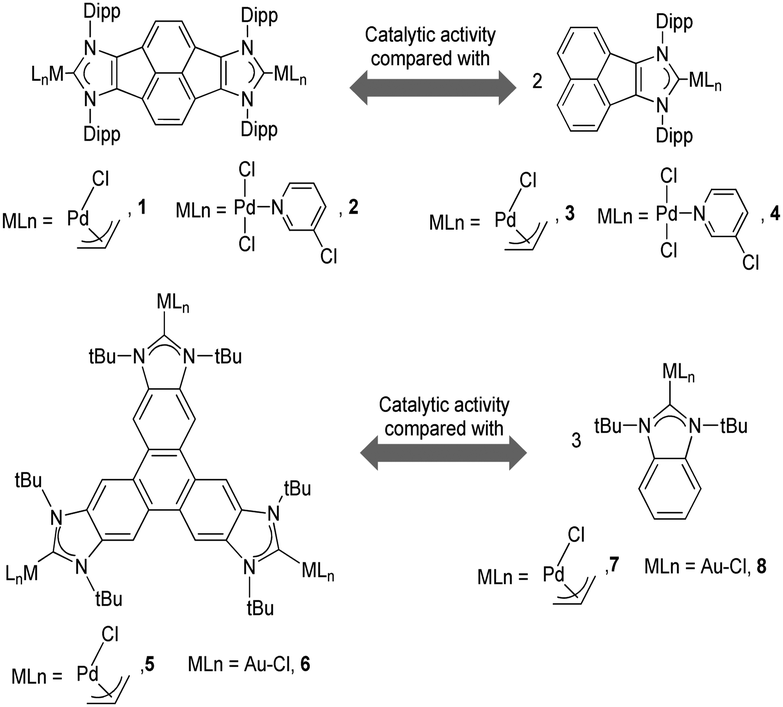 | ||
| Scheme 3 | ||
The catalytic activities of the palladium dimetallic complexes 1 and 2 were compared with the ones shown by the monometallic analogues, 3 and 4, in the Suzuki–Miyaura C–C coupling of arylboronic acids and arylhalides. As an illustrative example of this reaction, Fig. 1 shows the results for the coupling between p-methoxyphenylbromide and p-tolylboronic acid, where it can be clearly seen that the activity of the dimetallic complexes is higher than the one shown by the monometallic analogues. This type of behavior is repeated for all the substrates that we tested in this C–C coupling reaction.12 It is important to mention that, in order to make a proper comparison, the catalyst loadings used had the same substrate/metal ratio, and for this reason we used a double molar concentration of the monometallic complexes compared with the concentration of the dimetallic ones.
 | ||
| Fig. 1 Comparison of the catalytic activity of the dimetallic complexes 1 and 2, with the related monometallic analogues 3 and 4, in the C–C coupling of p-methoxyphenylbromide and p-tolylboronic acid. | ||
We first thought that the dimetallic nature of complexes 1 and 2 was responsible for the higher catalytic performances of these two complexes, compared to the activities given by the monometallic analogues. However, factors not related with the nuclearity of the catalyst may also be at play, and for that reason we performed a more detailed analysis of the results. Despite the topological similarities between the monodentate and bidentate ligands, their steric and electronic properties may not be the same, and this may easily justify the differences in the catalytic performances of their related metal complexes. The electronic properties of the pyracene-linked bis-imidazolylidene and the acetanaphtene-supported-NHC were proven to be very similar,8c according to the comparison of the CO stretching frequencies shown in the infrared spectra of the related IrCl(CO)2 complexes.13 Also, in spite of the connection of the two metals in 1 and 2 by the highly extended π-delocalized system, the cyclic voltammetry analysis of the related rhodium complexes with the pyracene-linked bis-imidazolylidene revealed that the two metals were essentially electronically disconnected,8c therefore the differences in the catalytic activity may not be attributed to the electronic connectivity between the metals. Also, despite the topological analogies of the two ligands, the analysis of the steric properties showed slight differences between them. The calculation of the percent of buried volume (%Vbur),14 performed by using the Sambvca program,15 revealed a %Vbur of 31.3 for the bidentate ligand (D), compared to 34.3 for the monodentate one (acetanaphtene–NHC), hence indicating a slightly higher degree of steric hindrance provided by the latter one. This subtle yet not negligible difference may solely be responsible for the differences observed in their catalytic activities, hence the initial hypothesis that this type of polymetallic complexes may provide benefits in homogeneous catalysis needed further studies.
The trimetallic complexes 5 and 6 (Scheme 3) with the triphenylene-based-tris-NHC ligand may be regarded as the exact combination of three molecules of the related monometallic benzimidazolylidene complexes 7 and 8 and, for this reason, constitutes an excellent opportunity to compare the activity of the trimetallic complexes with their closest related monometallic analogues. For the comparative study of the activities of the palladium complexes (5 and 7), we decided to study the α-arylation of arylalkyl ketones with aryl halides, and the Suzuki–Miyaura coupling between aryl halides and aryl boronic acids. As illustrated in Fig. 2a and b, the trimetallic complex 5 provided better performances than the monometallic complex 7, not only for the substrates shown in the figure, but also for all substrates used in the study.11a In order to find if this was a general behavior, not only shown by palladium-containing complexes but also for catalysts with other metals, we also compared the activity of the gold complexes 6 and 8, in the hydroamination of terminal alkynes with arylamines, where once more, the trimetallic complex (6) afforded higher activities than the monometallic analogue (8) (Fig. 2c). Again, this happened not only for the reaction shown in Fig. 2c, but also for all the substrates that were tested. All the required experiments to discard that any of these reactions could be heterogeneously catalysed were made,16 and hence the parameters governing the activity of the catalysts could be attributed to the nature of the molecular complexes.
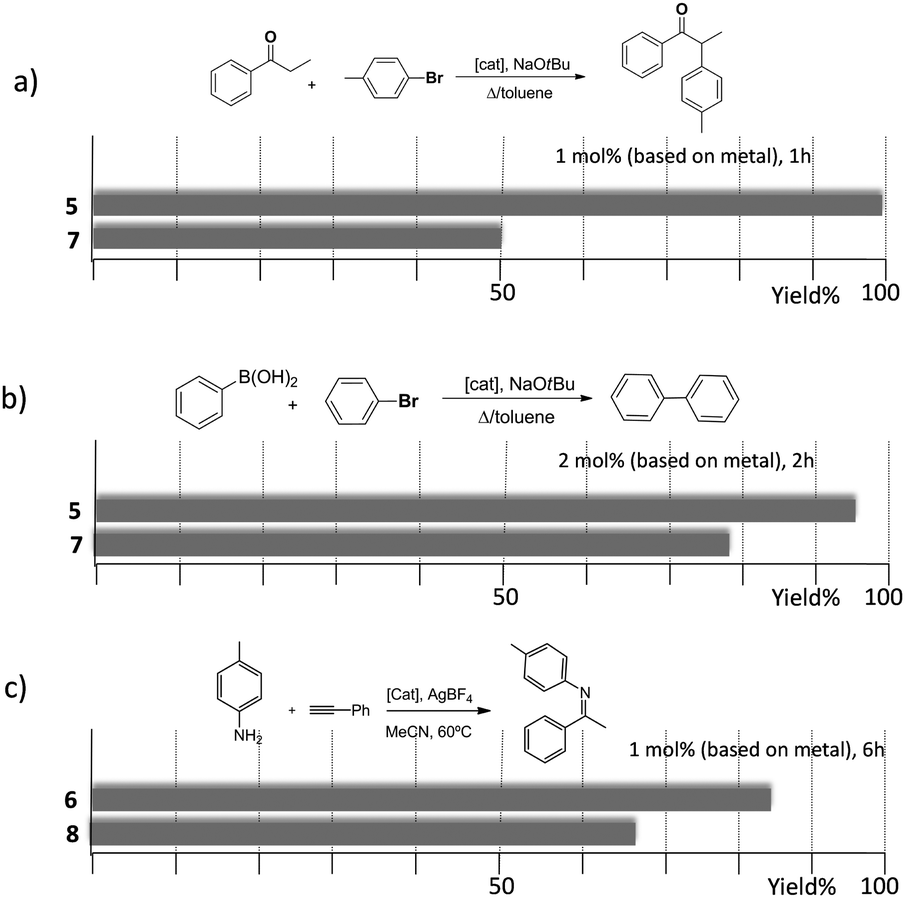 | ||
| Fig. 2 Comparison of the catalytic activities of the trimetallic complexes 5 and 6 with their monometallic analogues 7 and 8. The palladium complexes were tested in the α-arylation of alkylaryl ketones (a) and in the C–C Suzuki–Miyaura coupling of aryl halides with aryl boronic acids (b). The gold complexes were tested in the hydroamination of terminal alkynes with aryl amines (c). | ||
The higher catalytic activities shown by the trimetallic complexes may be due to reasons not directly connected to the trimetallic nature of the compounds. These reasons may be the following: (a) different stabilities of the trimetallic and monometallic complexes, under the reaction conditions of the catalytic experiments, and (b) subtle differences in the steric/electronic properties of the tridentate and monodentate ligands. The stabilities of the two complexes were proven to be similar, by 1H NMR spectroscopy monitoring the decomposition rates of the trimetallic and monometallic complexes in toluene-d8 at 100 °C. The analysis of the steric hindrance of the ligands, indicated that the trimetallic ligand showed a %Vbur = 36.1, while for the monodentate ligand the value was 36.4. The electron-donating character of the two ligands was determined by calculating the Tolman Electronic Parameter (TEP), which was 2057 cm−1 for the tridentate ligand, and 2057.2 cm−1, for the benzimidazolylidene. These findings clearly indicated that the differences in the catalytic performances of the complexes may not be attributed to changes in the steric or electronic properties of the ligands.
