
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synchronous intramolecular cycloadditions of the polyene macrolactam polyketide heronamide C†
Thomas J.
Booth
a,
Silke
Alt
a,
Robert J.
Capon
b and
Barrie
Wilkinson
*a
aDepartment of Molecular Microbiology, John Innes Centre, Norwich Research Park, Norwich NR4 7UH, UK. E-mail: barrie.wilkinson@jic.ac.uk
bInstitute for Molecular Bioscience, The University of Queensland, St. Lucia, Queensland, Australia 4072
First published on 31st March 2016
Abstract
A growing number of natural products appear to arise from biosynthetic pathways that involve pericyclic reactions. We show here that for the heronamides this can occur via two spontaneous pathways involving alternative thermal or photochemical intramolecular cycloadditions.
The heronamides are unusual polyketide metabolites produced by Streptomyces sp. CMB-0406 isolated from marine sediment collected off the coast of Heron Island, Australia.1 Since their discovery, the heronamides have undergone revisions to their stereochemistry.2–4 They represent an intriguing example of natural products for which transannular [6π+4π] and [6π+6π] intramolecular cycloadditions appear to occur during the biosynthesis of the congeners heronamide A (1) and B (2) from the co-isolated precursor heronamide C (3) (Fig. 1). Although 1 and 2 have no reported biological activity, 3 exhibits inhibitory activity against fission yeast, the basis of which involves unique targeting of the lipid membrane, probably through interaction with saturated lipids, and leads to changes in cell wall morphology.4 Raju et al. reported that human cancer cell lines exposed to 3 developed large intracellular structures that dissipated on removal of 3 from the cell media, with no apparent adverse effects.3 It is tempting to speculate that these changes may arise from the interaction of 3 with the lipid membranes of internal cellular organelles.
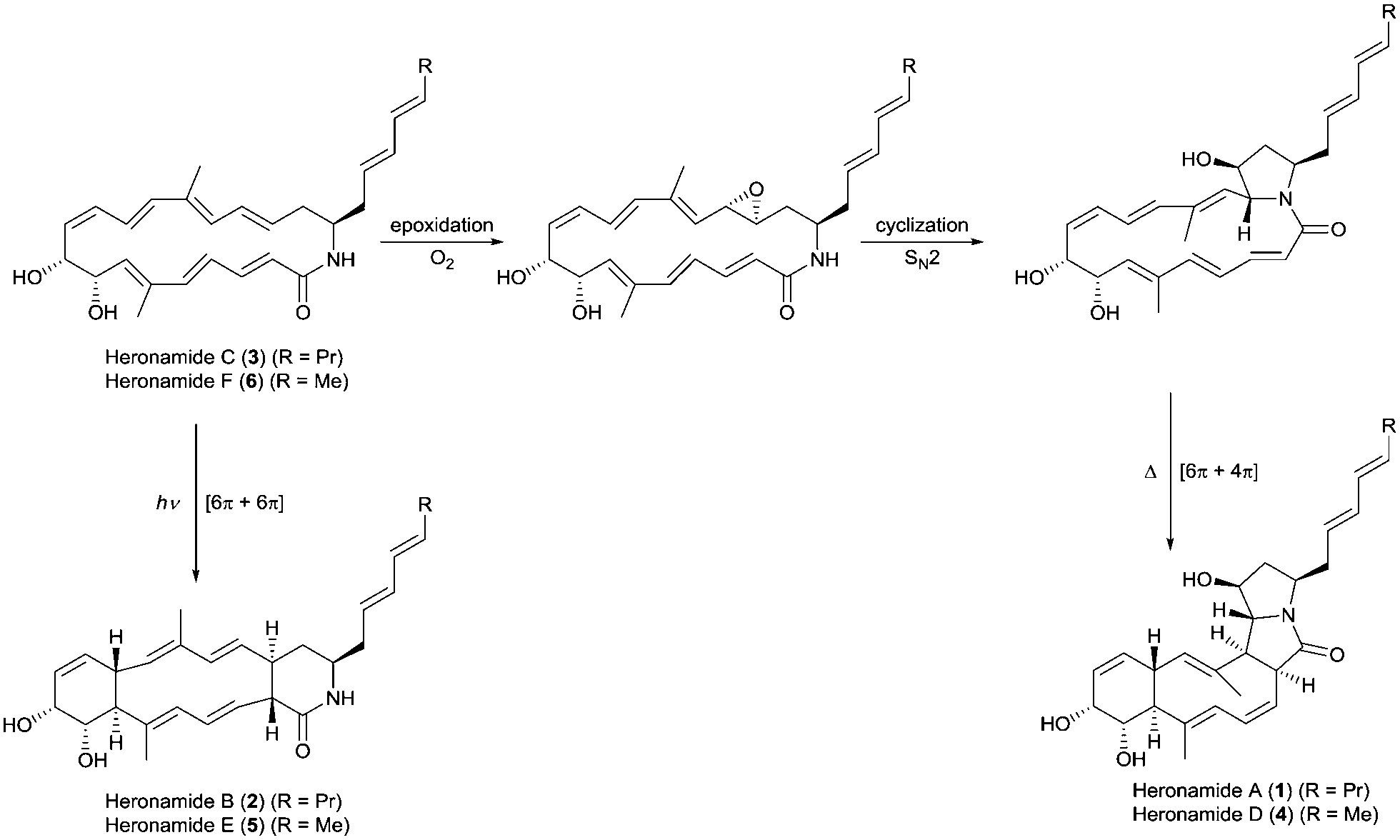 | ||
| Fig. 1 Proposed biosynthetic pathways for the heronamides A/B (1/2) and D/E (4/5) from heronamides C (3) and F (6). | ||