Other reasons related to the trimetallic nature of the complex were also considered. The possibility that the catalytic differences could be due to electronic communication between the metals was discarded, because the cyclic voltammetry studies revealed that the three metal centers were electronically decoupled.11b In general, polymetallic complexes may introduce some catalytic benefits if the metals are able to cooperate, but for this cooperation to occur, it is accepted that the metals need to be in close proximity (3.5–6 Å),17 although there are cases in which macromolecular catalysts show some cooperative effects at longer distances, due to the so-called ‘dendrimer effect’.18 This effect is often explained as a consequence of the higher nanolocal concentration of the catalytic active site in dendrimer-like catalysts. In our case the trimetallic complexes afford a high nanolocal concentration of the catalytic active sites within well-defined nanoscopic reaction volumes. In order to investigate this point, we decided to obtain the palladium and gold complexes with a triptycene-based tris-NHC ligand reported by Bielawski and co-workers in 2010.19 We were able to determine the molecular structure of the triptycene-based-tris(NHC) complex of AuCl (9, in Scheme 4), which showed a Au–Au distance of 13.9 Å, very similar to the Au–Au distance in the triphenylene–tris-NHC–AuCl complex 6 (13.4 Å),11a hence the molecular volumes of the trimetallic complexes derived from the triyptycene– and triphenylene–tris(NHCs) are roughly the same. Therefore, since both ligands have similar stereoelectronic properties and provide the same nanolocal concentration of the catalytic active sites, it could be assumed that their catalytic activities would be similar, but in fact the gold and palladium complexes 9 and 10, showed significant lower activities than the related triphenylene-derived-tris(NHC) complexes 5 and 6.11a
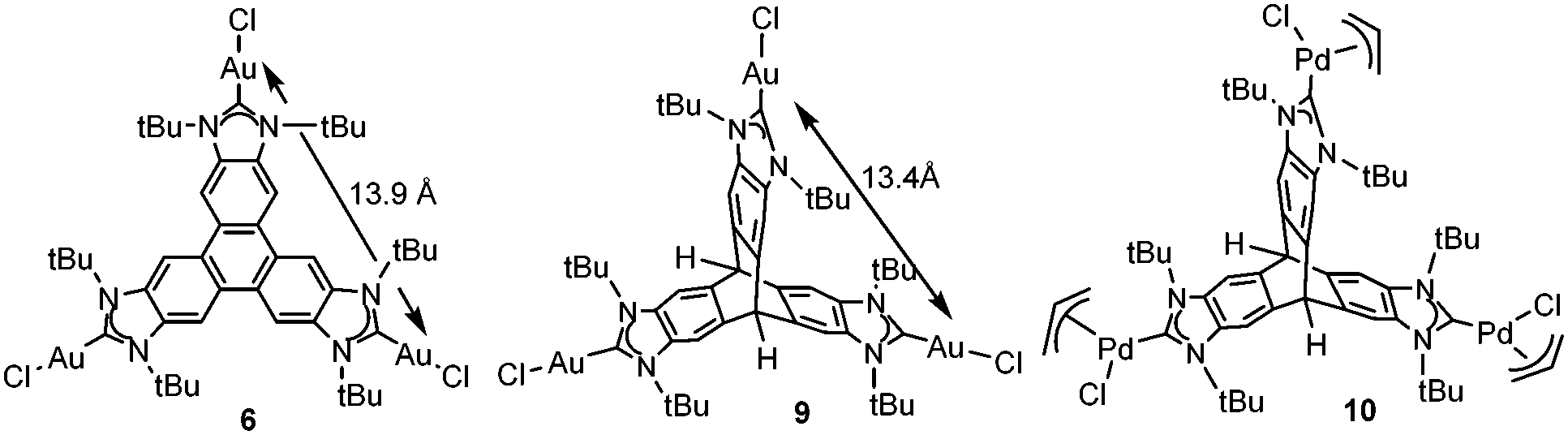 | ||
| Scheme 4 | ||
All these studies experimentally proved that the metal complexes with the triphenylene–tris(NHC) provide significant catalytic benefits compared to the trimetallic and monometallic complexes with formally the same stereoelectronic properties, benefits that are reproduced in three different catalytic reactions facilitated by two different metals. Once we got to this point, we thought that the catalytic benefits provided by the triphenylene-derived-tris(NHC) could be ascribed to supramolecular effects, because the triphenylene fragment should be prone to π–π stacking interactions with the aromatic substrates used in our catalytic experiments.
As a preliminary study about how π-stacking interactions may influence the activity of our catalysts, we decided to repeat the catalytic experiments with added catalytic amounts of pyrene or hexafluorobenzene (10 mol% with respect to the substrates), as external π-stacking additives. We expected that these additives, which are known to promote π-stacking interactions with polyaromatic surfaces, should have an effect on the catalytic performance of our complexes. We observed that the addition of any of the additives produced a significant reduction of the activity of 5 and 6 in all the reactions tested, but the activity of the benzimidazolylidene and triptiycene–tris(NHC) complexes remained practically unchanged.11a
These results prompted us to start a research line devoted to finding ways for studying how π-stacking interactions may influence the activity of catalysts containing rigid polyaromatic functionalities. In this regard, our next step was to obtain a series of di-palladium complexes connected by bi-phenylene-based di(NHCs). The N-substituents were methyl or methyl-pyrene groups. The relevant monometallic palladium complexes were also obtained. The objective of the work was to determine how the presence of the polyaromatic fragment (pyrene) attached to the ligand could influence the activity of the catalyst, and also to compare the activities of the dimetallic complexes with the related monometallic analogues20 (Scheme 5).
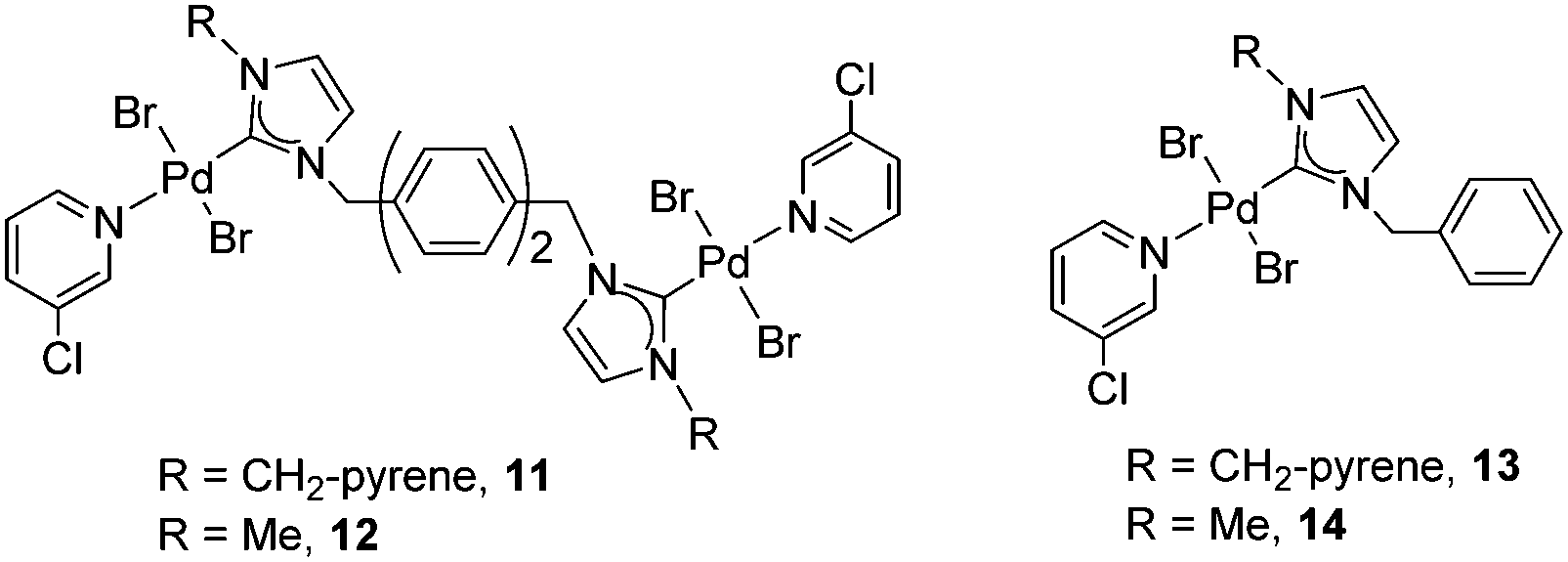 | ||
| Scheme 5 | ||
The catalytic properties of complexes 11–14 were tested in the acylation of arylhalides with hydrocinnamaldehyde, and also in the Suzuki–Miyaura coupling between arylhalides and arylboronic acids. Fig. 3 shows some illustrative examples in which the activity in the C–C Suzuki–Miyaura coupling between bromobenzene and p-tolylboronic acid is compared for all the catalysts.
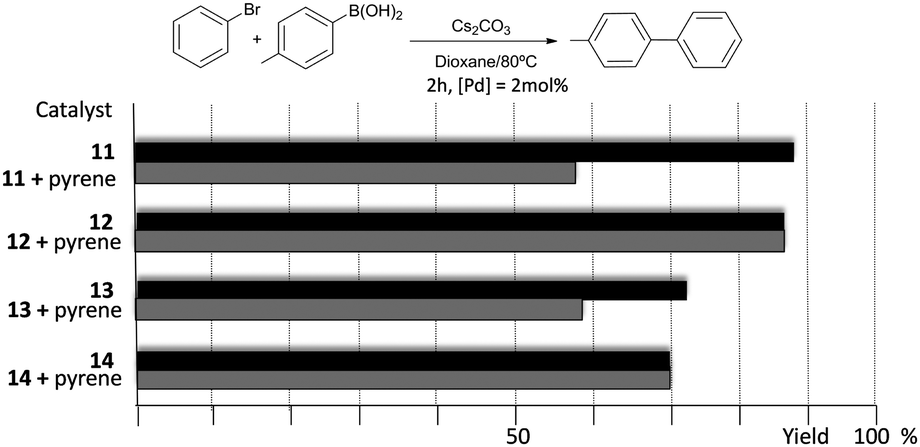 | ||
| Fig. 3 Comparison of the catalytic activities of complexes 11–14 in the C–C Suzuki–Miyaura coupling of bromobenzene and p-tolylboronic acid. Grey bars indicate the activities in the presence of 10 mol% of pyrene. | ||
The results illustrated in Fig. 3 indicate that the complexes bearing the pyrene tag are slightly more active than the ones with a methyl group. This observation becomes evident when the time-dependent reaction profiles are compared.20 The dimetallic complexes are also more active than their monometallic counterparts, yet the most interesting observation arises from the experiments carried out in the presence of catalytic amount of pyrene (10 mol% with respect to substrates), for which a partial inhibition of the catalytic activity is observed, but only for the two complexes bearing the pyrene functionalities (11 and 13), while the activity of the complexes with the N-methyl groups remains unchanged. A similar result was observed when naphthalene was added instead of pyrene, and similar results were observed when other substrates were used.
The interaction between the pyrene-containing monometallic complex 13 with pyrene was also studied by 1H NMR spectroscopy, by titrating a solution of 13 in CDCl3/CD3OD with increasing amounts of pyrene. The spectra clearly showed that the resonances due to the pyrene-tag in 13 were affected by the addition of pyrene, hence suggesting that a non-covalent interaction was taking place between the pyrene-tag and added pyrene.21
3. Ligand-additive interactions in metal complexes with polyaromatic NHC ligands. Nature and magnitude of the interaction
In order to design a more direct approach to the study of the interaction between external additives and the polyaromatic functionalities of NHC ligands, a series of monodentate ligands with extended polyaromatic backbones were obtained (Scheme 6). For comparative purposes, we also included in the study the well-known imidazolylidene and benzimidazolylidene ligands. The initial hypothesis was that the presence of the extended polyaromatic system should make these systems sensitive to the addition of π-stacking additives, such as pyrene or hexafluorobenzene.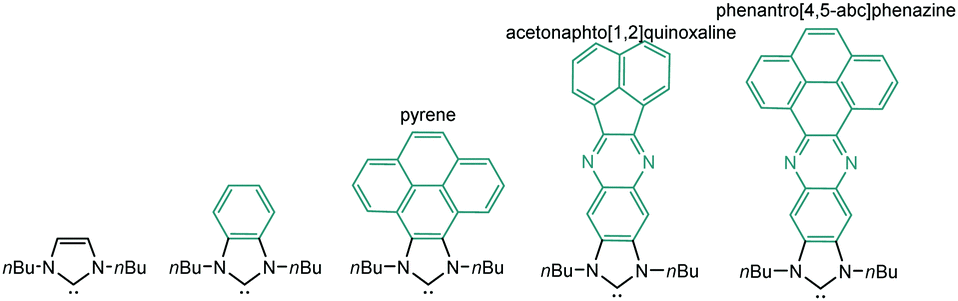 | ||
| Scheme 6 | ||
The π–π-stacking capabilities of the polyaromatic ligands depicted in Scheme 6 are well illustrated by the molecular structures of the metal complexes that were crystallographically characterized. Fig. 4 shows three representative molecular structures of complexes bearing pyrene-,22 acetonaphtoquinoxaline-23 and phenanthrophenazine-derived23 NHC ligands. In all three cases, aggregates formed by the antiparallel π-stacking interaction between two molecules.
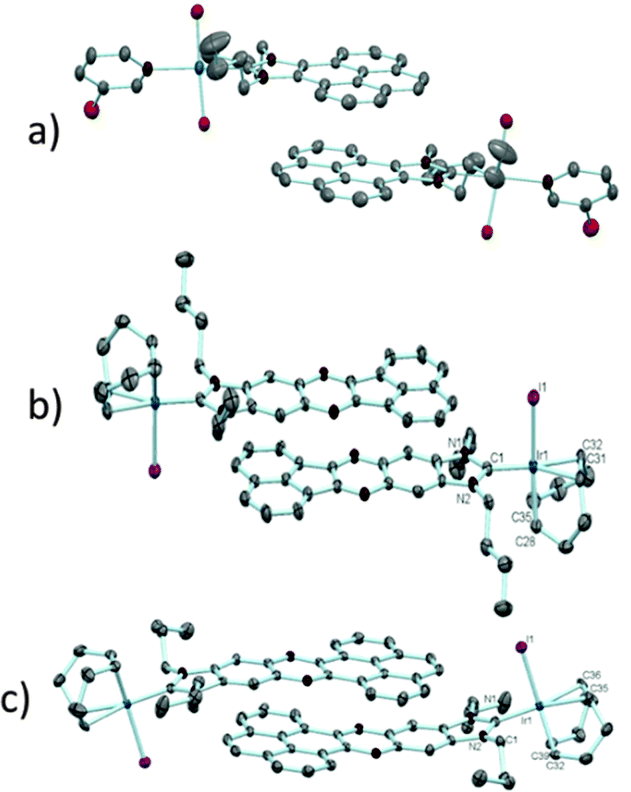 | ||
| Fig. 4 Three representative molecular diagrams showing the antiparallel π-stacking interactions in (a) [PdCl2(3-chloropyridine)(pyrene-NHC)], (b) [IrI(acetonaphtoquinoxaline-NHC)(1,5-COD)], and (c) [IrI(phenanthrophenazine-NHC)(1,5-COD)]. | ||
The study of the influence of the π-stacking interactions on the electronic properties of the ligands was performed using four different strategies: (i) electrochemical studies (cyclic voltammetry), (ii) DFT calculations, (iii) infrared spectroscopy, and (iv) 1H NMR spectroscopy.
For the electrochemical studies the [NiClCp(NHC)] (NHC = imidazolylidene, benzimidazolylidene and pyrene-imidazolylidene) complexes 15–17 were prepared (Fig. 5), primarily because this type of compounds are known to give reversible and well-behaved electrochemical responses.24 The half-wave potentials of the complexes bearing imidazolylidene and pyrene-imidazolylidene were identical, hence indicating a similar degree of electrondonating character for these two ligands. To determine if the electrochemical responses of these complexes were sensitive to the addition of an external π-stacking additive, such as pyrene, the cyclic voltammetry titrations of the three complexes were performed, by adding increasing amounts of pyrene in solutions of 15, 16 and 17.22
 | ||
| Fig. 5 Graphical representation of the E1/2 values of complexes 15 and 17 upon addition of different amounts of pyrene. Measurements performed in CH2Cl2, referenced to SCE by shifting ferrocene to 440 mV. | ||
As can be seen in the graphic shown in Fig. 5, the pyrene–NHC nickel complex 17 is very sensitive to the addition of pyrene, and a maximum positive shift of the potential (ΔE1/2 ≈ 80 mV) was achieved when one equivalent of pyrene had been added. For the experiments carried out with the imidazolylidene complex 15, a positive shift of the half-wave potential was also observed (ΔE1/2 ≈ 20 mV), but this time the shift was much smaller than for 17. For the experiments carried out with the benzimidazolylidene complex 16, also a small shift of ΔE1/2 ≈ 30 mV was detected (not shown in Fig. 5). These results indicate that, while all three complexes are sensitive to the addition of pyrene, 17 is the one that is significantly affected, most probably due to the π-staking of pyrene with the pyrene fragment of the NHC ligand.22
Although the results shown above indicated that the electron-donating character of the NHC ligand with the extended polyaromatic fragment may be affected by the addition of an external π-stacking additive, we decided to make a more detailed study in order to quantify this effect. For this study, we first calculated the TEP values of a model [Ni(NHC)(CO)3] complex, with NHC = pyrene-imidazolylidene, and then we compared the TEP values obtained for the optimized structures with two π-stacking additives (pyrene and hexafluorobenzene). The results indicated that the π-stacking results in a significant variation of the TEP value, by −3.8 and 2.3 cm−1, depending on whether pyrene or hexafluorobenzene is considered.23 This result indicates that, in theory, the electronic character of the ligand may be modified by 6.1 cm−1, by simply adding the suitable π-stacking additive. However, this result should be treated as qualitative, because the calculations were performed in the gas phase, and in the absence of solvent the non-covalent interactions are overexpressed.