The biosynthesis of 1 is likely to first involve epoxidation of 3 with subsequent intramolecular SN2 attack of the proximal amide nitrogen to yield an intermediate pyrrolidinol which can then undergo [6π+4π] cycloaddition (Fig. 1). Recent computational studies support this proposal and suggest a thermal reaction occurring through an ambimodal transition state that leads to a [4π+2π] adduct in addition to the observed [6π+4π] adduct.5 Interconversion of these adducts was predicted to occur via a facile Cope rearrangement leading to accumulation of the thermodynamically more stable [6π+4π] adduct (Fig. S1, ESI†). Preliminary support for this proposal was provided by a report in which 3 was dissolved in DMSO and left for an extended period with exposure to air.4 This led to production of 1 but no yields were given and limited characterization was reported.
Through consideration of the Woodward–Hoffmann rules we predict that the conversion of 3 into the second polycyclic congener 2 is likely to follow an allowed photochemical [6π+6π] cycloaddition (Fig. S2, ESI†). While no data have yet been reported to corroborate this it was recently reported that the [6π+6π] cycloaddition required for biosynthesis of the related metabolite ciromicin B from ciromicin A is photocatalysed (Fig. S3, ESI†).6 Our interest in heronamide biosynthesis was further piqued by the report of the metabolites ML-4497 and BE-141068 produced by Streptomyces species isolated from Norwegian marine sediments (Fig. S4, ESI†). ML-449 is suggested to have the same gross structure as 3 but with a cis-rather than trans-geometry about the C6–C7 double bond. BE-14106 has the same macrolactam structure as ML-449 but lacks an olefin unit in the side chain. Most notably there was no report of additional congeners of either compound that would correspond to 1 and 2, and on the basis of these observations we were intrigued to understand whether or not there might be a biological basis for the production of 1 and 2 by Streptomyces sp. CMB-0406.