In order to see if these results could be experimentally verified, a series of [IrCl(NHC)(CO)2] complexes were obtained, in which NHC represents the series of ligands displayed in Scheme 6. The purpose was to study the variation of the C–O stretching frequencies on the IR spectra of the complexes, upon addition of pyrene or hexafluorobenzene. The results are summarized in Fig. 6, were a plot of the average variation of the CO stretching frequency is represented for each of the complexes for the addition of each of the two additives.
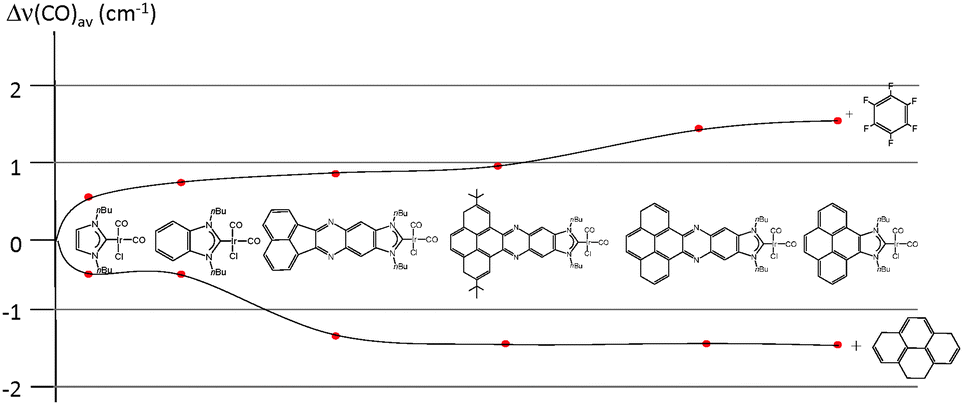 | ||
| Fig. 6 Representation of the variation of the average C–O stretching frequencies of a series of [IrCl(NHC)(CO)2] complexes. The data were taken from the IR spectra of the complexes in CH2Cl2, by adding 5 equivalents of the additive (pyrene or hexafluorobenzene). Solid lines are used only to guide the eye. | ||
From the results shown in Fig. 6, it can be seen that the variations are negligible for the imidazolylidene and benzimidazolylidene complexes, in agreement with the lower π-stacking ability of the complexes lacking a polyaromatic system. All other complexes showed larger shifts of the CO stretching frequencies, depending on the nature of the ligand. The ligands containing a pyrene fragment were the ones to display larger shifts, and among them, those with lower steric hindrance showed the largest Δ(CO)av.23 A maximum variation of 2.9 cm−1 was observed for the case of the iridium complex containing the pyrene-derived-NHC ligand, relative to the situations in which pyrene and hexafluorobenzene were added. This result is relatively small, although indicates a certain degree of modification of the electron-donating character of the ligand. As a reference for comparing this modification, it may be mentioned that the TEP difference of two classical NHC ligands that are considered electronically different, such as N,N′-dimethylimidazolylidene (IMe) and N,N′-dimethylbenzimidazolylidene, is exactly 2.9 cm−1.25 The same type of effect on the electronic properties of a bis-NHC ligand connected by a quinoxalinophenanthrophenazine core (E in Scheme 2) bound to two IrCl(CO)2 fragments was observed, upon addition of hexafluorobenzene and pyrene.8d
In order to shed some light into the nature of the non-covalent interactions between the π-stacking additive and the metal complexes with the polyaromatic ligands, 1H NMR titrations were performed. A complex bearing an acetonaphtoquinoxaline–NHC ligand bound to IrCl(CO)2 was chosen as the model complex. The 1H NMR titrations gave valuable information about both, the nature of the non-covalent interaction and the bonding energy. Fig. 7 shows a series of 1H NMR spectra of the titration of [IrI(phenanthrophenazine-NHC)(CO)2] with hexafluorobenzene. The analysis of this series revealed the downfield shift of the signals due to the internal hydrogens of the pyrene, and the resonances due to the hydrogens of the phenylene, hence indicating that the π–π-stacking interaction between the molecule of hexafluorobenzene and [IrI(phenanthrophenazine-NHC)(CO)2] is mainly produced above the pyrazine ring of the polyaromatic ligand, in the proximity of these two types of hydrogens.
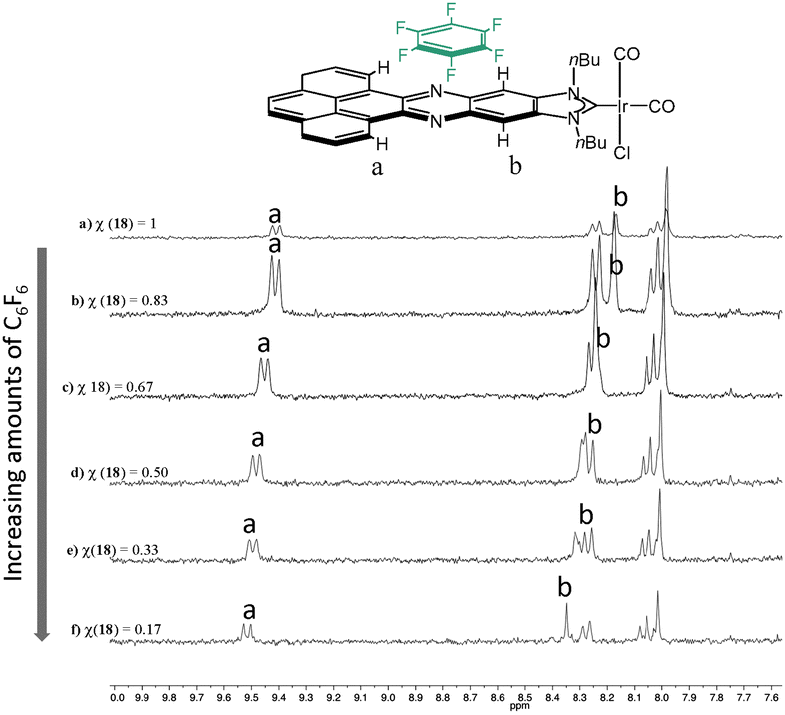 | ||
| Fig. 7 Aromatic region of the 1H NMR titration of [IrI(phenanthrophenazine-NHC)(CO)2] with hexafluorobenzene. χ represents the molar fraction of the iridium complex, relative to hexafluorobenzene. | ||
By using the same type of 1H NMR titrations, and applying the Method of Continuous Variations (MCV, also known as the method of Job),26 a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry of the aggregates was determined when both hexafluorobenzene and pyrene were used. Once the stoichiometry was determined, the association constants were estimated by using the Benesi–Hildebrand treatment.27 The association constants for the association of the π-stacking aggregate of [IrI(phenanthrophenazine-NHC)(CO)2] with pyrene and hexafluorobenzene were estimated as 2.21 and 0.13 M−1, respectively. These values are in agreement with the DFT calculations that predicted a higher affinity for pyrene compared to hexafluorobenzene in the case of the association with the pyrene-fused NHC bound to Ni(CO)3.23
:1 stoichiometry of the aggregates was determined when both hexafluorobenzene and pyrene were used. Once the stoichiometry was determined, the association constants were estimated by using the Benesi–Hildebrand treatment.27 The association constants for the association of the π-stacking aggregate of [IrI(phenanthrophenazine-NHC)(CO)2] with pyrene and hexafluorobenzene were estimated as 2.21 and 0.13 M−1, respectively. These values are in agreement with the DFT calculations that predicted a higher affinity for pyrene compared to hexafluorobenzene in the case of the association with the pyrene-fused NHC bound to Ni(CO)3.23
4. π-Stacking and catalysis. Some mechanistic studies