To investigate the biosynthetic potential of Streptomyces sp. CMB-0406, we obtained the genome sequence via the Pacific Biosciences (PacBio) RSII sequencing platform. The heronamide biosynthetic gene cluster was readily identified using antiSMASH9 and found to be essentially identical to the published clusters for ML-449 and BE-14106 apart from the expected difference of a single modular polyketide synthase (PKS) module in the latter case (Table S4, ESI†).7,8 Bioinformatics analysis of the heronamide, ML-449 and BE-14106 PKS genes showed that the β-ketoreductase domains of all modules except for module 4 of the macrolactam PKS contain sequence motifs corresponding to formation of the B-type stereochemistry,10 consistent with the 9R-hydroxyl group stereochemistry observed and the presence of trans-double bonds in all positions other than at C10–C11.11 This suggests that the structure of ML-449 is identical to that of 3, but does not explain why equivalent analogues of 1 and 2 were not reported to be co-produced. An explanation may lie in the fact that these compounds were isolated on the basis of antifungal activity (only 3 displays antifungal activity). Recently, the biosynthetic gene cluster for the heronamide congeners D–F (4–6) was reported and although the genetic architecture differs slightly to the heronamide A–C gene cluster (Fig. 2 and Table S4, ESI†), the same complement of biosynthetic genes appear to be present.12 Further comparison of these biosynthetic gene clusters to those for related polyene macrolactams did not suggest any unique genes that might be specific to the biosynthesis of 1 and 2 from 3 (or of 4 and 5 from 6) (ESI†).
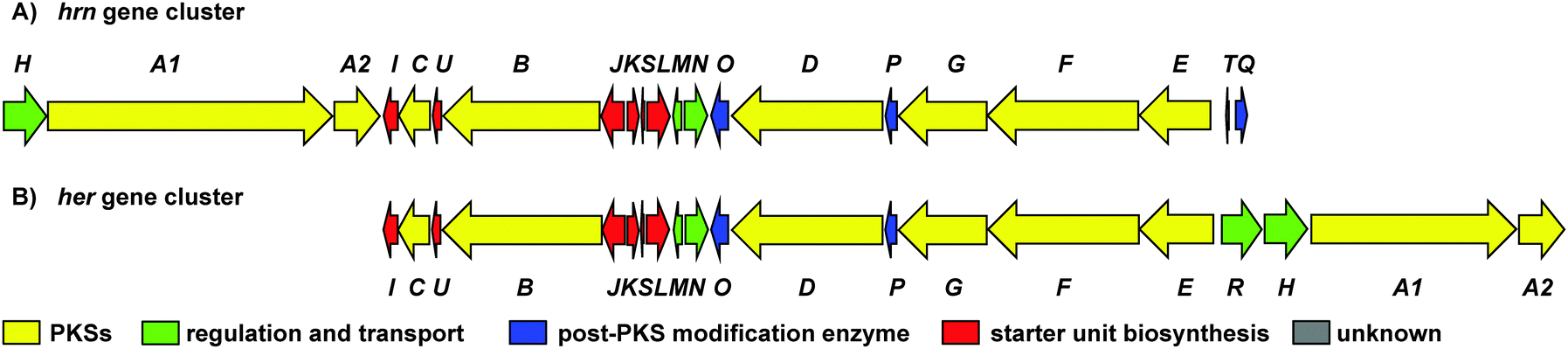 | ||
Fig. 2 (A) Organisation of the 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 166 bp hrn biosynthetic gene cluster in Streptomyces sp. CMB-0406. (B) Comparison of the hrn cluster organisation with the her cluster from Streptomyces SCSIO 03032. The function of each gene is listed in Table S4 (ESI†). 166 bp hrn biosynthetic gene cluster in Streptomyces sp. CMB-0406. (B) Comparison of the hrn cluster organisation with the her cluster from Streptomyces SCSIO 03032. The function of each gene is listed in Table S4 (ESI†). | ||
All of the biosynthetic gene clusters discussed contain a single monooxygenase gene (encoding a cytochrome P450 monooxygenase; cyp450) for introduction of the C8-hydroxyl group and mutational analysis of the BE-141068 and 4–612 producers is consistent with this. Notably, no gene is present in any of the clusters whose product can be attributed to epoxidation of the C16–C17 double bond. This was surprising as although polyenes are known to autoxidize in air13 it seemed unlikely that this might occur with both the regio- and stereoselectivity required to produce 1, especially as no additional stereoisomers of 1 or alternatively oxidized congeners of 3 have been reported. We therefore wondered if this step might be catalyzed by an enzyme encoded elsewhere in the genome. To determine the likelihood of biocatalysis, we performed a time course study following the production of 1–3 and dry cell mass over 10 days (Fig. S5, ESI†), in addition to experiments modifying fermentation media and conditions, growing in the presence of cyp450 inhibitors and co-culture with various Gram-positive and Gram-negative bacteria (data not shown). The results showed that biosynthesis of 3 correlates to cell growth but that the production of 1 and 2 do not, and do not appear to be linked to later induction of gene expression. These data, in addition to our genetic analysis suggested biological catalysis of C16–C17 epoxidation was unlikely.