Once the nature and the magnitude of the π-stacking interactions between complexes decorated with polyaromatic ligands and π-stacking additives were established, the next step was to perform kinetic studies in order to shed some light onto the influence that this type of interaction may have in the catalytic activity of the complexes. For these studies a set of Ir(I) complexes was obtained (Scheme 7). These complexes were chosen because they provided an excellent comparative pattern, with which relevant information could be obtained. Complex 18 is a dimetallic complex of iridium(I) with two pyrene tags connected to the imidazolylidene ligands. Complex 19 is the monometallic analogue of 18, and can be regarded as one half of the dimetallic complex. Finally, the monometallic complex 20 is similar to 19, except for the presence of a methyl group instead of the pyrene fragment.28 At this point it is important to mention, that both 18 and 19 have the pyrene tags electronically disconnected from the imidazolylidene ligands, and hence any non-covalent interaction of the pyrene group with a substrate or an additive is not expected to afford any electronic alteration of the metal fragment.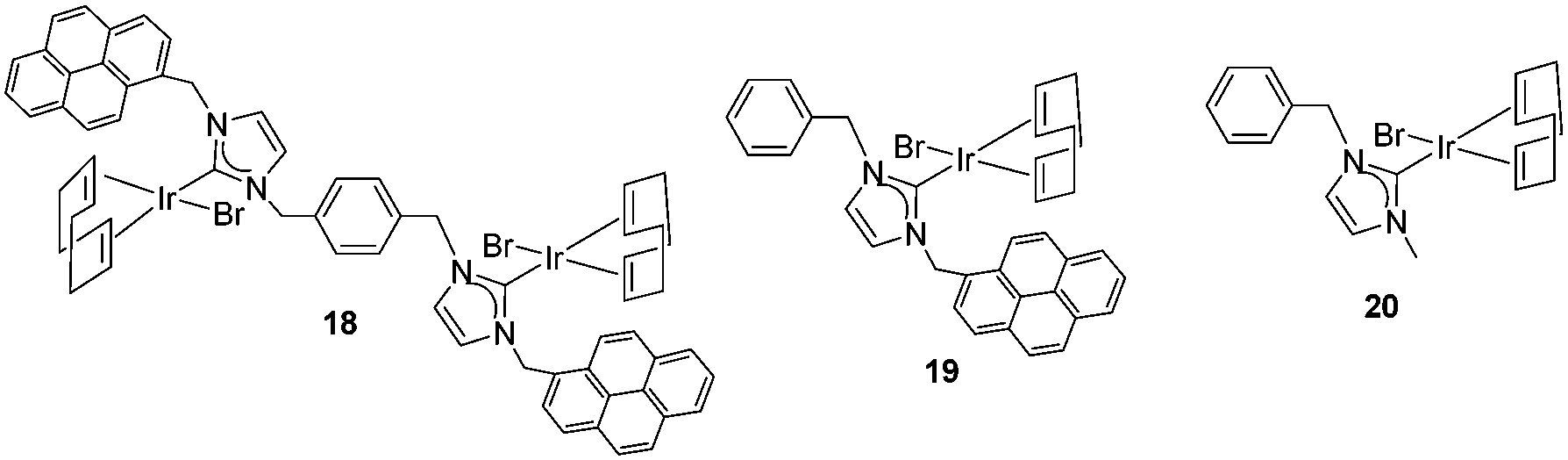 | ||
| Scheme 7 | ||
Complexes 18–20, were tested in two typical borrowing-hydrogen reactions, such as the reduction of ketones by transfer hydrogenation, and the β-alkylation of secondary alcohols with primary alcohols, two types of processes for which Ir(I) complexes have proven to be very effective catalysts.29 For the transfer hydrogenation process, we decided to study the reduction of two benchmark ketones, namely acetophenone and cyclohexanone. Fig. 8 illustrates some of the most representative results, from which interesting information was derived.
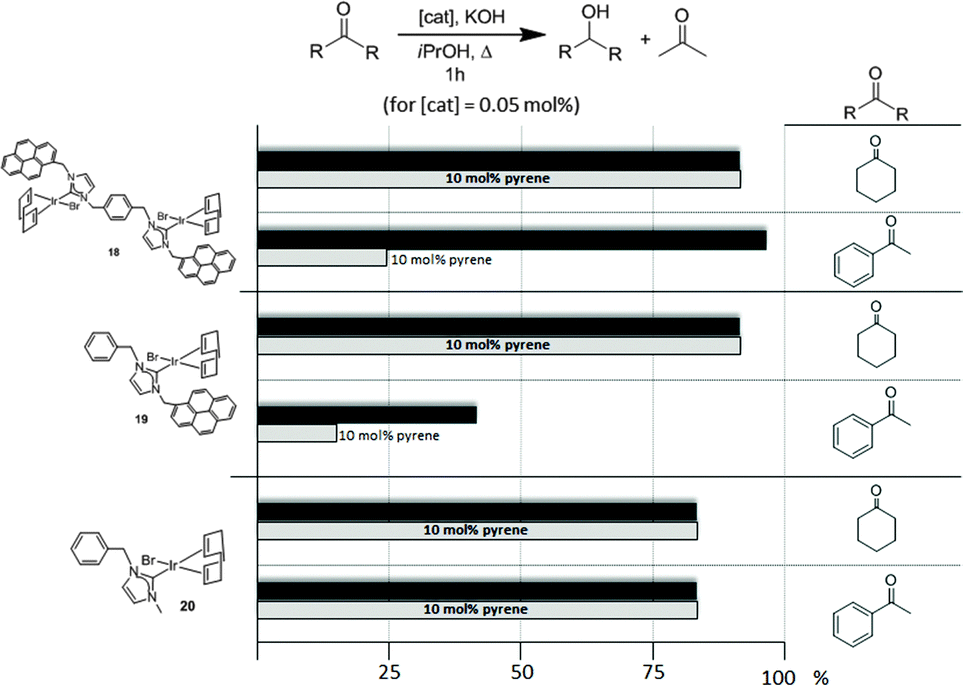 | ||
| Fig. 8 Comparison of the catalytic activities of complexes 18–20 in the reduction of acetophenone and cyclohexanone by transfer hydrogenation using isopropanol as hydrogen source. | ||
The reduction of cyclohexanone to cyclohexanol revealed that all three catalysts were almost equally active, affording very high yields to the final product. The addition of a catalytic amount of pyrene (10 mol% with respect to the substrate) did not produce any modification in the catalytic activity of all three catalysts. For the reduction of acetophenone to 1-phenylethanol, the dimetallic complex 18 was the most active catalyst. The addition of pyrene produced a substantial inhibition of the catalytic activity of both catalysts containing a pyrene tag (18 and 19), while the activity shown by the complex lacking this polyaromatic functionality (20) remained unchanged. These results clearly indicate that only when both, aromatic substrates and catalysts with polyaromatic tags are used, there is a supramolecular effect that has a significant influence on the activity of the catalyst, hence supporting the idea that π-stacking interactions are responsible for the alteration of the activity of the catalyst.
In order to shed some light on to the reaction mechanism, especially with regard to the interaction of the catalysts with the alcohols in the initial steps of the processes, kinetic studies were performed. The studies were carried out on the β-alkylation of secondary alcohols with primary alcohols. Scheme 8 summarizes the results that were obtained for the determination of the reaction orders with respect to the substrates for three combinations in which two catalysts (18 and 20) and two primary alcohols (benzyl alcohol and n-butanol) were used.
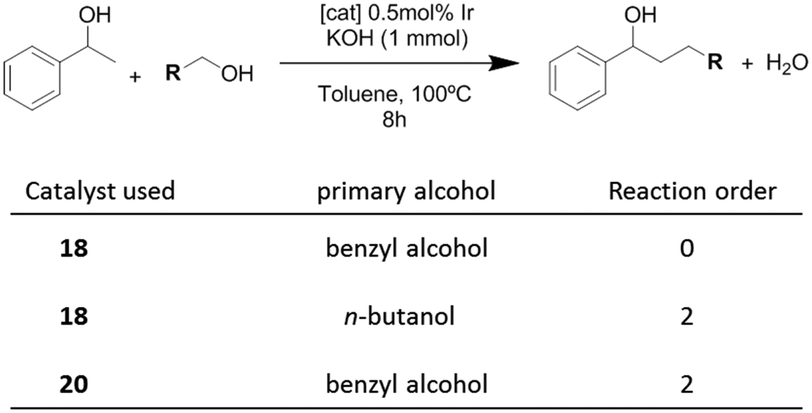 | ||
| Scheme 8 | ||
The time-dependent reaction profiles for the C–C coupling of 1-phenyl ethanol and benzyl alcohol using the dimetallic pyrene-tagged complex 18, followed a zeroth order rate in the substrate for all the experiments carried out at different concentrations of catalyst. This result, which resembles enzymatic catalysis, is strongly suggestive of a situation in which the catalyst is saturated by the substrate, hence only a portion of the substrate is in a location able to react. In this case, this situation may arise from the non-covalent interaction between the aromatic substrate and the pyrene-tag of the catalyst, thus saturating the catalyst, and affording a reaction rate which is non-dependent on the concentration of the substrate. For all other combinations of catalysts and substrates (aliphatic primary alcohol and/or catalyst lacking of the polyaromatic functionality, i.e.20), the reaction followed a second order rate, a kinetics that is fully consistent with the presence of two substrates that are reacting in a 1:1 stoichiometry.28 These results indicate that when the reaction is carried out with a catalyst containing a polyaromatic fragment, and the substrates are aromatic, the supramolecular interaction between the substrate and the catalyst is clearly influencing the kinetics of the reaction.