Thus, prompted by the earlier report of direct chemical conversion of 3 to 1 in DMSO,4 we decided to investigate the autoxidation of 3 and subsequent production of 1 in more detail. Aliquots of 3 (225 μM) dissolved in a range of solvents were used to assess the reaction in the presence of air or the oxidizing agents mCPBA and oxone. Light was excluded at all stages and these experiments were performed at several temperatures (4 °C, 30 °C and 60 °C) for up to 7 days and then analyzed by using HPLC and LCMS in comparison to authentic standards. In the presence of mCPBA and oxone a large number of products were observed at low levels in the LCMS chromatogram along with trace amounts of 1, reflecting the sensitivity of 3 to oxidation (data not shown). When exposed to air under ambient conditions we observed production of 1 at significantly higher levels than in the presence of chemical oxidants. The composition of solvent was also important with the most efficient conversion of 3 to 1 occurring in methanol containing 10% DMSO. This may reflect the mild oxidizing conditions or the accelerated rate of cycloaddition often observed in polar solvents. Despite giving higher yields of 1, it is worth noting that incubating at higher temperatures lead to the production of a number of other products accounting for an additional 30% of the molar yield at 60 °C (Fig. 3). With m/z values significantly lower than 3, it is likely these are products of degradation rather than pathway intermediates. Remarkably, on the basis of these results it appears that biosynthesis of 1 does indeed involve the autoxidation of 3 with facial selectivity at the C16–C17 double bond. Previous studies on the autoxidation of the polyene antifungal natural products filipin and lagosin showed that epoxide formation can occur with a strong regiochemical preference, although in these cases at the most substituted olefin, and that the process is likely to involve a radical addition process.13 Although the C16–C17 olefin of 3 is not the most substituted we presume the selectivity observed reflects either a higher reactivity of the C16–C17 double bond, a steric effect due to the solution conformation of 3, or some combination of both these factors. Although side products were formed in our experiments, the excellent molar conversion of 3 to 1, along with the lack of any significant pathway intermediates or alternative reaction products suggest that the autoxidation step may be rate limiting. This is consistent with the computational study of Houk and co-workers5 who calculated the theoretical half-life of the pyrrolidinol precursor as approximately three minutes. In all of these experiments we excluded light and observed no production of 2.