The determination of the rate orders with respect to the concentration of the catalysts also revealed very interesting information regarding the supramolecular interactions influencing the catalytic behavior of 18–20. For the study of the coupling between 1-phenyl ethanol and benzyl alcohol the dependence of the rate constants on the concentration of each catalyst revealed that pyrene-containing catalysts (18,19) followed a fractional reaction order (<1) with respect to the concentration of the catalyst. In particular, the reaction order with respect to catalyst 18 was 0.75, while the order with respect to 19 was 0.5. This result suggests self-association of the pyrene-tagged catalysts, and that the monomers are the active catalytic species.30 For example, in the case of complex 19, the half-order dependence arises from the concentration of the monomeric species, which may be expressed as [monomer] = ([dimer]/Keq)1/2, where Keq is the self-association constant. On the other hand, the use of the catalyst without the pyrene functionality (20) derived into a first order dependence on the concentration of the catalyst.28 This result, unveils important implications about the reaction mechanism, indicating the influence of self-association of the pyrene-tagged catalysts (ligand–ligand interaction) on the kinetics of the process. The results also illustrate how the presence of a polyaromatic functionality strongly modifies the reactivity of the catalyst, compared to otherwise identical metal complexes.
5. Immobilization of catalysts with pyrene tags onto graphene derivatives
Non-covalent interactions between the catalyst and the support constitute interesting alternatives to the more widely used covalent interactions because they avoid the functionalization of both, the catalyst and the surface, which may turn into the modification of the inherent properties of the catalyst, although some other problems may arise, such as leaching.31 In connection with the results described in the previous sections, we became interested in evaluating whether we could use the π-stacking abilities of pyrene-containing metal complexes for their non-covalent immobilization onto graphene derivatives. This aim was also supported by the fact that some pyrene-containing metal complexes had already been supported onto graphitized surfaces,32 and had been used in catalysis demonstrating interesting recyclability properties.33 Also, together with their inherent properties, graphene derivatives offer a unique opportunity to non-covalent modifications by π-stacking interactions with molecules containing polycyclic aromatic hydrocarbons,34 from which pyrene-functionalized systems constitute promising examples.35Our first approach to the immobilization of pyrene-containing complexes onto graphene derivatives was the grafting of complexes 21 and 22 onto reduce graphene oxide (rGO). The process only requires mixing the palladium or ruthenium complexes with rGO in a solution of CH2Cl2, stirring overnight at room temperature and filter the resulting black solid that can be characterized using UV/Vis, FTIR, Raman, SEM, HRTEM and ICP-MS analyses.36 The resulting solids, 21-rGO and 22-rGO, were tested in two benchmark reactions typically catalyzed by Pd and Ru. The palladium-containing material, 21-rGO, was tested in the hydrogenation of unsaturated organic substrates using molecular hydrogen,37 while the ruthenium containing material 22-rGO was tested in the dehydrogenation of alcohols (Scheme 9).38 Interestingly, the results indicated that the catalytic properties are improved in the hybrid materials, compared to the catalytic outcomes provided by the homogenous analogues (21 and 22), under the same reaction conditions. The analyses of the solids after their use in the catalytic reactions showed that the palladium materials formed nanoparticles, while the ruthenium material did not. The recyclability experiments allowed to confirm that both catalysts (21-rGO and 22-RGO), could be reused up to ten times without any measurable decrease in activity, affording quantitative yields of products.36
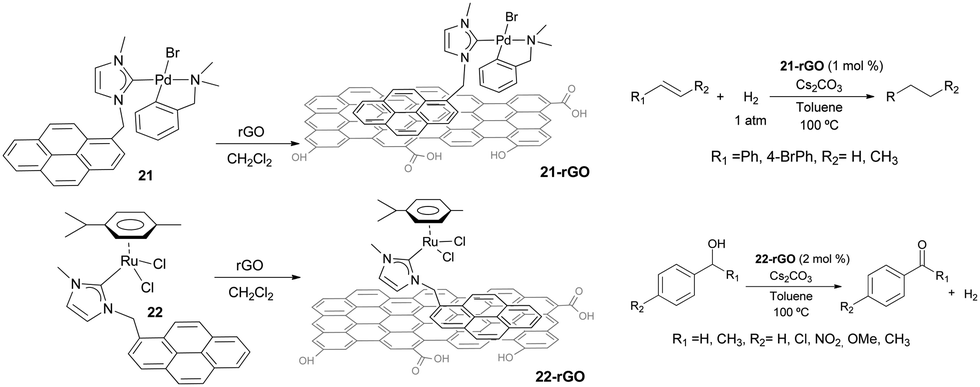 | ||
| Scheme 9 | ||
Once proved that both Pd and Ru complexes could be effectively immobilized onto rGO, we decided to immobilize both complexes onto the same solid, so that we could obtain a hybrid material containing both metals. Our interest in obtaining one solid material with both Pd and Ru catalysts, was based on our previous findings that mixed palladium/ruthenium complexes proved to be very effective in the hydrodefluorination of fluorinated aromatic and aliphatic fluorocarbons.39 The solid material was obtained by mixing equimolecular amounts of 21 and 22 with rGO in CH2Cl2 (Scheme 10).40
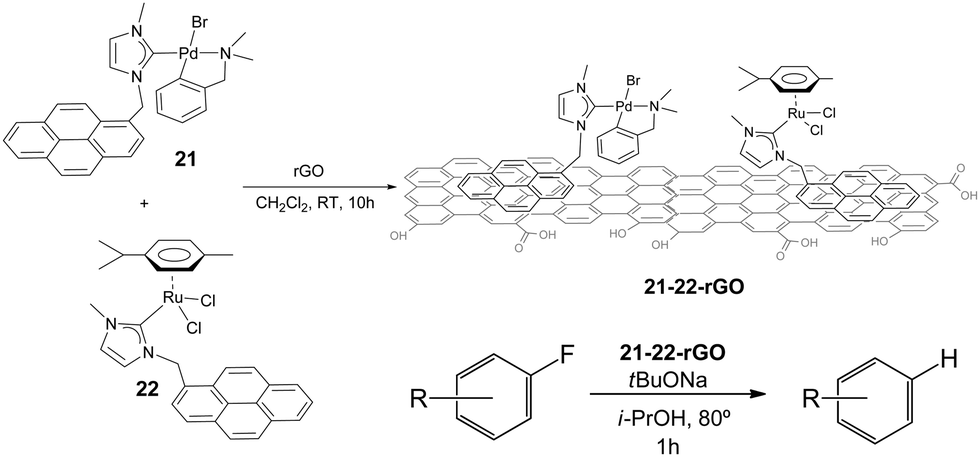 | ||
| Scheme 10 | ||
The hybrid catalyst formed (21–22-rGO) was very effective in the hydrodefluorination of a series of fluoroarenes, under the reaction conditions depicted in Scheme 10. However, the analysis of the catalyst before and after being used in the catalytic reaction indicated the formation of small nanoparticles presumably formed by palladium aggregates. This result suggested that these palladium nanoparticles, together with the ruthenium heterogeneised complex should form the active catalytic system. The catalyst could be recycled up to 12 runs in the hydrodefluorination of 4-fluorobenzene, without any measurable decrease of activity and quantitatively affording benzene,40 hence constituting one very rare example of a recyclable HDF catalyst.
Complexes 18 and 19 (Scheme 7) were also immobilized onto rGO by following the same procedure depicted in Scheme 9. We thought that the comparison of the activities provided by the two supported catalysts (18-rGO and 19-rGO) could give us valuable information about the influence of the mono- or dimetallic nature of the catalyst, and about the presence of one or two pyrene tags for the immobilization onto the solid surface. The two solids were tested in the β-alkylation of 1-phenyl ethanol and benzyl alcohol, where we paid special attention to the recyclability properties of the two catalysts.28 As can be seen from the results depicted in Fig. 9, a significant feature of the results is that the activity shown in the first run is significantly lower than the activity shown for the rest of the cycles, and this situation is common for 18-rGO and 19-rGO. This observation was explained as a consequence of the need of activation of the two catalysts along the first reaction run. More importantly, while 19-rGO (only one pyrene tag) was effectively recycled only three times, 18-rGO (two pyrene tags) was recycled up to twelve times without any detectable loss of activity. The determination of the metal content in both materials before and after being used in the catalytic experiments revealed that 19-rGO is largely desorbed, while 18-rGO was effectively retained by the solid (no leaching detected),28 thus indicating a more effective immobilization of the complex bearing two pyrene tags.