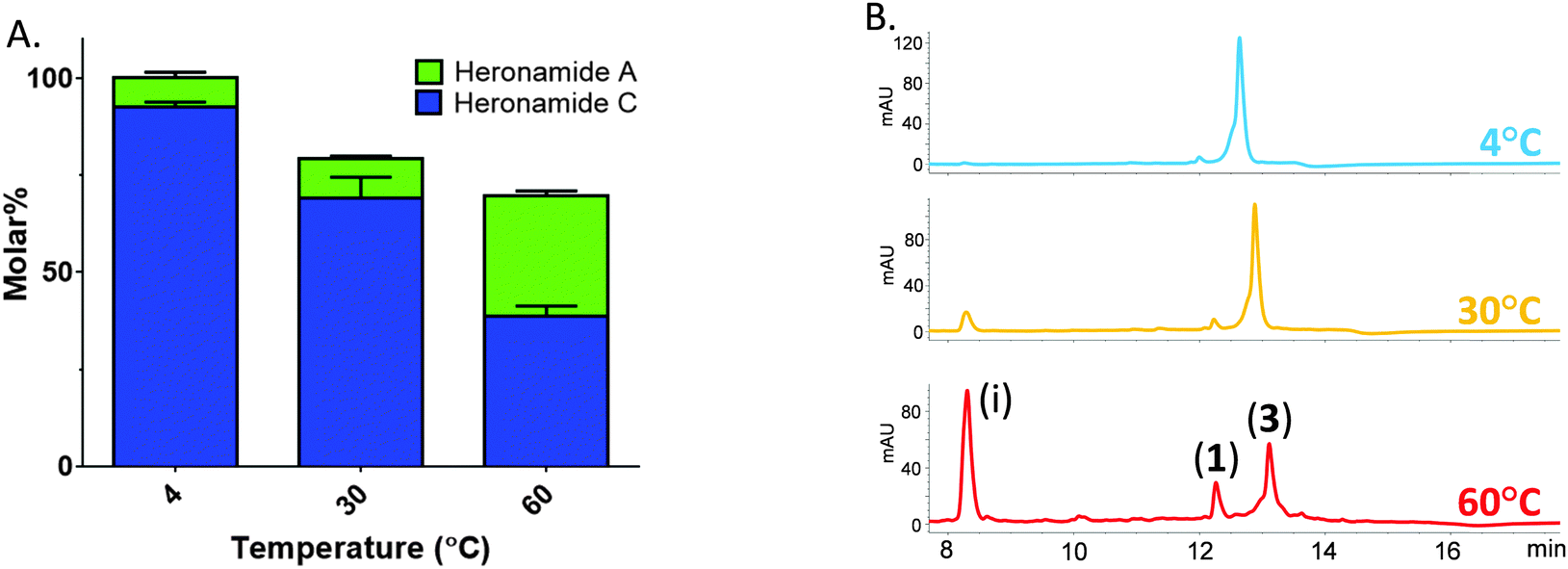 | ||
| Fig. 3 Temperature sensitive conversion of heronamide C (3) to heronamide A (1) after seven days of incubation: (A) the proportion of 1 and 3 as a molar percentage of the starting material; and (B) a section of the HPLC trace showing 1, 3 and the main decomposition product (i) (for full chromatograms see Fig. S6, ESI†). | ||
Based on their chemical structures the conversion of 3 into 2 should involve a [6π+6π] cycloaddition, which, according to the Woodward–Hoffman rules, is indicative of a photochemical reaction (Fig. S2, ESI†). To test this hypothesis purified 3 was dissolved in methanol (225 μM) and placed in a borosilicate vial. Replicate vials were prepared and one placed in a box to exclude light; both were then placed in direct bright sunlight for 1 h and subsequently analyzed by LCMS (Fig. S7, ESI†). The sample of 3 exposed to direct sunlight showed excellent molar conversion to 2 (approx. 60%) with little evidence of additional isomers or other side products, while the sample excluded from light contained only 3. Exposure of the second sample to sunlight for 1 h then gave similar conversion to 2 as observed for the first sample. The experiment was repeatable, and the identity of 2 was confirmed by comparison to a standard.
To characterize this reaction further samples of 3 dissolved in methanol (225 μM) were exposed to several wavelengths of light (330, 375 and 405 nm) using a narrowband (ca. 5 nm) LED UV for several time intervals (10 to 160 s). The photochemical consumption of 3 follows first-order kinetics with significantly faster rates towards the ultraviolet range (Fig. 4 and Table 1) yielding 2 as the major product at all monitored wavelengths. To ascertain that the product of this reaction was 2 the product was purified and compared to standard material. Apparent quantum yields (Φapparent) were calculated from the linear phase (Table 1) of the reaction. Φapparent was the greatest at 330 nm, presumably because photons absorbed by 3 at this wavelength are more likely to be absorbed by the conjugated polyene and hence trigger isomerization. Calculations of Φapparent are based on the assumption that the initial absorbance is constant throughout the reaction. Despite this, the results are comparable across our experiments and demonstrate the rapid conversion of 3 to 2 at 330 nm. Prolonged exposure to shorter wavelengths led to the production of additional side products of identical m/z to 2 and 3. These are likely to be isomers of 3, as 2 does not absorb at the wavelengths tested.
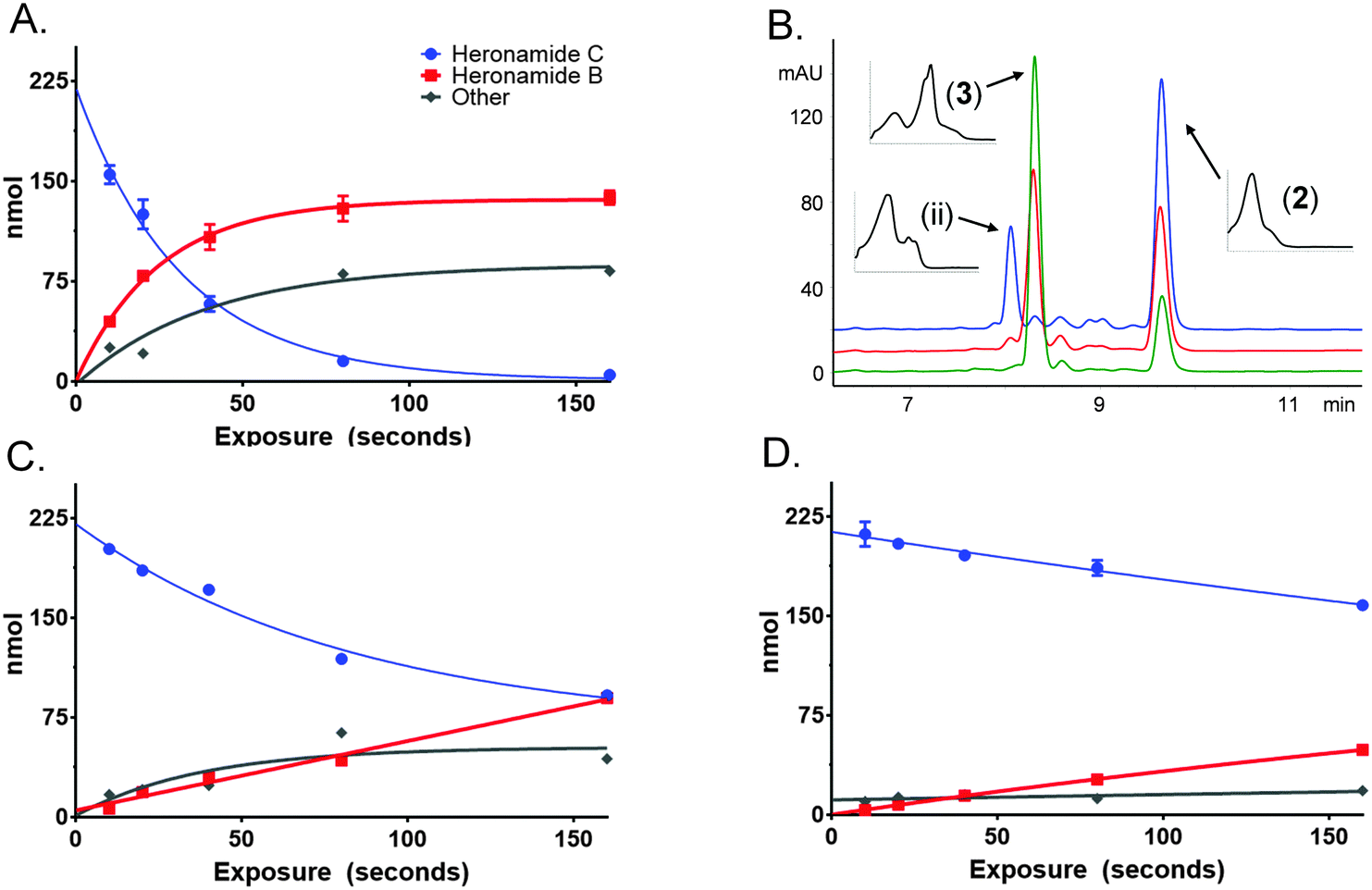 | ||
| Fig. 4 Photochemical conversion of heronamide C (3) to heronamide B (2) and the putative isomer (ii). The change in concentration of 2 (red) and 3 (blue) are shown over time at: (A) 330 nm, (C) 375 nm, and (D) 405 nm. Side products (grey) are inferred based on the amount of starting material. (B) HPLC trace following exposure of 3 for 160 s at 330 nm (blue), 375 nm (red) and 405 nm (green). | ||
| Wavelength, λ (nm) | k (s−1) | t 1/2 (s) | Photons delivered to cuvette, q (mol s−1) | Power delivered to cuvette, P (mW) | UV-Vis absorption 225 μM heronamide C, A | 1 × 10−A, χ | Time, t (s) | qχt, Nphotons (mol) | Heronamide B, Nmole (mol) | Φ apparent |
|---|---|---|---|---|---|---|---|---|---|---|
| 330 | 0.030 | 22 | 7.889 × 10−9 | 2.86 | 0.95 | 0.89 | 10 | 7.004 × 10−8 | 4.4775 × 10−8 | 0.6393 |
| 20 | 1.401 × 10−7 | 7.8975 × 10−8 | 0.5638 | |||||||
| 375 | 0.005 | 134 | 7.819 × 10−8 | 24.93 | 0.34 | 0.54 | 10 | 4.245 × 10−7 | 6.3 × 10−9 | 0.0148 |
| 20 | 8.490 × 10−7 | 1.8675 × 10−8 | 0.0220 | |||||||
| 405 | 0.002 | 337 | 7.114 × 10−7 | 210.02 | 0.31 | 0.51 | 10 | 3.630 × 10−6 | 3.6 × 10−9 | 0.0010 |
| 20 | 7.260 × 10−6 | 7.425 × 10−9 | 0.0010 |
The study of pericyclic reactions involved in biosynthetic pathways is an area of significant growing interest.14,15 Particular attention has been paid to the [4π+2π] Diels–Alder cycloaddition16 with the strongest data in support of a true Diels–Alderase being reported in 2011 for SpnJ,17 an enzyme required for the biosynthesis of the insecticidal spinosyn family of bacterial polyketides. It is plausible, however, that the majority of pericyclic reactions which occur during biosynthetic pathways occur spontaneously once a reactive intermediate structure has been assembled.14,15,18 It is likely that for these examples stereochemical induction occurs due to the steric constraints of an asymmetric environment, but without catalysis occurring in the sense of transition state stabilization and rate enhancement. It may be viewed that such processes provide a resource efficient mechanism for diversity orientated biosynthesis, and that the products themselves may be further modified by additional biosynthetic enzymes. Indeed, deliberately exposing natural or synthetic polyenes structurally related to 3 to a range of oxidative and photochemical conditions may represent a route for expanding molecular diversity.
A natural example of this logic is found for the marine polyketides heronamide A (1) and B (2) whose biosynthesis originates from the precursor molecule heronamide C (3). We have now shown that these reactions occur via pathways consistent with a thermal [6π+4π] cycloaddition to yield 1 or a photochemical [6π+6π] cycloaddition to yield 2 without the involvement of biological catalysts. The stereo- and regiospecificity of the olefin oxidation required during the biosynthesis of 1 is particularly noteworthy and may provide important insights for synthetic chemistry. Given the large number of polyene macrolactam natural products related to 3 which have been reported, we envisage that many additional compounds arising from similar reactions remain to be discovered.