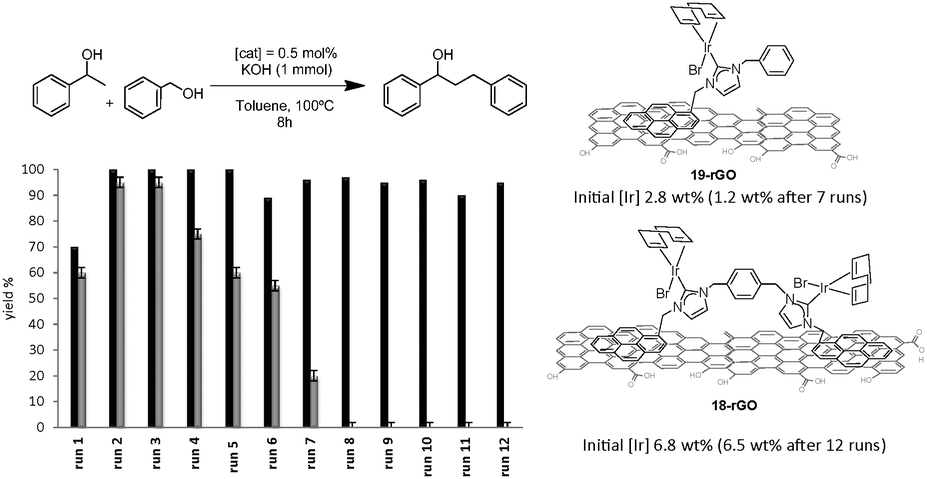 | ||
| Fig. 9 Recycling experiments of the β-alkylation of 1-phenylethanol with benzyl alcohol. Black bars indicate the activity of 18-rGO. Grey bars for the activity of 19-rGO. | ||
6. Conclusions and outlook
This account article describes a rational study about how the activity of catalysts bearing rigid polyaromatic functionalities is influenced by non-covalent interactions, mainly based on π–π-stacking. The work is centered in catalysts with NHCs, a type of ligands that we have been using for the last few years, but the conclusions are equally valid for any other types of ligands decorated with polyaromatic π-extended systems. The study allowed establishing well-supported conclusions on the nature of the π-stacking interactions produced, and on the catalytic consequences derived. Examples about ligand–ligand interactions, ligand–additive interactions and ligand–substrate interactions have been provided, justified and quantified for some key cases. The work demonstrates that π-stacking forces are of major importance for the design of effective homogeneous catalysts, but can also be effectively used for the preparation of heterogeneized catalysts. By performing a detailed analysis of the research of our group in the last four years, which is fundamentally based on the application of host–guest chemistry techniques to organometallic catalysis, the following important conclusions may be extracted:(1) The electronic properties of ligands having extended-polyconjugated systems, may be tuned (post-modified) if the proper π-stacking additives are chosen. This effect, which is a direct consequence of the ligand–additive interaction, has important consequences in the modification of the catalytic performance of metal complexes containing NHC ligands fused to polyaromatic fragments.
(2) The reactions in which aromatic substrates and catalysts with polyaromatic ligands are used follow a zeroth order dependence on the concentration of the substrates, as a consequence of the saturation of the catalyst with the substrate all along the reaction course. This situation, which strongly resembles enzymatic catalysis, arises from the π-stacking bonding between the substrate and the ligand (ligand–substrate interaction).
(3) The kinetic studies of the reactions in which catalysts with polyaromatic ligands are used indicate that the self-association of the catalysts is clearly influencing the catalytic performance (ligand–ligand interaction). The self-assembly properties of this type of catalysts are also demonstrated by their X-ray molecular structures, which in all cases show the formation of aggregates by π–π-stacking interactions.
(4) Non-covalent interactions allow the heterogeneization of catalysts with polyaromatic ligands onto graphene derivatives. The number of polyaromatic tags attached to the catalyst determines the efficiency of the immobilization. The simplicity in the design of these heterogeneous catalytic systems, in combination with great catalytic performance and recyclability, should be taken into account for the design of future heterogeneous catalysts.
In summary, this article sheds light onto the understanding of the nature and strength of the interaction between the catalyst and the substrate, and aims to improve the basis of the design of organometallic-based supramolecular catalysts. The results given have fundamental implications in the design of future catalysts containing rigid polyaromatic systems, and may inspire future researchers in the design of improved homogeneous catalysts, by taking into account that the activities of the metal complexes are strongly modified by supramolecular interactions.
Acknowledgements
I am very thankful to all the members of the research group, who enthusiastically contributed to the development of the research described in this review article. In particular, I have to sincerely thank the contributions of José A. Mata, Macarena Poyatos, Gregorio Guisado, Sergio Gonell, Sara Sabater, Amparo Prades, Candela Segarra, Hugo Valdés, Sheila Ruíz-Botella and Carmen Mejuto. I am also very thankful to Prof. Dmitry G. Gusev (Wilfrid Laurier University) for the DFT calculations performed for all the examples shown in this work, and for his continuous support to our research. I would like to very especially thank Prof. Robert H. Crabtree, who was the first to predict that π-stacking interactions were responsible for the better catalytic performance of complexes 5 and 6 with respect to the monometallic analogues. He really was the one to inspire all the work described in this article. Finally, I gratefully acknowledge financial support from the ‘Ministerio de Economía y Competitividad’ MINECO of Spain (CTQ2014-51999-P) and the Universitat Jaume I (P11B2014-02).Notes and references
- R. H. Crabtree, New J. Chem., 2011, 35, 18–23 RSC.
- Supramolecular Catalysis, ed. P. W. N. M. Van Leeuwen, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- (a) K. Hermann, Y. Ruan, A. M. Hardin, C. M. Hadad and J. D. Badjic, Chem. Soc. Rev., 2015, 44, 500–514 RSC; (b) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed; (c) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC; (d) J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC; (e) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC; (f) S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC; (g) P. Dydio and J. N. H. Reek, Chem. Sci., 2014, 5, 2135–2145 RSC; (h) P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, in Adv. Catal., ed. B. C. Gates and H. Knozinger, 2011, vol. 54, pp. 63–126 Search PubMed; (i) T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC.
- R. R. Knowles and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS PubMed.
- W. Tang, S. Johnston, J. A. Iggo, N. G. Berry, M. Phelan, L. Lian, J. Bacsa and J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 1668–1672 CrossRef CAS PubMed.
- (a) N. M. Goodey and S. J. Benkovic, Nat. Chem. Biol., 2008, 4, 474–482 CrossRef CAS PubMed; (b) L. Kovbasyuk and R. Kramer, Chem. Rev., 2004, 104, 3161–3187 CrossRef CAS PubMed; (c) A. S. T. Ribeiro and V. Ortiz, Chem. Rev., 2016 DOI:10.1021/acs.chemrev.1025b00543.
- J. A. Mata, F. E. Hahn and E. Peris, Chem. Sci., 2014, 5, 1723–1732 RSC.
- (a) E. Mas-Marza, J. A. Mata and E. Peris, Angew. Chem., Int. Ed., 2007, 46, 3729–3731 CrossRef CAS PubMed; (b) S. Gonell, M. Poyatos and E. Peris, Chem. – Eur. J., 2014, 20, 9716–9724 CrossRef CAS PubMed; (c) A. Prades, E. Peris and M. Alcarazo, Organometallics, 2012, 31, 4623–4626 CrossRef CAS; (d) H. Valdes, M. Poyatos and E. Peris, Organometallics, 2015, 34, 1725–1729 CrossRef CAS.
- D. G. Gusev and E. Peris, Dalton Trans., 2013, 42, 7359–7364 RSC.
- (a) D. M. Khramov, A. J. Boydston and C. W. Bielawski, Angew. Chem., Int. Ed., 2006, 45, 6186–6189 CrossRef CAS PubMed; (b) A. J. Boydston, J. D. Rice, M. D. Sanderson, O. L. Dykhno and C. W. Bielawski, Organometallics, 2006, 25, 6087–6098 CrossRef CAS; (c) A. J. Boydston and C. W. Bielawski, Dalton Trans., 2006, 4073–4077 RSC; (d) A. J. Boydston, K. A. Williams and C. W. Bielawski, J. Am. Chem. Soc., 2005, 127, 12496–12497 CrossRef CAS PubMed.
- (a) S. Gonell, M. Poyatos and E. Peris, Angew. Chem., Int. Ed., 2013, 52, 7009–7013 CrossRef CAS PubMed; (b) S. Gonell, R. G. Alabau, M. Poyatos and E. Peris, Chem. Commun., 2013, 49, 7126–7128 RSC; (c) C. Segarra, J. Linke, E. Mas-Marza, D. Kuck and E. Peris, Chem. Commun., 2013, 49, 10572–10574 RSC.
- G. Guisado-Barrios, J. Hiller and E. Peris, Chem. – Eur. J., 2013, 19, 10405–10411 CrossRef CAS PubMed.