T. J. B. is supported by a Norwich Research Park Biotechnology and Biological Sciences Research Council, UK (BBSRC) Doctoral Training Program Studentship BB/JO14524/1. S. A. is funded by the BBSRC via Institute Strategic Programme Grant BB/J004561/1 to the John Innes Centre. We thank Dr Lu Shin Wong, Professor Jason Micklefield and Dr Sarah Shepherd at the Manchester Institute of Biotechnology for access to the LED UV photoreactor and associated facilities and for helpful comments. We acknowledge The Genome Analysis Centre (TGAC, Norwich UK) for sequencing and assembly of the Streptomyces sp. CMB-0406 genome.
Notes and references
- R. Raju, A. J. Piggott, M. M. Conte and R. J. Capon, Org. Biomol. Chem., 2010, 8, 4682–4689 CAS.
- R. Sugiyama, S. Nishimura and H. Kakeya, Tetrahedron Lett., 2013, 54, 1531–1533 CrossRef CAS.
- K. Sankanishi, S. Itoh, R. Sugiyama, S. Nishimura, H. Kakeya, Y. Iwabuchi and N. Kanoh, Eur. J. Org. Chem., 2014, 1376–1380 CrossRef.
- R. Sugiyama, S. Nishimura, N. Matsumori, Y. Tsunematsu, A. Hattori and H. Kakeya, J. Am. Chem. Soc., 2014, 136, 5209–5212 CrossRef CAS PubMed.
- P. Yu, A. Patel and K. N. Houk, J. Am. Chem. Soc., 2015, 137, 13518–13523 CrossRef CAS PubMed.
- D. K. Derewacz, B. C. Covington, J. A. McLean and B. O. Bachmann, ACS Chem. Biol., 2015, 10, 1998–2006 CrossRef CAS PubMed.
- H. Jørgenson, K. F. Degnes, A. Dikiy, E. Fjærvik, G. Klinkenberg and S. B. Zotchev, Appl. Environ. Microbiol., 2009, 76, 283–293 CrossRef PubMed.
- H. Jørgenson, K. F. Degnes, H. Sletta, E. Fjærvik, A. Dikiy, L. Ferfindal, P. Bruheim, G. Klinkenberg, H. Bredholt, G. Nygård, S. O. Døskeland, T. E. Ellingsen and S. B. Zotchev, Chem. Biol., 2009, 16, 1109–1121 CrossRef PubMed.
- T. Weber, K. Blin, S. Duddela, D. Krug, H. U. Kim, R. Bruccoleri, S. Y. Lee, M. A. Fischbach, R. Muller, W. Wohlleben, R. Breitling, E. Takano and M. H. Medema, Nucleic Acids Res., 2015, 43, 237–243 CrossRef PubMed.
- P. Caffrey, ChemBioChem, 2003, 4, 654–657 CrossRef CAS PubMed.
- A. T. Keatinge-Clay, Nat. Prod. Rep., 2016, 33, 141–149 RSC.
- Y. Zhu, W. Zhang, Y. Chen, C. Yuan, H. Zhang, G. Zhang, L. Ma, Q. Zhang, X. Tian, S. Zhang and C. Zhang, ChemBioChem, 2015, 14, 2086–2093 CrossRef PubMed.
- R. W. Rickard, R. M. Smith and B. T. Golding, J. Antibiot., 1970, 23, 603–612 CrossRef.
- E. M. Stocking and R. M. Williams, Angew. Chem., Int. Ed., 2003, 42, 3078–3115 CrossRef CAS PubMed.
- C. M. Beaudry, J. P. Malerich and D. Trauner, Chem. Rev., 2005, 105, 4757–4778 CrossRef CAS PubMed.
- K. Klas, S. Tusukamoto, D. H. Sherman and R. M. Williams, J. Org. Chem., 2015, 80, 11672–11685 CrossRef CAS PubMed.
- H. J. Kim, M. W. Ruszczycky, S.-H. Choi, Y.-N. Liu and H.-W. Liu, Nature, 2011, 473, 109–112 CrossRef CAS PubMed.
- S. Alt and B. Wilkinson, ACS Chem. Biol., 2015, 10, 2468–2479 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental detail, additional data (tables and figures). See DOI: 10.1039/c6cc01930g |
| This journal is © The Royal Society of Chemistry 2016 |