- K. V. Vasudevan, R. R. Butorac, C. D. Abernethy and A. H. Cowley, Dalton Trans., 2010, 39, 7401–7408 RSC.
- R. Dorta, E. D. Stevens, N. M. Scott, C. Costabile, L. Cavallo, C. D. Hoff and S. P. Nolan, J. Am. Chem. Soc., 2005, 127, 2485–2495 CrossRef CAS PubMed.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759–1766 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536–1554 CrossRef CAS PubMed.
- E. K. van den Beuken and B. L. Feringa, Tetrahedron, 1998, 54, 12985–13011 CrossRef CAS.
- (a) J. N. H. Reek, S. Arevalo, R. Van Heerbeek, P. C. J. Kamer and P. Van Leeuwen, in Adv. Catal., ed. B. C. Gates and H. Knozinger, 2006, vol. 49, pp. 71–151 Search PubMed; (b) B. Helms and J. M. J. Frechet, Adv. Synth. Catal., 2006, 348, 1125–1148 CrossRef CAS; (c) D. Astruc, C. R. Chim., 2005, 8, 1101–1107 CrossRef CAS.
- K. A. Williams and C. W. Bielawski, Chem. Commun., 2010, 46, 5166–5168 RSC.
- S. Ruiz-Botella and E. Peris, Organometallics, 2014, 33, 5509–5516 CrossRef CAS.
- (a) W.-Y. Zhang, Y.-F. Han, L.-H. Weng and G.-X. Jin, Organometallics, 2014, 33, 3091–3095 CrossRef CAS; (b) R. Nagarajaprakash, D. Divya, B. Ramakrishna and B. Manimaran, Organometallics, 2014, 33, 1367–1373 CrossRef CAS; (c) A. Mishra, Y. J. Jeong, J.-H. Jo, S. C. Kang, H. Kim and K.-W. Chi, Organometallics, 2014, 33, 1144–1151 CrossRef CAS; (d) V. Vajpayee, Y. H. Song, Y. J. Jung, S. C. Kang, H. Kim, I. S. Kim, M. Wang, T. R. Cook, P. J. Stang and K.-W. Chi, Dalton Trans., 2012, 41, 3046–3052 RSC; (e) A. Mishra, H. Jung, J. W. Park, H. K. Kim, H. Kim, P. J. Stang and K.-W. Chi, Organometallics, 2012, 31, 3519–3526 CrossRef CAS PubMed.
- H. Valdes, M. Poyatos, G. Ujaque and E. Peris, Chem. – Eur. J., 2015, 21, 1578–1588 CrossRef CAS PubMed.
- H. Valdes, M. Poyatos and E. Peris, Inorg. Chem., 2015, 54, 3654–3659 CrossRef CAS PubMed.
- O. R. Luca, B. A. Thompson, M. K. Takase and R. H. Crabtree, J. Organomet. Chem., 2013, 730, 79–83 CrossRef CAS.
- D. G. Gusev, Organometallics, 2009, 28, 6458–6461 CrossRef CAS.
- (a) J. S. Renny, L. L. Tomasevich, E. H. Tallmadge and D. B. Collum, Angew. Chem., Int. Ed., 2013, 52, 11998–12013 CrossRef CAS PubMed; (b) V. M. S. Gil and N. C. Oliveira, J. Chem. Educ., 1990, 67, 473–478 CrossRef CAS.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- S. Ruiz-Botella and E. Peris, Chem. – Eur. J., 2015, 21, 15263–15271 CrossRef CAS PubMed.
- (a) M. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS; (b) M. G. Edwards, R. F. R. Jazzar, B. M. Paine, D. J. Shermer, M. K. Whittlesey, J. M. J. Williams and D. D. Edney, Chem. Commun., 2004, 90–91 RSC; (c) G. Guillena, D. J. Ramon and M. Yus, Angew. Chem., Int. Ed., 2007, 46, 2358–2364 CrossRef CAS PubMed; (d) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555–1575 CrossRef CAS; (e) P. J. Black, G. Cami-Kobeci, M. G. Edwards, P. A. Slatford, M. K. Whittlesey and J. M. J. Williams, Org. Biomol. Chem., 2006, 4, 116–125 RSC; (f) O. Saidi and J. M. J. Williams, in Iridium Catalysis, ed. P. G. Andersson, 2011, pp. 77–106 Search PubMed; (g) T. Suzuki, Chem. Rev., 2011, 111, 1825–1845 CrossRef CAS PubMed.
- (a) R. S. Srivastava and K. M. Nicholas, Organometallics, 2005, 24, 1563–1568 CrossRef CAS; (b) E. L. Hegg, S. H. Mortimore, C. L. Cheung, J. E. Huyett, D. R. Powell and J. N. Burstyn, Inorg. Chem., 1999, 38, 2961–2968 CrossRef CAS PubMed.
- (a) J. M. Fraile, J. I. Garcia and J. A. Mayoral, Chem. Rev., 2009, 109, 360–417 CrossRef CAS PubMed; (b) D. J. Cole-Hamilton, Science, 2003, 299, 1702–1706 CrossRef CAS PubMed.
- (a) A. Le Goff, B. Reuillard and S. Cosnier, Langmuir, 2013, 29, 8736–8742 CrossRef CAS PubMed; (b) J. A. Mann, J. Rodriguez-Lopez, H. D. Abruna and W. R. Dichtel, J. Am. Chem. Soc., 2011, 133, 17614–17617 CrossRef CAS PubMed.
- (a) M. Keller, V. Colliere, O. Reiser, A.-M. Caminade, J.-P. Majoral and A. Ouali, Angew. Chem., Int. Ed., 2013, 52, 3626–3629 CrossRef CAS PubMed; (b) S. Wittmann, A. Schaetz, R. N. Grass, W. J. Stark and O. Reiser, Angew. Chem., Int. Ed., 2010, 49, 1867–1870 CrossRef CAS PubMed; (c) P. Kang, S. H. Zhang, T. J. Meyer and M. Brookhart, Angew. Chem., Int. Ed., 2014, 126, 8853–8857 CrossRef; (d) L. Xing, J.-H. Xie, Y.-S. Chen, L.-X. Wang and Q.-L. Zhou, Adv. Synth. Catal., 2008, 350, 1013–1016 CrossRef CAS.
- (a) G. Eda and M. Chhowalla, Adv. Mater., 2010, 22, 2392–2415 CrossRef CAS PubMed; (b) R. Podeszwa, J. Chem. Phys., 2010, 132 Search PubMed; (c) B. Pan and B. S. Xing, Environ. Sci. Technol., 2008, 42, 9005–9013 CrossRef CAS PubMed; (d) J. Y. Chen, W. Chen and D. Zhu, Environ. Sci. Technol., 2008, 42, 7225–7230 CrossRef CAS PubMed; (e) S. Muller, K. U. Totsche and I. Kogel-Knabner, Eur. J. Soil Sci., 2007, 58, 918–931 CrossRef; (f) J. Balapanuru, J.-X. Yang, S. Xiao, Q. Bao, M. Jahan, L. Polavarapu, J. Wei, Q.-H. Xu and K. P. Loh, Angew. Chem., Int. Ed., 2010, 49, 6549–6553 CrossRef CAS PubMed.
- (a) Y. B. Sun, S. B. Yang, G. X. Zhao, Q. Wang and X. K. Wang, Chem. – Asian J., 2013, 8, 2755–2761 CrossRef CAS PubMed; (b) K. Yang, X. L. Wang, L. Z. Zhu and B. S. Xing, Environ. Sci. Technol., 2006, 40, 5804–5810 CrossRef CAS PubMed; (c) B. Chefetz, A. P. Deshmukh, P. G. Hatcher and E. A. Guthrie, Environ. Sci. Technol., 2000, 34, 2925–2930 CrossRef CAS; (d) X. Mao, H. Su, D. Tian, H. Li and R. Yang, ACS Appl. Mater. Interfaces, 2013, 5, 592–597 CrossRef CAS PubMed; (e) S. Qu, M. Li, L. Xie, X. Huang, J. Yang, N. Wang and S. Yang, ACS Nano, 2013, 7, 4070–4081 CrossRef CAS PubMed.
- S. Sabater, J. A. Mata and E. Peris, ACS Catal., 2014, 4, 2038–2047 CrossRef CAS.
- (a) S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS; (b) X. H. Cui and K. Burgess, Chem. Rev., 2005, 105, 3272–3296 CrossRef CAS PubMed.
- (a) M. Bertoli, A. Choualeb, A. J. Lough, B. Moore, D. Spasyuk and D. G. Gusev, Organometallics, 2011, 30, 3479–3482 CrossRef CAS; (b) M. Nielsen, A. Kammer, D. Cozzula, H. Junge, S. Gladiali and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 9593–9597 CrossRef CAS PubMed.
- S. Sabater, J. A. Mata and E. Peris, Nat. Commun., 2013, 4, 2553 Search PubMed.
- S. Sabater, J. A. Mata and E. Peris, Organometallics, 2015, 34, 1186–1190 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |